

Abstract
The crystal structure of the membrane-bound O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli (EcHyd-1) has been solved in three different states: as-isolated, H2-reduced, and chemically oxidized. As very recently reported for similar enzymes from Ralstonia eutropha and Hydrogenovibrio marinus, two supernumerary Cys residues coordinate the proximal [FeS] cluster in EcHyd-1, which lacks one of the inorganic sulfide ligands. We find that the as-isolated, aerobically purified species contains a mixture of at least two conformations for one of the cluster iron ions and Glu76. In one of them, Glu76 and the iron occupy positions that are similar to those found in O2-sensitive [NiFe]-hydrogenases. In the other conformation, this iron binds, besides three sulfur ligands, the amide N from Cys20 and one Oϵ of Glu76. Our calculations show that oxidation of this unique iron generates the high-potential form of the proximal cluster. The structural rearrangement caused by oxidation is confirmed by our H2-reduced and oxidized EcHyd-1 structures. Thus, thanks to the peculiar coordination of the unique iron, the proximal cluster can contribute two successive electrons to secure complete reduction of O2 to H2O at the active site. The two observed conformations of Glu76 are consistent with this residue playing the role of a base to deprotonate the amide moiety of Cys20 upon iron binding and transfer the resulting proton away, thus allowing the second oxidation to be electroneutral. The comparison of our structures also shows the existence of a dynamic chain of water molecules, resulting from O2 reduction, located near the active site.
Keywords: [4Fe-3S] cluster, membrane-bound hydrogenase, Mössbauer spectroscopy, QM/MM, structure/function relationships
[NiFe]- and [FeFe]-hydrogenases are enzymes involved in hydrogen uptake and evolution in microorganisms according to the reaction: H2↔2H+ + 2e- (1). They have attracted significant attention as sources of inspiration for developing fuel cells and solar-based H2 production (2, 3). Hydrogenase active sites contain iron, nickel (in one class), thiolates, CO and CN- (1), all abundant and inexpensive components, and their H2 uptake efficiencies compare well with those of expensive traditional catalysts like Pt (4). Impressive progress has been made recently in the field of bioinspired chemistry, and some synthetic catalysts equal or even surpass enzymes in hydrogen evolution activity (5). A recent example is a complex that combines features of both [NiFe]- and [FeFe]-hydrogenases, containing a nickel ion and pendant amines (6).
A general problem with hydrogenases is their sensitivity to attack by O2. In the case of the [NiFe]-hydrogenases, two oxidized species that give rise to electron paramagnetic resonance (EPR) spectral signatures called Ni-A and Ni-B have been identified in aerobically purified samples (7). The Ni-A species is difficult to activate, requiring elevated temperatures and a long reductive treatment, whereas a sample in the Ni-B state reactivates rapidly under reducing conditions. Accordingly, Ni-A and Ni-B have been termed unready and ready states, respectively. Although the Ni-B form should reactivate in vivo, that is unlikely to be the case for the Ni-A state. In the Desulfovibrio fructosovorans hydrogenase crystal structure corresponding to the Ni-B form a (hydr)oxo ligand bridges the Ni and the Fe ions of the active site (8). Conversely, in the oxidized as-isolated structure, we modeled the Ni-A state with a diatomic [NiFe] bridging ligand that we assigned as (hydro)-peroxo (8); however, such assignment has not been universally accepted (9). It appears that the formation of either Ni-A or Ni-B states depends on the number of electrons, either two or four, that can rapidly react with O2 at the active site upon air exposure (10). Therefore, resistance to inactivation by O2 may depend on the hydrogenase having enough electrons available to avoid formation of unready states (11).
During the evolution of [NiFe]-hydrogenases, at least three different strategies have emerged to neutralize the deleterious effects of their exposure to O2: (i) a narrow hydrophobic access to the active site that acts as a filter in the constitutive regulatory hydrogenases, discriminating between H2 and the bulkier O2 (12–14); (ii) one of the terminal Cys Ni ligands is changed to SeCys in O2-resistant [NiFeSe]-hydrogenases (15), which do not display the Ni-A EPR spectrum and are able to reduce protons in the presence of low levels of O2 (16); (iii) the presence of two supernumerary small subunit Cys residues in the coordination sphere of the proximal cluster of O2-tolerant membrane-bound [NiFe]-hydrogenases, which can oxidize hydrogen at atmospheric O2 levels (17–19). As reported for the O2-tolerant Aquifex aeolicus hydrogenase 1 (Aa-Hase 1), the proximal cluster undergoes two one-electron redox processes, and the higher potential redox transition is pH-independent between pHs 6.4 and 7.4 (20). Further adaptations for oxygen tolerance are a much lower Km for H2 than the Ki for O2 (21), and the more positive potentials of all metal centers relative to O2-sensitive [NiFe]-hydrogenases.
Mössbauer studies of the Aa-Hase 1 have indicated that the oxidized proximal cluster is more Fe(III)-rich than standard clusters. This observation is consistent with the proposition (20, 21) that the two oxidation processes associated with the proximal cluster of these enzymes may be assigned to 3Fe(II)-1Fe(III) → 2Fe(II)-2Fe(III) and 2Fe(II)-2Fe(III) → 1Fe(II)-3Fe(III) changes. Remarkably, the two redox couples lie within a narrow potential range when compared to that observed for the high-potential iron-sulfur protein (HiPIP), which is about 1 V and involves nonphysiological superreduction of the [4Fe-4S] cluster to the +1 oxidation level (22, 23).
Single and double exchanges of the supernumerary Cys19 and Cys120 of EcHyd-1 to Gly, the corresponding residue in O2-sensitive enzymes, have established that Cys19 is the crucial residue for O2 tolerance, with Cys120 playing a less important role (24). In the course of our X-ray crystallographic and theoretical studies of EcHyd-1, Fritsch et al. (25) and Shomura et al. (26) published the H2-reduced crystal structures of O2-tolerant hydrogenases from Ralstonia eutropha and Hydrogenovibrio marinus (Hm), respectively. Shomura et al. (26) also reported the chemically oxidized Hm hydrogenase structure, which shows that one of the Fe atoms has become coordinated by the peptide amide-N from Cys26. Our corresponding structures agree well with those reported by these authors but also provide further insights into the molecular principles for O2 tolerance. Using our EcHyd-1 crystal structures, we have (i) identified a glutamic acid carboxylate as the base involved in amide-N deprotonation and (ii) calculated and reproduced previously published Mössbauer and EPR data (20), allowing us to describe the electronic and conformational changes that take place during the superoxidation of the proximal cluster.
Results
The 1.67 Å Resolution Structure of As-Isolated EcHyd-1.
In this structure, obtained from an orthorhombic crystal (SI Appendix, Table S1), the hydrogenase is a dimer of heterodimers, including in each monomer a hydrophobic membrane-anchoring small subunit C-terminal domain and its bound His tag. Although there is clearly room for the latter two regions, they are disordered and consequently invisible in the electron density map. Our dimer of heterodimers (Fig. 1) is isostructural with that reported by Shomura et al. (26), supporting their proposition that this oligomeric arrangement is physiologically relevant. The proximity of the two distal clusters in these two dimeric enzymes suggests that intermonomer electron transfer could play a role in O2 tolerance, as previously observed for the “jump-start” activation of Hyd-1 in the Ni-B state by active enzyme (27).
Fig. 1.

Crystal structure of the dimeric [NiFe]-hydrogenase 1 from E. coli. The C-terminal anchor helices are not visible in the electron density maps. Electrons resulting from H2-oxidation are transferred to cytochrome b.
The active site of as-isolated EcHyd-1 closely resembles that of standard, O2-sensitive hydrogenases in the EPR active Ni-B state (8) (SI Appendix, Fig. S1A). However, both more reduced and more oxidized states of this site may also be present. The distal and medial clusters are of the standard [4Fe-4S] and [3Fe-4S] types, respectively. Conversely, as very recently observed for related enzymes (25, 26), the proximal cluster has one of the sulfide (S2-) iron ligands of a classical [4Fe-4S] cluster replaced by the bridging Cys19 Sγ; in addition, Cys120 Sγ forms a second protein ligand to Fe3 (Fig. 2 and SI Appendix, Fig. S2).
Fig. 2.

(A) As-isolated EcHyd-1 proximal cluster structure. Iron positions are depicted with their corresponding anomalous difference (Δanom) electron density peaks (Left) contoured at the 5σ level (purple). The omit map for Glu76 is depicted in light blue contoured at 8σ (the same color codes apply to (B) and (C). A schematic representation of the two conformations for Fe4 and Glu76 (black/gray and red) is shown on the right side of the figure. (B) H2-reduced cluster structure. The positions of Glu76 and Fe4 are as in O2-sensitive [NiFe]-hydrogenases. The peaks for the iron ions and the omit map are contoured at the 10 and 20σ levels, respectively. (C) The 4-OH-1,4-naphthoquinone/ferricyanide-oxidized structure. Glu76 has the alternative positions already observed in the as-isolated structure, whereas Fe4 is modeled with two nearby positions bound to S1, the Sγ and amide N from Cys20 and to either the Sγ of Cys19 or one Oϵ from Glu76. In both cases the iron coordination forms a distorted tetrahedron. The iron peaks and the omit map are contoured at the 6 and 9σ levels, respectively. Hydrogens have not been included in these figures.
One of the irons (Fe4) of the proximal cluster and Glu76 display two discernable positions, which we interpreted as corresponding to two discrete states (Fig. 2A). In one of these states, both Glu76 and Fe4 are positioned like their counterparts in the O2-sensitive [NiFe] hydrogenase from D. fructosovorans (8), with Fe4 tetrahedrally bound by Cys19, Cys20, and two cluster sulfido ions (SI Appendix, Fig. S2); one of these (S3) forms a 2.8-Å-long bond with Fe4. In the other state, the Fe4-S3 bond is broken and the iron is penta-coordinated by the thiolates of Cys19, Cys20, S1, the main chain N of Cys20, and one carboxylate Oϵ from Glu76, forming a distorted trigonal bipyramid (Fig. 2A; red conformation). The coordination spheres of these two proximal cluster states suggest that Fe4 is more oxidized in the latter because a deprotonated peptide-N is a very strong donor ligand. Shomura et al. (26) reached a similar conclusion. Because the protein sample was exposed to air during purification, it is unlikely that a significant fraction of the proximal cluster would be fully reduced. Consequently, we have called these two putative proximal cluster structures PC2 and PC3, by analogy with the reported +2 and +3 [4Fe-4S] cluster redox states in HiPIP. However, these states will be more appropriately defined if the charges of all its S-ligands, including the two extra thiolates that replace one of the inorganic sulfides, are taken into account (28, 29). Accordingly, the [4Fe-3S-6SCys] cluster in PC2 and PC3 will be described as having charges of −2 and −1, respectively (Table 1).
Table 1.
Nomenclatures of the proximal cluster
| Generic designation | PC3 | PC2 | PC1 |
| Core oxidation level | [4Fe-3S]5+ | [4Fe-3S]4+ | [4Fe-3S]3+ |
| Equivalent oxidation level in conventional [4Fe-4S] core | [4Fe-4S]3+ | [4Fe-4S]2+ | [4Fe-4S]+ |
| Cluster charge with all S-bonds | [4Fe-3S-6SCys]- | [4Fe-3S-6SCys]2- | [4Fe-3S-6SCys]3- |
| Calculated models in this study | 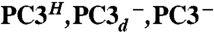 |
PC2- | N/A |
| Glu76 charge | 0 −1* −1 | −1* | N/A |
| Cys20 N charge | −1 −1 −1 | 0 | N/A |
| Overall PC charge | −2 −2 −3 | −2 | N/A |
*Distal Glu76 is not included in the overall PC charge count.
H2-Treated Reduced EcHyd-1 Structure.
In the 1.47-Å resolution structure of EcHyd-1 obtained from a crystal exposed to eight bars of H2 for 10 min (SI Appendix, Table S1 and Fig. 2B), Fe4 and Glu76 are positioned as in our PC2 model and in the reduced hydrogenase structures from Fritsch et al. (25) and Shomura et al. (26). From our crystallographic experiment it is not possible to determine unambiguously the oxidation level of the proximal cluster; both −2 and −3 redox states are possible. H2-induced reduction was confirmed by the absence of the [NiFe]-bridging (hydr)oxo ligand at the active site, which shortens the Ni-Fe distance from 2.9 to 2.6 Å. This site may now be in a mixture of Ni-C and Ni-R states (SI Appendix, Fig. S1B).
Redox-Mediator + K3Fe(CN)6-Treated Oxidized EcHyd-1 Structures.
We obtained a 2.0-Å resolution EcHyd-1 structure from a crystal of as-isolated enzyme aerobically soaked in 2 mM 4-OH-1,4-naphtoquinone (OH-NQ) and 10 mM K3Fe(CN)6 (SI Appendix, Table S1). The active site showed the characteristic [NiFe]-bridging ligand of the Ni-B species. Our initial assignment of the −1 proximal cluster state to PC3 proved to be only partially correct: Although in this purposely oxidized structure Fe4 is near the PC3 position, Glu76 adopts the two conformations found in the PC2/PC3 mixture of the as-isolated EcHyd-1 form, with similar occupancies (Fig. 2C). We will use the subscript d (as in PC3d) to denote the structure that has Glu76 oriented as in the H2-reduced structure (distal from Fe4) and Fe4 bound to the amide N of Cys20. Furthermore, in this structure the elongated electron density peak corresponding to Fe4 suggests that the metal occupies two juxtaposed PC3 positions with about 50% occupancy each. We modeled this structure with Fe4 bound to either Glu76 Oϵ or Cys19 Sγ (Fig. 2C). By comparison, Shomura et al. (26) modeled their ferricyanide-oxidized structure with only one Fe2 position and one conformation for Glu82 (Fe4 and Glu76 in EcHyd-1). However, we found that their model only partially explains the Hydrogenovibrio marinus (H. marinus) hydrogenase proximal cluster electron density, with the ellipsoids corresponding to the anisotropic temperature factors of Fe2 and Glu82 being unusually elongated (SI Appendix, Fig. S3A). After additional refinement of this structure, we conclude that the H. marinus enzyme contains the two conformations of Glu82 and Fe2 that we observe for Glu76 and Fe4 in oxidized EcHyd-1 (SI Appendix, Fig. S3B).
Calculations.
To assign electronic structures to PC2 and PC3 and determine the respective Glu76 protonation states, invisible in our X-ray structures, we geometry-optimized several candidates using hybrid quantum mechanical (QM)/molecular mechanical (MM) potentials (30, 31) and performed pure QM calculations on smaller derived models. Mössbauer and EPR parameter calculations were carried out with in-house codes (see the SI Appendix for details).
Structural optimizations.
Using the numbering of Fig. 3, we constructed the six possible broken symmetry (BS) electronic states for PC3 (1Fe(II) and 3Fe(III), Table 1), corresponding to the spin configurations BS13 (-4/2, +5/2, -5/2, +5/2), BS24 (+5/2,-5/2, +5/2, -4/2), BS34 (+5/2, +5/2, -5/2, -4/2), BS23 (+5/2, -4/2, -5/2, +5/2), BS12 (-4/2, -5/2, +5/2, +5/2), and BS14 (-5/2, +5/2, +5/2, -4/2). The QM/MM optimizations of these BS states hypothesized for the PC3 states with (i) protonated Glu76, PC3H; (ii) deprotonated Glu76, PC3-; and (iii) deprotonated Glu76 in the distal position it adopts in PC2 (Fig. 2A), hydrogen-bonded to Trp11, 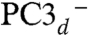 ; led to very similar structures (see SI Appendix for details). Key computed distances for all models in the subsequently favored BS13 electronic state are presented in Table 2 and compared to experimental values. The two Fe4 positions in the oxidized EcHyd-1 X-ray structure (Fig. 2C) could be explained by the PC3- state where Fe4 binds the deprotonated Glu76 (Fig. S3) and alternative states where Glu76 is either protonated and still close to Fe4 (PC3H) or farther away (
; led to very similar structures (see SI Appendix for details). Key computed distances for all models in the subsequently favored BS13 electronic state are presented in Table 2 and compared to experimental values. The two Fe4 positions in the oxidized EcHyd-1 X-ray structure (Fig. 2C) could be explained by the PC3- state where Fe4 binds the deprotonated Glu76 (Fig. S3) and alternative states where Glu76 is either protonated and still close to Fe4 (PC3H) or farther away (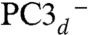 ). In the latter case, Cys19 Sγ completes the tetrahedral Fe4 coordination (Table 2). In going from the PC3 states to PC2- (with a deprotonated distal Glu76; see SI Appendix), the Oε-Fe4 distance increases by more than 2 Å in agreement with the X-ray-defined PC2 and reduced structures (Fig. 2
A and B). From these calculations alone, we could not structurally discriminate between the various electronic states of PC2 and PC3.
). In the latter case, Cys19 Sγ completes the tetrahedral Fe4 coordination (Table 2). In going from the PC3 states to PC2- (with a deprotonated distal Glu76; see SI Appendix), the Oε-Fe4 distance increases by more than 2 Å in agreement with the X-ray-defined PC2 and reduced structures (Fig. 2
A and B). From these calculations alone, we could not structurally discriminate between the various electronic states of PC2 and PC3.
Fig. 3.

Schematic representation of the pattern of exchange pathways within the unusual proximal [4Fe-3S] cluster (red lines) in the PC3 state. Labels 1–4 represent Fe atoms (numbered as in Fig. 2B). There is no direct exchange interaction between Fe3 and Fe4. By contrast, within a standard [4Fe-4S] pseudocubane cluster, each iron ion magnetically interacts with all the others through superexchange couplings.
Table 2.
Key distances of experimental proximal cluster models and quantum mechanical/molecular mechanical models in the BS13 electronic state
| Models Distances (Å) | Df PC2X | Ec as-isolated PC2X/PC3X | Ec reduced PC1X | PC2- | Ec oxidized PC3X | PC3- | PC3H | 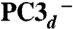 |
| Fe4-S1 | 2.3 | 2.3/2.3 | 2.3 | 2.35 | 2.4/2.4 | 2.47 | 2.42 | 2.40 |
| Fe4-S3 | 2.3 | 2.8/4.1 | 2.4 | 2.47 | 4.3/4.0 | 4.67 | 4.20 | 4.15 |
| Fe4-SγC19(S4)* | (2.3) | 2.2/2.7 | 2.4 | 2.38 | 2.8/2.3 | 2.74 | 2.46 | 2.43 |
| Fe4-NC20 | 3.7 | 3.1/2.1 | 3.3 | 3.26 | 2.1/2.2 | 1.99 | 1.98 | 1.95 |
| Fe4-SγC20 | 2.3 | 2.3/2.3 | 2.3 | 2.29 | 2.4/2.4 | 2.36 | 2.30 | 2.31 |
| Fe4-OεE76 | 5.1 | 4.9/2.4 | 4.7 | 4.61 | 2.1/3.9 | 2.05 | 2.40 | 3.79 |
| NC20-OεE76 | 3.3 | 3.8/2.7 | 3.1 | 2.90 | 2.8/3.7 | 2.86 | 2.90 | 3.39 |
| OεE16-OεE76 | 3.9 | 3.3/3.6 | 3.8 | 3.84 | 3.6/3.0 | 3.04 | 3.36 | 3.59 |
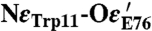 |
2.9 | 2.8/4.0 | 2.9 | 2.89 | 3.9/3.0 | 4.20 | 4.13 | 2.83 |
Subscript X stands for X-ray models.
*SγC19 substitutes S4 in Desulfovibrio fructosovorans (Df) [NiFe]-hydrogenase; all non-shown Fe-S distances are standard for [FeS] clusters at 2.30 + /-0.05 Å.
Mössbauer parameter calculations.
We performed three series of calculations, corresponding to the following models: (i) PC3H (SI Appendix, Table S2), (ii) PC3- (SI Appendix, Table S3), and (iii) 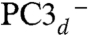 (SI Appendix, Table S4). Based on both the relative energies (in kJ/mol) for the six BS states and the computed quadrupole splittings ΔEQ for the four iron atoms, we favor the BS13 spin arrangement in the PC3H or
(SI Appendix, Table S4). Based on both the relative energies (in kJ/mol) for the six BS states and the computed quadrupole splittings ΔEQ for the four iron atoms, we favor the BS13 spin arrangement in the PC3H or 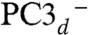 states (see SI Appendix and Table 3).
states (see SI Appendix and Table 3).
Table 3.
Comparison of the PC3 computed quadrupole splittings ΔEQ(Fei)i=1–4 (mm/s) with those obtained experimentally for Aa-Hase 1 (20)
| Electronic state | ΔEQ(Fe1) | ΔEQ(Fe2) | ΔEQ(Fe3) | ΔEQ(Fe4) |
| Experimental values* | 2.41 | 0.60 | 0.60 | 1.23 |
| BS13 PC3H | 2.36 | 0.31 | −0.31 | 0.96 |
| BS13 PC3- | 2.42 | 0.43 | −0.32 | 0.79 |
BS13 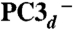
|
2.36 | 0.27 | −0.31 | −0.92 |
*Iron assignments as proposed in main text.
Pandelia et al. (20) assigned the 2.41 (×1) and 1.23( × 1) mm/s splittings to the mixed-valence pair and the 0.60( × 2) mm/s ones to the ferric pair. However, following our favored electronic BS13 state, an alternative assignment would be 2.41 (×1) and 0.60( × 1) mm/s to the localized mixed-valence pair 1–3, and 0.60 (×1) and 1.23( × 1) mm/s to the ferric pair 2–4, in that order, where Fe4 most probably attracts extra electronic charge from the amido ligand.
EPR (g-tensor) calculations.
To lend support to our assignment of the BS13 electronic state to PC3, we computed the g-tensor predicted for the spin-coupled S = 1/2 PC3H, PC3-, and 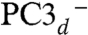 models from both BS13 and BS34 spin-uncoupled states. A spin-coupling procedure was specifically designed for each, taking into account the fact that there is no magnetic exchange pathway between Fe3 and Fe4 (Fig. 3) (see details in SI Appendix). This procedure led to the g-tensor components presented in SI Appendix, Table S6. The Δg anisotropy value computed from BS13 of 0.036 is significantly closer to the experimental value of 0.025 (20) than the one predicted from BS34 (0.047). Again, this favors the BS13 electronic state. SI Appendix, Table S6 shows that PC3H is a better model than PC3- or
models from both BS13 and BS34 spin-uncoupled states. A spin-coupling procedure was specifically designed for each, taking into account the fact that there is no magnetic exchange pathway between Fe3 and Fe4 (Fig. 3) (see details in SI Appendix). This procedure led to the g-tensor components presented in SI Appendix, Table S6. The Δg anisotropy value computed from BS13 of 0.036 is significantly closer to the experimental value of 0.025 (20) than the one predicted from BS34 (0.047). Again, this favors the BS13 electronic state. SI Appendix, Table S6 shows that PC3H is a better model than PC3- or 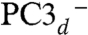 ; however, this prediction is less robust than in the case of the Mössbauer parameter calculations because it strongly depends on a tentative spin-coupling procedure as unusual as this cluster is.
; however, this prediction is less robust than in the case of the Mössbauer parameter calculations because it strongly depends on a tentative spin-coupling procedure as unusual as this cluster is.
Orbital characterization of the PC2- and the PC3H structures in the BS13 state.
There was little difference between the possible BS optimized structures of PC2-. We found that the PC3H BS13 state with Fe1 = -4/2, Fe2 = +5/2, Fe3 = -5/2, and Fe4 = +5/2 satisfies structural and spectroscopic experimental data for the most oxidized state of the cluster. Thus, in the absence of additional experimental data, we intuitively select the BS state for PC2- that results in the least electronic change relative to PC3 (in bold): BS13 with Fe1 = -4/2, Fe2 = +5/2, Fe3 = -5/2, and Fe4 = +4/2. This reasoning implies that Fe4 is the site that is oxidized from ferrous in PC2 to ferric in PC3, eliciting the structural changes observed in the proximal cluster (Fig. 2). In the PC3H BS13 model, the singly occupied molecular orbital (SOMO) shows that Fe1 is the ferrous ion (Fig. 4A). In addition, the lowest unoccupied molecular orbital (LUMO) (Fig. 4B) and the next one above (LUMO+1) (SI Appendix, Fig. S4) clearly indicate that out of the three ferric sites in PC3, Fe4 is the one that will preferentially receive the electron leading back to PC2. The SOMOs α and β of the PC2- model show the localized mixed-valence pair on Fe1 (SI Appendix, Fig. S5A, similar to the SOMO of PC3H in Fig. 4A), whereas the second one is delocalized over all the Fe atoms (SI Appendix, Fig. S5B). Similar results were obtained for the 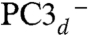 model in the BS13 state.
model in the BS13 state.
Fig. 4.

Molecular orbitals of the favored PC3H model in the BS13 state: (A) SOMO α and (B) LUMO β; both belong to their respective minority spin orbital set (32). Atoms are represented according to standard color codes.
Pathways for Proton Transfer and Water Escape.
In order to locate the most significant structural differences between the as-isolated and H2-reduced structures, we calculated a twofold averaged (Fas-isolated-FH2-reduced) electron density map (SI Appendix, Fig. S6A). If the active site and proximal cluster are not taken into account, two groups of clustered spherical peaks are observed with interpeak distances that in most cases are compatible with H-bonding interactions. The first group of peaks represents water molecules near the active site (Fig. 5). Some of them could result from the full reduction of O2, as they seem to be trapped in the hydrophobic tunnel through which both H2 and O2 are thought to diffuse. Water escape may involve the movement of Asp574L to the position observed in the reduced EcHyd-1 X-ray structure (see also ref. 25); however, our structures do not show a clear pathway from there to the molecular surface. Molecular dynamics studies will be necessary to probe this. The second group of peaks includes a putative dynamic H-bonding network that connects Glu76 to Thr21 from where one branch leads to Asp45 via a water molecule (SI Appendix, Fig. S6B). A second longer branch leads, via His13 and four successive water molecules, to Asp46, which is located close to the molecular surface. These structural changes might result from protons being transferred between the proximal cluster and the solvent, which would require side chain rotations and water movements.
Fig. 5.

Partial water escape pathway from the active site. Light-gray semitransparent surfaces show hydrophobic cavities that may be accessible to O2 and H2. Small and large subunit residues are labeled in blue and black (italics), respectively. Putative waters, resulting from O2 reduction, are labeled in red for the as-isolated structure, which is shown as a color-coded ball-and-stick model, and in green for the model corresponding to the H2-reduced structure shown in light gray. Arrows indicate probable displacements of water molecules upon reduction. The waters labeled O1, O3, O4, O5, and OH- are only present in the structure of the as-isolated enzyme. All the dashed lines are compatible with hydrogen-bonding interactions.
Discussion
A remarkable and unexpected feature of the proximal cluster of O2-tolerant [NiFe]-hydrogenases is its plasticity. During normal hydrogen uptake catalysis the cluster oscillates between −3 and −2 redox states, corresponding to +1 and +2 in standard [4Fe-4S] centers. However, when the enzyme is exposed to molecular oxygen, and assuming the active site is in the Ni-C state, the first redox step should be Ni(III)-H--Fe(II) + O2 → Ni(III)-O(OH)-Fe(II). In order to avoid damage, the transient (hydro)peroxo ligand must be immediately reduced to H2O plus OH- by two electrons coming from the proximal cluster. The structure of as-isolated EcHyd-1 shows four partially occupied H2O molecules close to the [NiFe] active site (Fig. 5) that are likely to correspond to O2-reduction products that need to be evacuated, possibly through the hydrophobic tunnel. The first −3 to −2 cluster oxidation is a classical one. But the second oxidation generating a cluster containing 3 Fe(III) and 1 Fe(II) (Table 1) is extraordinary for a hydrogenase FeS cluster. Our calculations indicate that the second electron comes directly from Fe4, which oxidizes from +2 to +3 (Fig. 4 and SI Appendix, Figs. S4 and S5 and Results). This reaction causes the facile migration of Fe4 from the position seen in PC2 toward the amide N of Cys20 and the carboxylate of Glu76. We speculate that the electrostatic attraction between the ferric iron and Glu76 induces the disruption of the hydrogen bond between this residue and Trp11, allowing its carboxylate moiety to modulate the approach of Fe4 to the amide-N of Cys20 (Fig. 2C). This approach decreases the double character of the C(O)-N bond and causes pyramidalization of N and acidification of the amide proton (33). Because the amide-N can become an Fe ligand only after deprotonation (34), we propose that Glu76 acts as the base to extract this proton. Formation of a Fe(III)-N(amide) bond simultaneously with amide deprotonation affords a way to produce and stabilize higher oxidation states of FeS centers (35): In this case, it stabilizes the superoxidized proximal cluster. The ability of the proximal cluster to undergo two oxidations in succession means that two electrons are immediately available for transfer to the active site when O2 attacks (11).
The difference in the orientation of Glu76 observed in our EcHyd-1 PC3 structures (Fig. 2
A and C) is caused by either a change in its overall protonation or in the position of the proton in the carboxylate moiety. These protonation changes should not affect the reduction potential of the proximal cluster, as indicated by the results of Pandelia et al. (19). A protonation/deprotonation role for the equivalent Glu76 in the O2-sensitive hydrogenase from D. fructosovorans has been postulated (36). This residue, along with the Ni ion Cys576 ligand (EcHyd-1 numbering), Glu28L, Thr18, and Glu16, is thought to be involved in a proton transfer pathway from the active site to the molecular surface (SI Appendix, Fig. S6B) (37, 38). In our calculations, the best agreement to the spectroscopic data was obtained for PC3H followed by PC3d (Tables 2 and 3). A deprotonated Glu76 bound to Fe4 (PC3-) did not reproduce the spectroscopic experimental data at the present level of analysis. Consequently, the proximal cluster of Aa-Hase 1, the subject of the Mössbauer and EPR spectroscopic experiments by Pandelia et al. (19, 20), was probably in the PC3H and/or 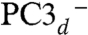 states. PC3H and
states. PC3H and 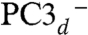 could therefore represent the first and second intermediates in the transfer of the Cys20 amide proton to the active site. When the amido ligand is taken into account, the overall charge is −2 for both PC3H and
could therefore represent the first and second intermediates in the transfer of the Cys20 amide proton to the active site. When the amido ligand is taken into account, the overall charge is −2 for both PC3H and 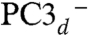 , which is also the charge of PC2- (Table 1). This makes the second oxidation of the proximal cluster thermodynamically favorable. In PC3-, the overall charge is −3; therefore, protonation of the Glu76 carboxylate should be necessary to cleave the short Fe4-Oε bond. During the reduction of molecular oxygen to water, two protons have to be provided to the active site (assuming that the oxidation of hydride in the Ni-C active site form yields a third proton). As discussed above and evoked by Shomura et al. (26), one of these protons could originate from the amide N of Cys20. This implies that the proton transfer pathway, which in this H2 uptake enzyme normally directs protons to the molecular surface, must now function in the opposite direction, i.e., it delivers them to the active site (Fig. 5 and SI Appendix, Fig. S6B).
, which is also the charge of PC2- (Table 1). This makes the second oxidation of the proximal cluster thermodynamically favorable. In PC3-, the overall charge is −3; therefore, protonation of the Glu76 carboxylate should be necessary to cleave the short Fe4-Oε bond. During the reduction of molecular oxygen to water, two protons have to be provided to the active site (assuming that the oxidation of hydride in the Ni-C active site form yields a third proton). As discussed above and evoked by Shomura et al. (26), one of these protons could originate from the amide N of Cys20. This implies that the proton transfer pathway, which in this H2 uptake enzyme normally directs protons to the molecular surface, must now function in the opposite direction, i.e., it delivers them to the active site (Fig. 5 and SI Appendix, Fig. S6B).
We conclude that as long as the active site Ni stays in the Ni-B form and the enzyme does not oxidize H2, there will be neither electrons nor protons available to change the proximal cluster from PC3, a state where the Fe4-amido bond protects its integrity, to catalytically active PC2/PC1 states, requiring the reprotonation of the amide N of Cys20. In PC3, the enzyme is inactive but also relatively resistant to O2-induced damage. As expected from the crystal structure of the oxidized form, there is a good correlation between the ready species and the EPR Ni-B signal. Conversely, there is no correlation between the unready species resulting from O2 exposure and the Ni-A signal (24), suggesting alternative inactive species involving S-O bonds (SI Appendix, Fig. S1A).
It is now possible to interpret the results of functional studies on Cys19 and Cys120 mutants (21, 24) within a structural framework. The fact that Re-MBH C19G variant is 30 times more active than the C19S variant (21) suggests that in the former case a sulfide replaces the missing cysteine thiolate, whereas in the latter, the sulfide may be replaced by the Oγ atom of serine, which does not have the same iron coordinating properties. From a structural and chemical viewpoint, the bridging Cys19 is crucial because the long Fe4-S3 bond in PC2 (Table 2) is easily ruptured, allowing Fe4 to become ligated instead by the hard amido ligand, favoring an Fe(III) state. Besides playing a role in the overall stability of the PC3 state, Cys120 may also have some influence on the cleavage of the Fe4-S3 bond (Fig. 2 A and C), because S3 is also a ligand to Fe3, which is coordinated by this cysteine thiolate (Fig. 2A). In the case of the C120G variant, the Fe4-S3 bond may be shorter and stronger, as observed in standard [4Fe-4S] clusters (SI Appendix, Fig. S2 and Table 2), interfering with Fe4 migration toward the N amide atom of Cys20. Our results imply that mutation of Glu76 will affect O2 tolerance, although alternative amide deprotonation mechanisms could still operate.
In conclusion, O2-tolerance in [NiFe]-hydrogenases mostly arises from the unusual structure of its proximal cluster, which can provide two successive electrons to the active site. Combined with the [NiFe] bridging hydride, expected to be present in reduced states of the enzyme (39), EcHyd-1 can provide four electrons to reduce molecular oxygen to H2O and OH-. This process bypasses the formation of either a stable (hydro)peroxide ligand or reactive oxygen species, or both, that may inflict serious or even irreversible damage to the active site in the case of O2-sensitive [NiFe]-hydrogenases. Structure-based computational analysis of the available Mössbauer and EPR spectroscopic results indicates that the proximal cluster second oxidation at high potential directly involves the unusual Fe4, which is ferrous in the PC2 state and ferric in the PC3 state. Our favored models maintain electroneutrality in the transition between these two states. Furthermore, because Glu76 deprotonates the amide N from Cys20, an amido-Fe4 bond is formed in a reaction that stabilizes the high-potential form of the cluster (35). During O2 reduction to water, the O2-tolerant [NiFe]-hydrogenases, which are usually hydrogen uptake enzymes, operate in reverse mode, sending both electrons and protons to the active site.
Materials and Methods
Orthorhombic and triclinic crystal forms of EcHyd-1 were obtained using a Gryphon robot (ArtRobins) operating in a customized glove box. Data were collected from crystals grown from as-isolated aerobically purified protein as well as from crystals exposed to high H2 pressure and oxidizing redox mediators. The structures were solved by the molecular replacement method. Hybrid QM/MM calculations on the enzyme models were performed with the QSite code in the Schrödinger Suite (40), whereas the ADF2009 code (41) was used on smaller QM models extracted from these QM/MM optimized structures. More detailed descriptions are available in the SI Appendix.
Supplementary Material
Acknowledgments.
We thank Yvain Nicolet for discussions and the staff of the European Synchrotron Radiation Facility beamlines ID23-1 and ID23-2 for providing a reliable setup for X-ray data collections. We appreciate the help from the staff of the computing facility provided by the Commissariat à l’Energie Atomique (CEA/DSV/GIPSI), Saclay, France, and the Centre de Calcul Recherche et Technologie (CEA/CCRT). The Metalloproteins unit thanks the CEA and Centre National de la Recherche Scientifique for institutional funding. A.P. thanks Merton College, Oxford, for her Junior Research Fellowship. The Biological and Biotechnological Sciences Research Council (Grant H003878-1) (F.A.A.) and the Engineering and Physical Sciences Research Council (Grant Supergen 5-EP/D047943/1) also provided funding.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3UQY, 3USC, and 3USE).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1119806109/-/DCSupplemental.
References
- 1.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 2.Friedrich B, Fritsch J, Lenz O. Oxygen-tolerant hydrogenases in hydrogen-based technologies. Curr Opin Biotechnol. 2011;22:358–364. doi: 10.1016/j.copbio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Vincent KA, et al. Electricity from low-level H2 in still air-an ultimate test for an oxygen tolerant hydrogenase. Chem Commun. 2006;48:5033–5035. doi: 10.1039/b614272a. [DOI] [PubMed] [Google Scholar]
- 4.Jones AK, Sillery E, Albracht SPJ, Armstrong FA. Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem Commun. 2002;8:866–867. doi: 10.1039/b201337a. [DOI] [PubMed] [Google Scholar]
- 5.Le Goff A, et al. From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 6.Helm ML, Stewart MP, Bullock RM, DuBois MR, DuBois DL. A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s-1 for H2 production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez VM, Hatchikian EC, Cammack R. Properties and reactivation of 2 different deactivated forms of Desulfovibrio gigas hydrogenase. Biochim Biophys Acta. 1985;832:69–79. [Google Scholar]
- 8.Volbeda A, et al. Structural differences between the ready and unready oxidized states of [NiFe] hydrogenases. J Biol Inorg Chem. 2005;10:239–249. doi: 10.1007/s00775-005-0632-x. [DOI] [PubMed] [Google Scholar]
- 9.Ogata H, Kellers P, Lubitz W. The crystal structure of the [NiFe] hydrogenase from the photosynthetic bacterium Allochromatium vinosum: Characterization of the oxidized enzyme (Ni-A state) J Mol Biol. 2010;402:428–444. doi: 10.1016/j.jmb.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 10.Lamle SE, Albracht SPJ, Armstrong FA. Electrochemical potential-step investigations of the aerobic interconversions of [NiFe]-hydrogenase from Allochromatium vinosum: Insights into the puzzling difference between unready and ready oxidized inactive states. J Am Chem Soc. 2004;126:14899–14909. doi: 10.1021/ja047939v. [DOI] [PubMed] [Google Scholar]
- 11.Cracknell JA, Wait AF, Lenz O, Friedrich B, Armstrong FA. A kinetic and thermodynamic understanding of O2 tolerance in [NiFe]-hydrogenases. Proc Natl Acad Sci USA. 2009;106:20681–20686. doi: 10.1073/pnas.0905959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volbeda A, Montet Y, Vernede X, Hatchikian EC, Fontecilla-Camps JC. High-resolution crystallographic analysis of Desulfovibrio fructosovorans [NiFe] hydrogenase. Int J Hydrogen Energy. 2002;27:1449–1461. [Google Scholar]
- 13.Buhrke T, Lenz O, Krauss N, Friedrich B. Oxygen tolerance of the H2-sensing [NiFe] hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the active site. J Biol Chem. 2005;280:23791–23796. doi: 10.1074/jbc.M503260200. [DOI] [PubMed] [Google Scholar]
- 14.Duche O, Elsen S, Cournac L, Colbeau A. Enlarging the gas access channel to the active site renders the regulatory hydrogenase HupUV of Rhodobacter capsulatus O2 sensitive without affecting its transductory activity. FEBS J. 2005;272:3899–3908. doi: 10.1111/j.1742-4658.2005.04806.x. [DOI] [PubMed] [Google Scholar]
- 15.Garcin E, et al. The crystal structure of a reduced [NiFeSe] hydrogenase provides an image of the activated catalytic center. Struct Fold Des. 1999;7:557–566. doi: 10.1016/s0969-2126(99)80072-0. [DOI] [PubMed] [Google Scholar]
- 16.Parkin A, Goldet G, Cavazza C, Fontecilla-Camps JC, Armstrong FA. The difference a Se makes? Oxygen-tolerant hydrogen production by the [NiFeSe]-hydrogenase from Desulfomicrobium baculatum. J Am Chem Soc. 2008;130:13410–13416. doi: 10.1021/ja803657d. [DOI] [PubMed] [Google Scholar]
- 17.Cracknell JA, et al. Enzymatic oxidation of H2 in atmospheric O2: The electrochemistry of energy generation from trace H2 by aerobic microorganisms. J Am Chem Soc. 2008;130:424–425. doi: 10.1021/ja078299+. [DOI] [PubMed] [Google Scholar]
- 18.Lenz O, et al. H2 conversion in the presence of O2 as performed by the membrane-bound [NiFe]-hydrogenase of Ralstonia eutropha. Chemphyschem. 2010;11:1107–1119. doi: 10.1002/cphc.200901002. [DOI] [PubMed] [Google Scholar]
- 19.Pandelia ME, et al. Membrane-bound hydrogenase I from the hyperthermophilic bacterium Aquifex aeolicus: Enzyme activation, redox intermediates and oxygen tolerance. J Am Chem Soc. 2010;132:6991–7004. doi: 10.1021/ja910838d. [DOI] [PubMed] [Google Scholar]
- 20.Pandelia ME, et al. Characterization of a unique [FeS] cluster in the electron transfer chain of the oxygen tolerant [NiFe] hydrogenase from Aquifex aeolicus. Proc Natl Acad Sci USA. 2011;108:6097–6102. doi: 10.1073/pnas.1100610108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goris T, et al. A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase. Nat Chem Biol. 2011;7:310–318. doi: 10.1038/nchembio.555. [DOI] [PubMed] [Google Scholar]
- 22.Cammack R. “Super Reduction” of chromatium high-potential iron-sulphur protein in the presence of dimethyl sulphoxide. Biochem Biophys Res Commun. 1973;54:548–554. doi: 10.1016/0006-291x(73)91457-5. [DOI] [PubMed] [Google Scholar]
- 23.Heering HA, Bulsink YBM, Hagen WR, Meyer TE. Reversible super reduction of the cubane [4Fe-4S](3+,2+,1+) in the high-potential iron-sulfur protein under non-denaturating conditions. Eur J Biochem. 1995;232:811–817. [PubMed] [Google Scholar]
- 24.Lukey MJ, et al. Oxygen-tolerant [NiFe]-hydrogenases: the individual and collective importance of supernumerary cysteines at the proximal Fe-S cluster. J Am Chem Soc. 2011;133:16881–16892. doi: 10.1021/ja205393w. [DOI] [PubMed] [Google Scholar]
- 25.Fritsch J, et al. The crystal structure of an oxygen tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature. 2011;479:249–252. doi: 10.1038/nature10505. [DOI] [PubMed] [Google Scholar]
- 26.Shomura Y, Yoon KS, Nishihara H, Higuchi Y. Structural basis for [4Fe-3S] cluster in the oxygen-tolerant membrane-bound [NiFe]-hydrogenase. Nature. 2011;479:253–256. doi: 10.1038/nature10504. [DOI] [PubMed] [Google Scholar]
- 27.Wait AF, Parkin A, Morley GM, dos Santos L, Armstrong FA. Characteristics of enzyme-based hydrogen fuel cells using an oxygen-tolerant hydrogenase as the anodic catalyst. J Phys Chem C. 2010;114:12003–12009. [Google Scholar]
- 28.Benning MM, Meyer TE, Rayment I, Holden HM. Molecular structure of the oxidized high-potential iron-sulfur protein isolated from Ectothiorhodospira vacuolata. Biochemistry. 1994;33:2476–2483. doi: 10.1021/bi00175a016. [DOI] [PubMed] [Google Scholar]
- 29.Carter CW, et al. Comparison of Fe4S4 clusters in high-potential iron protein and in ferredoxin. Proc Natl Acad Sci USA. 1972;69:3526–3529. doi: 10.1073/pnas.69.12.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin H, Truhlar DG. QM/MM: What have we learned, where are we, and where do we go from here? Theor Chem Acc. 2007;117:185–199. [Google Scholar]
- 31.Senn HM, Thiel W. Atomistic Approaches in Modern Biology: from Quantum Chemistry to Molecular Simulations. Vol 268. Berlin: Springer; 2007. QM/MM methods for biological systems; pp. 173–290. Topics in Current Chemistry. [Google Scholar]
- 32.Noodleman L, Peng CY, Case DA, Mouesca JM. Orbital interactions, electron delocalization and spin coupling in iron sulfur clusters. Coord Chem Rev. 1995;144:199–244. [Google Scholar]
- 33.Nemirovskiy OV, Gross ML. Complexes of iron(II) with cysteine-containing peptides in the gas phase. J Am Soc Mass Spectrom. 1996;7:977–980. doi: 10.1016/1044-0305(96)00054-2. [DOI] [PubMed] [Google Scholar]
- 34.Sigel H, Martin RB. Coordinating properties of the amide bond-stability and structure of metal-ion complexes of peptides and related ligands. Chem Rev. 1982;82:385–426. [Google Scholar]
- 35.Sharp CR, Duncan JS, Lee SC. [Fe4S4]q cubane clusters (q = 4+, 3+, 2+) with terminal amide ligands. Inorg Chem. 2010;49:6697–6705. doi: 10.1021/ic100742c. [DOI] [PubMed] [Google Scholar]
- 36.Volbeda A, Fontecilla-Camps JC. Structure-function relationships of nickel-iron sites in hydrogenase and a comparison with the active sites of other nickel-iron enzymes. Coord Chem Rev. 2005;249:1609–1619. [Google Scholar]
- 37.Galvan IF, Volbeda A, Fontecilla-Camps JC, Field MJ. A QM/MM study of proton transport pathways in a [NiFe] hydrogenase. Proteins. 2008;73:195–203. doi: 10.1002/prot.22045. [DOI] [PubMed] [Google Scholar]
- 38.Teixeira VH, Soares CM, Baptista AM. Proton pathways in a [NiFe]-hydrogenase: A theoretical study. Proteins. 2008;70:1010–1022. doi: 10.1002/prot.21588. [DOI] [PubMed] [Google Scholar]
- 39.Brecht M, van Gastel M, Buhrke T, Friedrich B, Lubitz W. Direct detection of a hydrogen ligand in the [NiFe] center of the regulatory H2-sensing hydrogenase from Ralstonia eutropha in its reduced state by HYSCORE and ENDOR spectroscopy. J Am Chem Soc. 2003;125:13075–13083. doi: 10.1021/ja036624x. [DOI] [PubMed] [Google Scholar]
- 40. (a) Maestro version 9.2 (b) QSite version 5.7 (c) Jaguar version 7.8 S, LLC, New York, 2011.
- 41.Te Velde G, Baerends EJ. Numerical integration for polyatomic systems. J Comput Phys. 1992;99:84–98. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.