

Abstract
Movement, cell division, protein biosynthesis, electron transfer against an electrochemical gradient, and many more processes depend on energy conversions coupled to the hydrolysis of ATP. The reduction of metal sites with low reduction potentials (E0′ < -500 mV) is possible by connecting an energetical uphill electron transfer with the hydrolysis of ATP. The corrinoid-iron/sulfur protein (CoFeSP) operates within the reductive acetyl-CoA pathway by transferring a methyl group from methyltetrahydrofolate bound to a methyltransferase to the [Ni-Ni-Fe4S4] cluster of acetyl-CoA synthase. Methylation of CoFeSP only occurs in the low-potential Co(I) state, which can be sporadically oxidized to the inactive Co(II) state, making its reductive reactivation necessary. Here we show that an open-reading frame proximal to the structural genes of CoFeSP encodes an ATP-dependent reductive activator of CoFeSP. Our biochemical and structural analysis uncovers a unique type of reductive activator distinct from the electron-transferring ATPases found to reduce the MoFe-nitrogenase and 2-hydroxyacyl-CoA dehydratases. The CoFeSP activator contains an ASKHA domain (acetate and sugar kinases, Hsp70, and actin) harboring the ATP-binding site, which is also present in the activator of 2-hydroxyacyl-CoA dehydratases and a ferredoxin-like [2Fe-2S] cluster domain acting as electron donor. Complex formation between CoFeSP and its activator depends on the oxidation state of CoFeSP, which provides evidence for a unique strategy to achieve unidirectional electron transfer between two redox proteins.
Energy transduction is fundamental for life. Aerobic and anaerobic organisms depend on coupling ATP hydrolysis to movement, activation of metabolites, or peptide bond formation, among others. Several metal-containing enzymes, such as nitrogenase, radical-dependent β,α-dehydratases, the related benzoyl-CoA reductases, and different cobalamin-dependent methyltransferases are able to convert unreactive molecules by acting in a low-potential regime. The highly energetic electrons required for these reactions (1–3) are injected by ATPases that enable the transfer of electrons against the redox potential gradient, driven by the hydrolysis of ATP. Three different types of reductive metallo-ATPase have been described so far.
The enzyme nitrogenase is by reducing dinitrogen with six electrons to ammonia at the heart of the global nitrogen cycle (1, 4, 5). Nitrogenase consists of the dinitrogenase, also called MoFe protein for the predominant Mo-containing variant, and the dinitrogenase reductase, called Fe protein (1, 4–6). The Fe protein is a homodimer covalently linked through a [4Fe-4S] cluster bound within the dimer interface. Both monomers are able to bind and hydrolyze ATP in a cleft containing a P loop. For electron transfer (ET) between Fe and MoFe proteins to occur, the reduced Fe protein binds MgATP and forms a complex with the MoFe protein positioning the electron-donating [4Fe-4S] cluster and electron-accepting P cluster within the typical limits for physiological ET (< 15 Å) (1, 4, 7, 8). Hydrolysis of two ATP molecules initiates a one-electron transfer between both partners (9, 10). Conformational changes of the Fe protein induced by ATP hydrolysis are believed to act as switches for the association/dissociation of the Fe:MoFe protein complex and the delivery of electrons (8–11). The Fe protein is bifunctional and also acts as a molybdate/homocitrate insertase during the maturation of nitrogenase (5, 12).
Benzoyl CoA reductases and 2-hydroxyacyl CoA dehydratases rely on homologous metallo-ATPases to catalyze the reduction of benzoyl-CoA or the β/α-dehydration of 2-hydroxyacyl-CoA compounds via formation of ketyl radicals (2). The structure of the homodimeric activator of 2-hydroxyglutaryl-CoA dehydratase revealed a [4Fe-4S] cluster covalently linking the two monomers, on a first glance resembling the Fe protein (13). The structure of the activator also showed it to be a member of the ASKHA (acetate and sugar kinases/heat shock protein 70/actin) superfamily. ASKHA proteins catalyze phosphoryl transfers or hydrolysis of ATP in a variety of biological contexts and are distinct from the P loop containing switch-type NTPases to which the Fe protein of nitrogenase belongs (13, 14). The binding of two MgATP molecules to the reduced activator is supposed to induce a conformational change and drives formation of the complex with the dehydratase. ATP hydrolysis likely increases the reducing power of the reduced [4Fe-4S] cluster of the activator, enabling the one-electron transfer to the low-potential [4Fe-4S] cluster of the dehydratase (2, 15). Unlike the 2-hydroxyacyl-CoA dehydratase system, the reduction of benzoyl-CoA is a two-step ET requiring a stoichiometric consumption of ATP (3).
Recently, a third class of electron-transferring metallo-ATPases has been discovered (16–18). This enzyme class belongs to the COG3894 protein family and has been termed reductive activases for corrinoid enzymes (RACE) (17). The genome of several anaerobic microorganisms, which encode corrinoid-dependent methyltransferases and enzymes of the reductive acetyl-CoA pathway, also encode for proteins homologous to the two investigated RACE proteins with their characteristic binding motifs for one Fe/S cluster (17, 18). Bacterial RACE proteins typically show [2Fe-2S] cluster-binding-motifs, as in the veratrol O-demethylase system of Acetobacterium dehalogenans (16), whereas in archaea, as in the activator of the methylamine methyltransferase of the methanogenic archaeon Methanosarcina barkeri [4Fe-4S] cluster-binding motifs are more abundant (17, 18).
The anaerobic hydrogenogenic bacterium Carboxydothermus hydrogenoformans is able to convert CO2 into cellular carbon compounds via the reductive acetyl-CoA pathway (also known as the Wood–Ljungdahl pathway) (19–21). The corrinoid/iron-sulfur protein (CoFeSP) connects the methyl and carbonyl branch of this pathway by accepting a methyl group from methyltetrahydrofolate bound to a methyltransferase and donating it to the Ni,Fe-containing acetyl-CoA synthase (22, 23). Three redox states are known for the corrinoid cofactor of CoFeSP: The nucleophilic Co(I) acts as a methyl-acceptor, Co(II) is an oxidized inactive state, and CH3 - Co(III) acts as the methyl donor of acetyl-CoA synthase (22, 23). The occasional oxidation of Co(I) to Co(II) inactivates CoFeSP, which has to be reactivated by a one-electron reduction (23, 24). The low midpoint potential needed to reduce Co2+ to Co1+ (< -504 mV at pH 7.4) (25) can be achieved in vitro using either chemical reducing agents such as Na-dithionite (DT), Ti3+ citrate, via photoreduction with deazariboflavin as a catalyst or enzymatically with electrons generated by the oxidation of CO to CO2 by carbon monoxide dehydrogenase (22, 26). An ATP-dependent reactivation of CoFeSP has not been reported so far.
An open reading frame (orf7), situated between the structural genes coding for the CoFeSP subunits CfsA and CfsB of Moorella thermoacetica (27), codes for a member of the COG3894 protein family and contains the putative [2Fe-2S] cluster-binding motif CX5CX2CXnC (17, 18). The genome of C. hydrogenoformans contains a similar arrangement of genes coding for enzymes of the reductive acetyl-CoA pathway as M. thermoacetica, including a homolog of Orf7 (CHY_1224 assigned as COG3894). To test whether an ATP-dependent reductive activator is operative in the reductive acetyl-CoA pathway, we established the heterologous production of the Orf7 homolog and investigated its activity, structure, and selective complex formation with CoFeSP. Furthermore, we reveal its relationship to known ATPases including the activator of 2-hydroxyacyl-CoA dehydratases and compare its strategy to achieve unidirectional electron transport with the other types of ATP-dependent activators/reductases.
Results
Biochemical Characterization.
The Orf7 homolog from C. hydrogenoformans was heterologously produced in Escherichia coli and purified to homogeneity using three chromatography steps under anoxic conditions (Fig. S1). The UV-visible (UV-vis) spectrum of the as-isolated protein shows the typical spectrum of an oxidized [2Fe-2S] cluster containing ferredoxin with maxima at 350, 410, and 460 nm, and a shoulder at 550 nm (Fig. S2, solid line). Bleaching of the [2Fe-2S] cluster spectrum was observed in the presence of 2 mM DTT. The reduction was completed within 10 min (Fig. S2, dashed line). The non-heme iron was determined to be approximately 1–2 mol of iron per mole of monomer protein, depending on preparation. Disappearance of the characteristic features of the [2Fe-2S] cluster was observed after exposing the protein to air for 2 d.
ATPase Activity.
ATP hydrolysis by the Orf7 homolog was followed under anoxic conditions using a coupled ATPase assay (28). The ATPase activity was dependent on the presence of Mg2+ and K+ ions in the assay buffer. The steady-state kinetic constants for the catalyzed ATP hydrolysis of the wild-type and active site mutants (D212A, D377A, and E404A) were determined by measuring the rate of hydrolysis at various ATP concentrations (Fig. S3). The Michaelis constant Km for the wild-type protein was determined to be 0.39 ± 0.07 mM with a Vmax of 6.4 ± 0.3 μmol min-1 (mg protein)-1, corresponding to a kcat of 7.24 ± 0.30 s-1. The rate of ATP hydrolysis was not influenced by the presence of CoFeSP or air in the ATPase assay. The catalytic activity of the variant E404A was approximately 145-fold lower (kcat = 0.048 ± 0.002 s-1), whereas the Km increased slightly (Km = 0.81 ± 0.11 mM). The other two mutations, D212A and D377A, diminished the rate of hydrolysis with minor changes in the Km value (D212A: kcat = 0.16 ± 0.01 s-1; Km = 0.83 ± 0.10 mM; D377A: kcat = 0.37 ± 0.02 s-1, Km = 0.53 ± 0.04 mM).
ATP-Dependent Reduction of Co(II)–CoFeSP.
UV-vis spectroscopy was used to follow the reduction of the inactive Co(II) to the active Co(I) state of CoFeSP by the Orf7 homolog (Fig. 1). An addition of 2 mM DTT, a condition under which reduction of CoFeSP does not occur, was sufficient to reduce the Orf7 homolog. Electron transfer from the reduced Orf7 homolog to Co(II)-CoFeSP was not observed in the absence of ATP, but was initiated by adding ATP to the reaction mixture. Upon reduction of Co(II) to Co(I), the absorption at 475 nm decreased (Co2+) and a distinct band at 390 nm, characteristic for Co1+, appeared (22, 29) (Fig. 1A). The reduction of CoFeSP by the Orf7 homolog was strictly dependent on the presence of ATP and conditions suitable for ATP hydrolysis. Both Mg2+ and K+ had to be present in the assay to enable ET. At least equimolar concentrations of the Orf7 homolog were necessary for a complete reduction of CoFeSP. No formation of Co(I) was detectable when only catalytic amounts of the Orf7 homolog were used. The rate of ET increased with the ATP concentration up to 2 mM ATP (Fig. 1B). No ATP-induced reduction of Co(II)-CoFeSP was detectable when the Orf7 homolog variants (D212A, D377A, and E404A) were used in the ET assay. The rate of ET may be too slow to be detected by our optical assay.
Fig. 1.

ATP-dependent one-electron reduction of Co(II)-CoFeSP by its reductive activator. Assay solutions containing Co(II)-CoFeSP and the activator were prepared as described in SI Materials and Methods. (A) ET from DTT-reduced RACo to Co(II)-CoFeSP was monitored by UV-vis spectroscopy. To initiate the ET, 0.5 mM ATP was added. The reduction of Co(II)- to Co(I)-CoFeSP is indicated by the decrease in absorption at 475 nm and the increase in absorption at 390 nm. (B) ATP dependency of Co(I)-CoFeSP formation. The ET was initiated by adding different concentrations of ATP. Observed rate constants for ET were determined by single-exponential fits to be 0.089 min-1 with 0.1 mM ATP, 0.150 min-1 with 0.5 mM ATP, and 0.288 min-1 with 2 mM ATP.
As the Orf7 homolog is able to activate Co(II)-CoFeSP, in the following text we named it reductive activator of CoFeSP (RACo) or “activator” for short.
Formation of the Activator:CoFeSP Complex.
Analytical size-exclusion chromatography was used to investigate the interaction between CoFeSP and RACo. As shown in Fig. 2A, the individual CoFeSP and RACo preparations showed distinct peaks at defined elution volumes. CoFeSP eluted at 77.5 mL (86 kDa) corresponding to a heterodimer composed of the CfsA and CfsB subunits. The molecular size of RACo was determined to be 158 kDa (elution volume of 68.3 mL), consistent with a homodimeric state in solution.
Fig. 2.

Complex formation between CoFeSP and its activator. (A) Gel filtration profiles of CoFeSP (11 µmol) and RACo (29 µmol). The elution profiles show peaks with molecular masses of 158 (RACo) and 86 kDa (CoFeSP). (B) Redox state-dependent complex formation. Different oxidation states of CoFeSP were generated (see SI Materials and Methods) and mixed with RACo. Dashed line, Co(II)-CoFeSP with activator; solid line, Co(I)-CoFeSP with activator; dotted line, methylated CoFeSP (Co(III)-CH3) with activator. (C) SDS-PAGE analysis of the main peak fractions. The first lane shows the molecular mass marker with the indicated molecular masses given in kilodaltons. Peak fractions and corresponding protein bands are labeled.
We then investigated the formation of a stable activator:CoFeSP complex by analytical gel filtration after incubating the Co(II) state of CoFeSP with DTT-reduced RACo. A new symmetrical peak at an elution volume of 60.3 mL was observed, which is clearly separated from the peaks obtained for the individual proteins (Fig. 2B, dashed-line). The elution volume of this peak corresponds to a molecular weight of 280 kDa. For further investigation, fractions of the distinct peaks were collected and analyzed by SDS-PAGE, as shown in Fig. 2C. The fractions corresponding to the peak with an elution volume of 60.3 mL clearly display three bands representing the activator (68 kDa monomer) and the two subunits of CoFeSP (48 and 34 kDa), whereas the other two peaks with elution volumes of 68.3 and 77.5 mL contain mainly the individual proteins. Densitometric analysis of SDS-PAGE gels indicates that approximately one molecule of RACo forms a complex with one molecule of CoFeSP. The results obtained from SDS-PAGE and size-exclusion chromatography (Fig. 2 B and C) are thus in excellent agreement with a 2∶2∶2 stoichiometry of CfsA, CfsB, and RACo with a theoretical molecular mass of 300 kDa. To check whether complex formation discriminates between active and inactive states of CoFeSP, we performed gel filtration experiments of RACo with the two active states of CoFeSP, namely the methylated (CH3-Co(III)-CoFeSP) and the reduced Co(I) state. When CH3-Co(III)-CoFeSP was incubated with the DTT-reduced RACo, only about 14% of the complex observed with Co(II)-CoFeSP (Fig. 2B, dashed line) was detected (Fig. 2B, dotted line). We cannot exclude that the sensitive Co-carbon bond became partly photolysed and the small amount of complex observed may be due to a conversion of CH3-Co(III)-CoFeSP to Co(II)-CoFeSP.
After incubating RACo with Co(I)-CoFeSP, prepared by reducing Co(II)CoFeSP with 1 mM DT, no peak in the region of 60 mL indicating complex formation was observed (Fig. 2B, solid line). These results show that the formation of a stable complex critically depends on the redox state of CoFeSP, which is strongly favored for the inactive Co(II)-CoFeSP state.
Furthermore, we tested whether the formation of the RACo:CoFeSP complex depends on the redox state of RACo or nucleotides present in the assay (Fig. S4). Complex formation between RACo and Co(II)-CoFeSP was observed irrespectively of the redox state of RACo (Fig. S4B, dashed line). The presence or absence of the nucleotides ATP and ADP did not have any influence on complex formation (Fig. S4B, dotted and solid lines, respectively).
Overall Structure of RACo.
We solved the crystal structure of the activator by single isomorphous replacement combined with anomalous scattering (SIRAS) methods using a HgCl2-derivatized crystal diffracting to 2.35 Å with space group P212121. A first protein model was built and refined (Table S1), however this model lacked the N-terminal region (residues 1–108) for both monomers present in the asymmetric unit (ASU). Another crystal form with an approximately 12-Å longer a axis having the same crystal symmetry was obtained and refined at 2.2 Å to a crystallographic R factor of 22% and Rfree factor of 26% (Table S2). From the two molecules found in the ASU, one molecule could be built and refined, including residues 3–630 containing the N-terminally bound [2Fe-2S] cluster (referred to as subunit A), whereas in the other molecule, residues 1–108 were again disordered (referred to as subunit B) (Fig. 3). The structure of RACo can be divided into four domains: the N-terminal domain (residues 3–94) binding the [2Fe-2S] cluster, a linker domain (residues 95–125), the middle domain (residues 126–206), and the large C-terminal domain (residues 207–630) as illustrated in Fig. 3A.
Fig. 3.

Crystal structure of the activator. (A) Overall structure. The two molecules of RACo in the asymmetric unit of the crystal are presented as cartoons. The left molecule (subunit A) contains all domains of the activator including the N-terminal domain (green), the linker domain (red), the middle domain (yellow), and the ASKHA domain (blue). The second monomer (subunit B) is colored in light-pink and does not include the N-terminal domain in its structure. Magnesium and phosphate ions are indicated as green and gray spheres, respectively. The [2Fe-2S] cluster is shown as spheres colored red for iron and yellow for sulfur. The distance between the ATP-binding site and the [2Fe-2S] cluster is indicated by an orange arrow. (B) Cartoon-representation of the ASKHA domain (residues 207–630) is shown on the left. The conserved βββαβαβα topology of the ASKHA family is illustrated with β-strands in magenta and α-helices in marine. Surface representation of the four lobes of the ASKHA domain (IA, IB, IIA, and IIB) in analogy to the ASKHA family proteins is given on the right. (C) Nucleotide-binding site. Mg2+ and  ions are shown as spheres and stick model. Five water molecules completing the octahedral coordination of the Mg2+ ion are displayed as red spheres. Residues in H-bonding distance (< 3.6 Å) are colored as in B (Left) for main-chain atoms. The steady-state kinetic constants, Km and kcat, for ATP hydrolysis of the wild type and the variants are shown.
ions are shown as spheres and stick model. Five water molecules completing the octahedral coordination of the Mg2+ ion are displayed as red spheres. Residues in H-bonding distance (< 3.6 Å) are colored as in B (Left) for main-chain atoms. The steady-state kinetic constants, Km and kcat, for ATP hydrolysis of the wild type and the variants are shown.
A first search for structural homologs of RACo using the DALI server (30) did not reveal any obvious hits. But when only the C-terminal domain was used, several similar proteins were identified, including the heat shock protein 70 (Hsp70) [Protein Data Bank (PDB) ID 3L6Q], the cell division protein FtsA (PDB ID 1E4G), the anhydro-N-acetylmuramic acid kinase (PDB ID 3QBW), the chaperone DnaK (PDB ID 2V7Y), the heat shock cognate 70 protein (Hsc70) (PDB ID 3L6Q), actin (PDB ID 1H1V), and the reductive activator of (R)-2-hydroxyglutaryl CoA dehydratase (PDB ID 1HUX), with DALI Z scores of 12–14 (Table S3). All of these structural homologs are members of the ASKHA superfamily, demonstrating that RACo is a member of the ASKHA family. We therefore term the C-terminal domain of RACo the ASKHA domain. The structure of the N-terminal domain containing the [2Fe-2S] cluster of the activator resembles the plant-type ferredoxins with DALI Z scores of 13 and 14 (Table S4). No homologous structures were found for the middle domain.
N-Terminal and Linker Domain.
The N-terminal domain binding the [2Fe-2S] cluster stands apart from the other three domains and shows the characteristic topology of the β-grasp found in plant-type [2Fe-2S] cluster ferredoxins (31), consisting of a β-sheet with five β-strands (S1–S3, S5, and S7) and an α-helix (H1) on top of the sheet (Fig. S5A). Two antiparallel β-strands (S4 and S6), one α-helix (H2), and a short 310 helix (H3) surround the central core. The position of the [2Fe-2S] cluster was confirmed using the anomalous signal from the iron atoms (Fig. S5A). The [2Fe-2S] cluster is located at the outer surface of the N-terminal domain and is in a distance of approximately 40 Å from the ATP-binding site of the ASKHA domain (Fig. 3A). The conserved cysteine motif C38-X5-C44-X2-C47-X29-C77 coordinates the two iron atoms, where three cysteines (Cys38, Cys44, and Cys47) are present in the loop region and Cys77 is part of the 310 helix (H3). The N-terminal domain displays a weaker electron density and on average higher B factors than the rest of the protein (Table S2), indicating a higher mobility of this domain.
The linker domain connects the N-terminal domain to the middle domain (Fig. S5C). Hydrogen bonds centered around Glu105 stabilize H4 by interaction with S8 of the linker domain (Fig. S5C). Residues Glu113 and Glu118 interact directly with residues of the ASKHA domain.
Middle Domain and Dimerization.
The middle domain includes a β-sheet of three antiparallel β-strands (S9, S11, and S12), an α-helix (H5), a short β-strand (S10) on top of the β-sheet, and two α-helices (H6 and H7) in close proximity (Fig. S6A). Dimerization is primarily mediated by residues from two α-helices (H6 and H7) of the middle domain with subdomain I of the ASKHA domain (Fig. S6B). Additionally, Glu163 of H6 is interacting with Tyr188 of the middle domain from subunit B. Nineteen hydrogen-bonding interactions between side chains and backbone atoms and via water molecules are possible (Fig. S6B). Only 7% of the overall accessible surface area of 45,487 Å2 are involved in dimerization of the activator.
Nucleotide Binding Site Within the ASKHA Domain.
Despite the low-sequence identity shared between members of the ASKHA family, they show a common central fold consisting of two domains with βββαβαβα topology (14). The architecture of the ASKHA domain resembles the structure of the common core motif of this superfamily, which forms an interdomain cleft where nucleotide binding and hydrolysis occur (Fig. 3B, Left and Fig. S7). Following the nomenclature originally proposed for Hsc70 (14), we divide the ASKHA domain of RACo into two large subdomains, I and II (Fig. 3B, Right). Each of the two subdomains consists of a five-stranded β-sheet surrounded by three α-helices with a β1β2β3α1β4α2β5α3 topology. As in Hsc70, the subdomains I and II can be further divided into the four lobes IA, IB, IIA, and IIB (Fig. 3B, Right). The two lobes IA and IB are separated by functionality rather than structure. Whereas lobe IA is a part of the core fold responsible for nucleotide binding and hydrolysis, lobe IB comprises a short region between the C terminus of H12 and the N terminus of H14 (26 residues) responsible for dimerization (Fig. 3B). Lobe IIB is positioned near the N terminus of H21 and consists of two nearly perpendicular two-stranded antiparallel β-sheets (S22–S26), surrounded by the four α-helices H17–H20 (Fig. 3B and Fig. S7).
We observed strong additional electron density in the cleft between subdomain I and II, which was modeled as a phosphate ion associated with a Mg2+ ion with its typical octahedral coordination by water molecules (Fig. 3C). The oxygen atoms of the phosphate ion are in hydrogen-bonding distance to the NH2 group of Asn298 and backbone atoms of Thr215, Asn380, and Ile381. The water molecules coordinating the magnesium ion are in hydrogen-bonding distance to the carboxylate groups of Asp212, Asp356, Asp377, Glu404, and the carbonyl oxygen of Asn572 (Fig. 3C). Asp, Asn, Glu, and Gln residues are well conserved in the core of the ASKHA proteins, where they interact with the phosphates of ATP and the bound Mg2+ ions (14).
Discussion
CoFeSP is an unusual corrinoid containing proteins because it occurs in the base-off/His-off coordination and has no protein ligand coordinating the cobalt ion (22, 32). The absence of protein ligands is the probable reason why the redox potential of the Co2+/Co1+ couple of CoFeSP is about 50–100 mV more positive than in other corrinoid containing methyltransferases (25, 33), rendering it reducible by electrons generated from the oxidation of CO by carbon monoxide dehydrogenases (26). The necessity for an ATP-dependent reductive activator has therefore not been anticipated. Genes coding for central enzymes of the reductive acetyl-CoA pathway are found in the genomes of various anaerobic microbes including firmicutes (mostly clostridia), δ-proteobacteria, and methanogenic euryarchaeotes. RACo and CoFeSP encoding genes are typically found in close proximity in the firmicutes and δ-proteobacteria, which include the acetogenic, hydrogenogenic, and sulfate-reducing bacteria. In euryarchaeota, RACo homologs are either found not near the CoFeSP genes or, as in the species belonging to Methanosaeta, Methanothermobacter, and Methanocaldococcus, are missing in the genome, which indicates that, although most microbes using the reductive acetyl-CoA pathway rely on a RACo-related ATPase to reactivate Co(II)-CoFeSP, other ways of CoFeSP reactivation probably exist. Alternative mechanisms to generate low-potential electrons may be operative in these bacteria, like a membrane-associated reversed electron transport (34) or a flavin-based electron bifurcation (35–38).
Our biochemical studies revealed RACo not only to be a homolog of RamA from Methanosarcina barkeri (18) and the activator of veratrol-O-demethylase from Acetobacterium dehalogenans (16, 17), but it is also functionally related to these two proteins and acts as an ATP-dependent reductive activator for CoFeSP. The structure of the activator presented here demonstrates that the RACE proteins belong to the ASKHA-type ATPases. Despite the structural homology, the activator does not show obvious sequence homologies to other members of the ASKHA family. Amino acid alignments based on a superimposition of selected members including RACo reveal sequence identities between 10–15%, which are judged as unreliable to identify homology based upon pairwise sequence comparison without the aid of structural information (14) (Table S3). In contrast, the homology of the N-terminal domain carrying the [2Fe-2S] cluster with plant-type [2Fe-2S] cluster ferredoxins is not only obvious from sequence identities (28–30%) but also from structural conservation (rmsd values for Cα atoms: 1.9–2.1 Å) (Table S4). This modular architecture, which separates the ATP-binding site and the iron-sulfur cluster domain is presumably also to be found in RamA and related enzymes with iron-sulfur cluster-binding motifs in their C-terminal domain (18).
One major challenge for any ATPase-coupled uphill ET is to assure that the transfer of the electron(s) is unidirectional. To achieve unidirectional ET from the Fe protein to the MoFe protein in nitrogenase, the Fe protein uses a molecular choreography based on nucleotide-dependent switch regions (39, 40). In the presence of ATP or the putative transition state analog 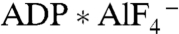 the electron-donating [4Fe-4S] cluster on the Fe protein shifts by 5 Å closer to the electron-accepting P cluster on the MoFe protein. The nucleotide-dependent complex formation between the Fe protein and the MoFe protein combined with this switch is the probable driving force in N2 reduction by nitrogenase (11, 39, 40). Complex formation between 2-hydroxyacyl-CoA dehydratases and their activators has been observed in the presence of
the electron-donating [4Fe-4S] cluster on the Fe protein shifts by 5 Å closer to the electron-accepting P cluster on the MoFe protein. The nucleotide-dependent complex formation between the Fe protein and the MoFe protein combined with this switch is the probable driving force in N2 reduction by nitrogenase (11, 39, 40). Complex formation between 2-hydroxyacyl-CoA dehydratases and their activators has been observed in the presence of 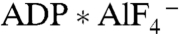 but not in the presence of ADP (41), and additional conformational changes in analogy to nitrogenase have been proposed (2). Thus nucleotide-dependent adaptations of the structure are linked to complex affinities, which likely drive the unidirectional ET in nitrogenase and the 2-hydroxyacyl-CoA dehydratases.
but not in the presence of ADP (41), and additional conformational changes in analogy to nitrogenase have been proposed (2). Thus nucleotide-dependent adaptations of the structure are linked to complex affinities, which likely drive the unidirectional ET in nitrogenase and the 2-hydroxyacyl-CoA dehydratases.
The presence or absence of nucleotides does not influence the formation of the complex between CoFeSP and RACo. In contrast, RACo has a high affinity for its substrate [Co(II)-CoFeSP] but a low affinity for the product [Co(I)-CoFeSP], and thus uses substrate specificity to favor one direction of the reaction. The apparent strict dependence on the oxidation state of CoFeSP to rule complex formation appears as a unique way to drive electron transfer from reduced RACo to Co(II)-CoFeSP. The back ET from Co(I)-CoFeSP to the oxidized RACo is avoided by complex dissociation, separating the electron donor–acceptor couple.
Redox-dependent complex formation distinguishes RACo from the other known ATP-dependent activators. As the three oxidation-states of the cobalamin will only differ slightly in the structure of the corrin ring, a direct interaction of RACo with the corrinoid cofactor of CoFeSP appears mandatory for the specific recognition of Co(II)-CoFeSP. The crystal structure of Co(II)-CoFeSP shows that the corrin ring is sandwiched between the C-terminal domain of the large subunit and the small subunit with both axial coordination sites of Co shielded off by the protein matrix (22, 32). A readout of the oxidation state appears to require a direct interaction of RACo with the corrin ring, making conformational changes of CoFeSP necessary when forming the encounter complex with RACo. This readout must determine whether the encounter complex progresses into the formation of a stable complex, if Co(II)-CoFeSP specific interactions can be established. Conformational changes are thus likely at the heart of complex formation and ET. Linker regions typically control the flexibility of domains and a long stretch is found between the N-terminal domain of RACo and the ASKHA domain. This linker domain is only present in RACE protein sequences with the N-terminal [2Fe-2S] cluster domain and is absent in the RamA-like RACE proteins (14, 15), suggesting that the linker domain and the N-terminal domain form one functional unit.
The CoFeSP activator is also distinct from the Fe protein and the dehydratase activator by its modular architecture (2, 13). In these proteins, the electron-donating [4Fe-4S] clusters are not found in an isolated domain like in RACo, but are directly linked to the ATPase site. The dehydratase activator and RACo are both members of the ASKHA-type ATPases (13), but have only the ASKHA core domain in common. The [4Fe-4S] clusters of the activator of dehydratases and the nitrogenase reductase are symmetrically coordinated by both monomers within the dimer interface, and ATP-dependent movements of the monomers against each other are believed to change the potential of the iron-sulfur cluster and trigger the ET (2). The CoFeSP activator is also a homodimer, but harbors not one but two iron-sulfur clusters, which appear to act independently from each other. A movement of the two RACo monomers is therefore unlikely to change the potential of the [2Fe-2S] cluster. Although in the nitrogenase reductase and the activator of dehydratases the dimers appear to be the functional unit, in RACo both monomers seem to act independently. This difference between a dimer versus a monomer as functional unit is also reflected in the stoichiometry between ATPase and electron-accepting protein. Whereas RACo forms a ternary complex with two CoFeSP molecules per activator dimer, one ATPase dimer of nitrogenase reductase/dehydratase activator binds to and activates one target electron acceptor. Nitrogenase reductase and the dehydratase activator are able to bridge a potential gap of 400–500 mV by hydrolyzing two ATP molecules, whereas the RACE ATPases only need to overcome a barrier of 200–300 mV, and thus the energy provided by hydrolysis of one ATP molecule per electron would be sufficient. The electron/ATP stoichiometry has not yet been determined, but we anticipate a 1∶1 ratio based on the architecture of the monomers. We clearly established that ET is initiated by ATP addition and our mutational studies further revealed that ATP hydrolysis is necessary for ET and that ATP binding alone will not suffice. However, the rate of ATP hydrolysis in our assays is about three orders of magnitude higher than the rate of ET, indicating a high nonproductive ATP hydrolysis, which appears wasteful and uncoupled from ET. We thus have not yet established the proper conditions for a coupled and efficient in vitro ET.
Our biochemical and structural characterization presented above revealed an ATP-dependent reductive activator with unique properties and redox-selective partner recognition to achieve unidirectional ET. The long distance between the ATP-binding site and the [2Fe-2S] cluster makes a direct coupling of ATP hydrolysis and ET unlikely and thus the major question, how ATP hydrolysis is coupled to ET, remains currently unresolved. Further experiments will have to clarify how the energy of ATP is used to enable uphill ET and to show the route the electrons take from the [2Fe-2S] cluster of the activator to the Co2+-corrinoid of CoFeSP.
Materials and Methods
The gene encoding RACo was cloned from genomic DNA of C. hydrogenoformans and RACo was produced in E. coli and purified with classical chromatographic steps under anoxic conditions. ATPase activity was determined in a coupled enzyme assay. ATP-dependent ET from RACo to Co(II)FeSP was initiated by ATP addition and followed absorption changes at 475 and 390 nm. Complex formation of RACo and CoFeSP was followed by gel filtration chromatography. The structure of RACo crystal was determined using SIRAS phasing and refined using standard crystallographic methods. See SI Materials and Methods for additional details.
Supplementary Material
Acknowledgments.
Scientists at beamline BL14.2 (BESSY-II, Berlin, Germany) are acknowledged for help in data collection. The German Research Foundation funded the project (DFG–DO 785/5-1).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank Europe, www.ebi.ac.uk/pdbe/ (PDB ID code 3ZYY).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1117126109/-/DCSupplemental.
References
- 1.Seefeldt LC, Hoffman BM, Dean DR. Mechanism of Mo-dependent nitrogenase. Annu Rev Biochem. 2009;78:701–722. doi: 10.1146/annurev.biochem.78.070907.103812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buckel W, Hetzel M, Kim J. ATP-driven electron transfer in enzymatic radical reactions. Curr Opin Chem Biol. 2004;8:462–467. doi: 10.1016/j.cbpa.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Boll M, Fuchs G. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur J Biochem. 1995;234:921–933. doi: 10.1111/j.1432-1033.1995.921_a.x. [DOI] [PubMed] [Google Scholar]
- 4.Hu Y, Ribbe MW. Decoding the nitrogenase mechanism: The homologue approach. Acc Chem Res. 2010;43:475–484. doi: 10.1021/ar900254x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rees DC, et al. Structural basis of biological nitrogen fixation. Philos Transact A Math Phys Eng Sci. 2005;363:971–984. doi: 10.1098/rsta.2004.1539. [DOI] [PubMed] [Google Scholar]
- 6.Schwarz G, Mendel RR, Ribbe MW. Molybdenum cofactors, enzymes and pathways. Nature. 2009;460:839–847. doi: 10.1038/nature08302. [DOI] [PubMed] [Google Scholar]
- 7.Barney BM, et al. Breaking the N2 triple bond: Insights into the nitrogenase mechanism. Dalton Trans. 2006;21:2277–2284. doi: 10.1039/b517633f. [DOI] [PubMed] [Google Scholar]
- 8.Igarashi RY, Seefeldt LC. Nitrogen fixation: The mechanism of the Mo-dependent nitrogenase. Crit Rev Biochem Mol Biol. 2003;38:351–384. doi: 10.1080/10409230391036766. [DOI] [PubMed] [Google Scholar]
- 9.Danyal K, Mayweather D, Dean DR, Seefeldt LC, Hoffman BM. Conformational gating of electron transfer from the nitrogenase Fe protein to MoFe protein. J Am Chem Soc. 2010;132:6894–6895. doi: 10.1021/ja101737f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard JB, Rees DC. Nitrogenase: A nucleotide-dependent molecular switch. Annu Rev Biochem. 1994;63:235–264. doi: 10.1146/annurev.bi.63.070194.001315. [DOI] [PubMed] [Google Scholar]
- 11.Schindelin N, Kisker C, Schlessman JL, Howard JB, Rees DC. Structure of ADP AIF4-stabilized nitrogenase complex and its implications for signal transduction. Nature. 1997;387:370–376. doi: 10.1038/387370a0. [DOI] [PubMed] [Google Scholar]
- 12.Hu Y, et al. Nitrogenase Fe protein: A molybdate/homocitrate insertase. Proc Natl Acad Sci USA. 2006;103:17125–17130. doi: 10.1073/pnas.0602651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Locher KP, et al. Crystal structure of the Acidaminococcus fermentans 2-hydroxyglutaryl-CoA dehydratase component A. J Mol Biol. 2001;307:297–308. doi: 10.1006/jmbi.2000.4496. [DOI] [PubMed] [Google Scholar]
- 14.Hurley JH. The sugar kinase/heat shock protein 70/actin superfamily: Implications of conserved structure for mechanism. Annu Rev Biophys Biomol Struct. 1996;25:137–162. doi: 10.1146/annurev.bb.25.060196.001033. [DOI] [PubMed] [Google Scholar]
- 15.Knauer SH, Buckel W, Dobbek H. Structural basis for reductive radical formation and electron recycling in (R)-2-hydroxyisocaproyl-CoA dehydratase. J Am Chem Soc. 2011;133:4342–4347. doi: 10.1021/ja1076537. [DOI] [PubMed] [Google Scholar]
- 16.Siebert A, Schubert T, Engelmann T, Studenik S, Diekert G. Veratrol-O-demethylase of Acetobacterium dehalogenans: ATP-dependent reduction of the corrinoid protein. Arch Microbiol. 2005;183:378–384. doi: 10.1007/s00203-005-0001-8. [DOI] [PubMed] [Google Scholar]
- 17.Schilhabel A, et al. The ether-cleaving methyltransferase system of the strict anaerobe Acetobacterium dehalogenans: Analysis and expression of the encoding genes. J Bacteriol. 2009;191:588–599. doi: 10.1128/JB.01104-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson T, et al. RamA, a protein required for reductive activation of corrinoid-dependent methylamine methyltransferase reactions in methanogenic archaea. J Biol Chem. 2009;284:2285–2295. doi: 10.1074/jbc.M807392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu M, et al. Life in hot carbon monoxide: The complete genome sequence of Carboxydothermus hydrogenoformans Z-2901. PLoS Genet. 2005;1:e65. doi: 10.1371/journal.pgen.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Svetlitchnyi V, Peschel C, Acker G, Meyer O. Two membrane-associated NiFeS-carbon monoxide dehydrogenases from the anaerobic carbon-monoxide-utilizing eubacterium Carboxydothermus hydrogenoformans. J Bacteriol. 2001;183:5134–5144. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Svetlitchnyi V, et al. A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc Natl Acad Sci USA. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goetzl S, Jeoung JH, Hennig SE, Dobbek H. Structural basis for electron and methyl-group transfer in a methyltransferase system operating in the reductive acetyl-CoA pathway. J Mol Biol. 2011;411:96–109. doi: 10.1016/j.jmb.2011.05.025. [DOI] [PubMed] [Google Scholar]
- 23.Ragsdale SW, Pierce E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta. 2008;1784:1873–1898. doi: 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menon S, Ragsdale SW. The role of an iron-sulfur cluster in an enzymatic methylation reaction. Methylation of CO dehydrogenase/acetyl-CoA synthase by the methylated corrinoid iron-sulfur protein. J Biol Chem. 1999;274:11513–11518. doi: 10.1074/jbc.274.17.11513. [DOI] [PubMed] [Google Scholar]
- 25.Harder SR, Lu WP, Feinberg BA, Ragsdale SW. Spectroelectrochemical studies of the corrinoid/iron-sulfur protein involved in acetyl coenzyme A synthesis by Clostridium thermoaceticum. Biochemistry. 1989;28:9080–9087. doi: 10.1021/bi00449a019. [DOI] [PubMed] [Google Scholar]
- 26.Menon S, Ragsdale SW. Role of the [4Fe-4S] cluster in reductive activation of the cobalt center of the corrinoid iron-sulfur protein from Clostridium thermoaceticum during acetate biosynthesis. Biochemistry. 1998;37:5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- 27.Loke HK, Lindahl PA. Identification and preliminary characterization of AcsF, a putative Ni-insertase used in the biosynthesis of acetyl-CoA synthase from Clostridium thermoaceticum. J Inorg Biochem. 2003;93:33–40. doi: 10.1016/s0162-0134(02)00457-9. [DOI] [PubMed] [Google Scholar]
- 28.Lindsley JE. Use of a real-time, coupled assay to measure the ATPase activity of DNA topoisomerase II. Methods Mol Biol. 2001;95:57–64. doi: 10.1385/1-59259-057-8:57. [DOI] [PubMed] [Google Scholar]
- 29.Liptak MD, Brunold TC. Spectroscopic and computational studies of Co1+ cobalamin: Spectral and electronic properties of the “superreduced” B12 cofactor. J Am Chem Soc. 2006;128:9144–9156. doi: 10.1021/ja061433q. [DOI] [PubMed] [Google Scholar]
- 30.Holm L, Rosenstrom P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zanetti G, Binda C, Aliverti A. Encyclopedia of Inorganic and Bioinorganic Chemistry. New York: Wiley; 2011. The [2Fe-2S] Ferredoxins. [Google Scholar]
- 32.Svetlitchnaia T, Svetlitchnyi V, Meyer O, Dobbek H. Structural insights into methyltransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis. Proc Natl Acad Sci USA. 2006;103:14331–14336. doi: 10.1073/pnas.0601420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragsdale SW, Lindahl PA, Munck E. Mossbauer EPR and optical studies of the corrinoid/iron-sulfur protein involved in the synthesis of acetyl coenzyme A by Clostridium thermoaceticum. J Biol Chem. 1987;262:14289–14297. [PubMed] [Google Scholar]
- 34.Schmehl M, et al. Identification of a new class of nitrogen fixation genes in Rhodobacter capsulatus: A putative membrane complex involved in electron transport to nitrogenase. Mol Gen Genet. 1993;241:602–615. doi: 10.1007/BF00279903. [DOI] [PubMed] [Google Scholar]
- 35.Wang S, Huang H, Moll J, Thauer RK. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 2010;192:5115–5123. doi: 10.1128/JB.00612-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaster AK, Moll J, Parey K, Thauer RK. Coupling of ferredoxin and heterodisulfide reduction via electron bifurcation in hydrogenotrophic methanogenic archaea. Proc Natl Acad Sci USA. 2011;108:2981–2986. doi: 10.1073/pnas.1016761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li F, et al. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol. 2008;190:843–850. doi: 10.1128/JB.01417-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herrmann G, Jayamani E, Mai G, Buckel W. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J Bacteriol. 2008;190:784–791. doi: 10.1128/JB.01422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tezcan FA, et al. Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science. 2005;309:1377–1380. doi: 10.1126/science.1115653. [DOI] [PubMed] [Google Scholar]
- 40.Rees DC, Howard JB. Structural bioenergetics and energy transduction mechanisms. J Mol Biol. 1999;293:343–350. doi: 10.1006/jmbi.1999.3005. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Lu Y, Buckel W. ATP- and redox-induced conformational changes in the activator of the radical enzyme 2-hydroxyisocaproyl-CoA dehydratase. C R Chim. 2007;10:742–747. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.