

Abstract
The Ras gene is frequently mutated in cancer, and mutant Ras drives tumorigenesis. Although Ras is a central oncogene, small molecules that bind to Ras in a well-defined manner and exert inhibitory effects have not been uncovered to date. Through an NMR-based fragment screen, we identified a group of small molecules that all bind to a common site on Ras. High-resolution cocrystal structures delineated a unique ligand-binding pocket on the Ras protein that is adjacent to the switch I/II regions and can be expanded upon compound binding. Structure analysis predicts that compound-binding interferes with the Ras/SOS interactions. Indeed, selected compounds inhibit SOS-mediated nucleotide exchange and prevent Ras activation by blocking the formation of intermediates of the exchange reaction. The discovery of a small-molecule binding pocket on Ras with functional significance provides a new direction in the search of therapeutically effective inhibitors of the Ras oncoprotein.
Keywords: small G protein, guanine nucleotide exchange, nuclear magnetic resonance, crystal structure, small-molecule inhibitors
Ras is a small GTP-binding protein that functions as a nucleotide-dependent switch for central growth signaling pathways (1, 2). In response to extracellular signals, Ras is converted from a GDP-bound (RasGDP) to a GTP-bound (RasGTP) state, as catalyzed by guanine nucleotide exchange factors (GEFs), notably the SOS1 protein. Active RasGTP mediates its diverse growth-stimulating functions through its direct interactions with effectors including Raf, PI3K, and Ral guanine nucleotide dissociation stimulator. The intrinsic GTPase activity of Ras then hydrolyzes GTP to GDP to terminate Ras signaling. The Ras GTPase activity can be further accelerated by its interactions with GTPase-activating proteins (GAPs), including the neurofibromin 1 tumor suppressor (2).
Ras, a human oncogene identified and characterized over 30 y ago, is mutated in more than 20% of human cancers. Among the three Ras isoforms (K, N, and H), KRas is most frequently mutated (2). Mutant Ras has a reduced GTPase activity, which prolongs its activated conformation, thereby promoting Ras-dependent signaling and cancer cell survival or growth (1, 2).
Mutations of Ras in cancer are associated with poor prognosis (2). Inactivation of oncogenic Ras in mice results in tumor shrinkage. Thus, Ras is widely considered an oncology target of exceptional importance. However, development of small-molecule inhibitors against Ras has thus far proven unsuccessful. Given the picomolar affinity between guanine nucleotides and Ras and the high cytosolic concentration of guanine nucleotides, it is very challenging to develop a conventional inhibitor competitive against nucleotide binding (1, 2). Outside of the nucleotide-binding pocket, the Ras protein does not contain obvious cavities for small-molecule binding. A number of small molecules have been reported to bind to Ras (3–7), but their mechanisms of action and the structural basis to achieve Ras inhibition remain elusive.
Fragment-based lead discovery (FBLD) has provided a starting point to develop a number of chemical entities for targets previously considered “undruggable,” notably Bcl2 (reviewed in ref. 8). This approach has benefited in particular from the use of NMR and surface plasmon resonance for ultrasensitive detection of interactions between ligand and protein, even at millimolar affinities. Aside from the identification of ligands for a known binding site, a fragment-based screen can also reveal unique binding sites in proteins. Central to this discovery process is the ability to obtain high-resolution structures of the ligand-protein complex. Characterization of the site through a combination of structural studies and biophysical and biochemical examination form the basis for the hit-to-lead optimization process. We have applied FBLD to probe oncogenic KRas, and report here the discovery of a unique small-molecule binding pocket on Ras and the characterization of the effect of the ligands on Ras activation.
Results
A Fragment Screen Identifies Ras-Binding Small Molecules.
An NMR-based saturation transfer difference (STD) assay (9) was used to detect ligand binding to the recombinant KRas4B-G12D, KRasm (Fig. S1A). A screen of a 3,300-compound library was performed (using pools of up to six fragments each) by measuring compound binding to a 1∶1 mixture of 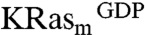 and KRasm bound to GDP or guanosine-5′-[(β, γ)-methyleno]triphosphate (GMPPCP) proteins. Representative screening data and their deconvolution are shown in Fig. 1A and Fig. S1B. Binders were identified that showed an STD signal/noise ratio greater than five. Candidate fragments from positive pools were retested as single compounds for their binding to
and KRasm bound to GDP or guanosine-5′-[(β, γ)-methyleno]triphosphate (GMPPCP) proteins. Representative screening data and their deconvolution are shown in Fig. 1A and Fig. S1B. Binders were identified that showed an STD signal/noise ratio greater than five. Candidate fragments from positive pools were retested as single compounds for their binding to 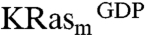 and
and 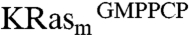 separately, yielding 240 primary hits. These hits were further validated by comparing 2D 1H15N heteronuclear single quantum coherence (HSQC) spectra of isotopically labeled
separately, yielding 240 primary hits. These hits were further validated by comparing 2D 1H15N heteronuclear single quantum coherence (HSQC) spectra of isotopically labeled 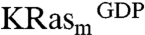 in the presence or absence of 2 mM compounds. Of these, 25 compounds produced chemical shift perturbations that can be mapped to a contiguous site on the KRasm structure and were thus classified as confirmed hits (Fig. S1A). Intriguingly, each of the 25 confirmed hits showed chemical shift perturbations for residues V8, T74/G75 and L56/D57 (Fig. 1B and Fig. S1C). Some compounds also perturbed additional resonances corresponding to residues close to the region of D57.
in the presence or absence of 2 mM compounds. Of these, 25 compounds produced chemical shift perturbations that can be mapped to a contiguous site on the KRasm structure and were thus classified as confirmed hits (Fig. S1A). Intriguingly, each of the 25 confirmed hits showed chemical shift perturbations for residues V8, T74/G75 and L56/D57 (Fig. 1B and Fig. S1C). Some compounds also perturbed additional resonances corresponding to residues close to the region of D57.
Fig. 1.

NMR-based fragment screen identifies Ras-binding compounds. (A) One-dimensional proton spectra showing representative screening results. (i) The 1D reference spectrum of a compound pool containing DCAI. Arrows point to the peaks of DCAI. (ii) The STD spectrum of the DCAI-containing pool. (B) These panels show the 1H15N HSQC of 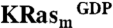 at a concentration of 100 μM titrated with 0, 250, 500, 1,000, 1,500, and 4,000 μM DCAI (from black to purple). Residues V8, T74, G75, L56, and D57 make up one contiguous site in the Ras protein structure. Side-chain resonances are labeled “sc.”
at a concentration of 100 μM titrated with 0, 250, 500, 1,000, 1,500, and 4,000 μM DCAI (from black to purple). Residues V8, T74, G75, L56, and D57 make up one contiguous site in the Ras protein structure. Side-chain resonances are labeled “sc.”
High-Resolution Cocrystal Structures Reveal a Unique Binding Pocket on Ras.
To understand the molecular basis of Ras interaction with these compounds, we determined the first full-length KRasm crystal structure in the presence of GDP or nonhydrolyzable GTP analogs (GMPPCP) at 1.75- and 2.05-Å resolution, respectively. Within the GTPase domain (amino acids 1–169), the KRasm structure (Fig. 2A) is similar to the structure of truncated KRas4B-Q61H [amino acids 1–169 with residue 61 mutated from glutamine to histidine; Protein Data Bank (PDB) code 3GFT; Structural Genomics Consortium]. Excluding the switch regions, the root-mean-square deviation in Cα positions between these two structures is 0.5 Å. In addition, they share a high degree of similarity to the truncated HRas (10) and NRas (PDB code 3CON; Structural Genomics Consortium) structures. The switch I and II regions display conformational heterogeneity among different Ras structures because they are sensitive to the nucleotide state and, to some extent, influenced by crystal packing interactions.
Fig. 2.

Ras-ligand cocrystal structures reveal an expandable small-molecule binding pocket in KRas. (A) Structure of KRasm bound to GTPγS-Mg2+. The switch I/II regions are colored in green and orange, respectively. The spheres indicate residues with chemical shift perturbations upon DCAI binding in the NMR experiments. The bound GTPγS is shown as sticks and Mg2+ as a blue dot. The disordered region of KRasm is drawn as a dotted line. Hypothetic cytoplasmic membrane is shown to illustrate its potential interactions with the KRasm C-terminal domain. (B) The small-molecule binding pocket on KRas. Surface of KRasm in complex with BZDN (orange) and BZIM (cyan) is colored by electrostatic potential with red being negative and blue being positive. (C) The DCAI-binding pocket on KRasm. D54 and R41 side chains are shown in rotamer conformation with (cyan) and without (blue) DCAI bound. Arrows indicate the direction of their rotation upon DCAI binding. Residues with chemical shift perturbations in the NMR spectra are colored in gray. (D) A close-up surface rendering of the DCAI 4-chlorine atom (in green) and its interacting surface on KRasm (in blue) in comparison with the KRasm surface with no ligand bound (in brown).
Our full-length KRasm structure reveals additional features pertaining to the C-terminal domain (CTD) (Fig. 2A). Aside from the C-terminal CAAX farnesylation motif, the CTD sequence is hypervariable across K, N, and HRas. The KRasm CTD contains a unique hexalysine stretch for direct association with cytoplasmic membrane. In the crystal structure, helix α5 from the GTPase domain is extended by one turn after which the CTD makes a π-turn followed by an extended loop containing the hexalysine stretch. This extended conformation is likely to promote interaction of the lysine stretch with negatively charged lipid head groups from the membrane (Fig. 2A).
Cocrystals of ligands and KRasm were obtained by soaking the confirmed fragment hits into the KRasm crystals. Fig. S2 A–C depicts the complexes with benzamidine (BZDN), benzimidazole (BZIM), and 4,6-dichloro-2-methyl-3-aminoethyl-indole (DCAI), respectively. In all three cases, compounds bind to a similar site on KRasm.
This small-molecule binding site (Fig. 2A and Fig. S2 A–D) resides between helix α2 and the core β-sheet, β1–β3. The compounds show amphipathic interactions centering on a hydrophobic pocket of approximately 7 × 7 Å at the opening and 5-Å deep (Fig. 2B). This binding pocket has a size sufficiently large to accommodate a benzyl and a chloro group. For comparison, a typical ATP-binding site found at kinase active sites is about 20 × 10 × 10 Å [as an example, see the p38 MAP kinase (PDB code 1WFC)].
Residues surrounding the binding pocket in Ras include K5, L6, V7, I55, L56, and T74 (Fig. S2D). This observation is largely consistent with the mapping from the NMR spectrum perturbations because all of the perturbed residues (Figs. 1B and 2 A–C, and Figs. S1C and S2 A–D) are located in or around the binding pocket. The surface area outside the pocket is more hydrophilic and engages in specific interactions with the polar portion of the compounds. For example, KRasm D54 at the rim of the binding pocket forms hydrogen bonds with the NH group in BZDN and BZIM (Fig. 2B). H, N, and KRas have identical amino acid sequences in the binding site, which suggests that the compounds would likely bind to all three isoforms. We confirmed that DCAI and BZIM bind to HRas in the STD assay (Fig. S2 E and F).
The Small-Molecule Binding Pocket on Ras Can Be Expanded upon DCAI Binding.
The crystal structure of 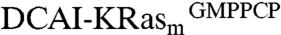 reveals a high degree of plasticity in the binding pocket (Fig. 2
C and D). The chloro-substituent at the six-position of DCAI anchors the ligand into the hydrophobic pocket. DCAI is larger than BZDN and BZIM, and the 4-chloro group of DCAI is accommodated in the binding pocket through reorienting its indole core by 65° relative to BZIM. Additionally, the side chain of D54 of KRasm moves away from the binding site by 2.5 Å (Fig. 2C). D54 normally forms a salt bridge with R41 in the absence of compound binding. This compound-induced rotamer conformation of D54 effects an outward flipping (7.5 Å) of R41 and preserves the salt bridge (Fig. 2C). Overall, the pocket surface retreats by about 3.5 Å upon DCAI binding (Fig. 2D), rendering the ligand binding site opening to a size of 7 × 10.5 Å, although the extended region is relatively shallow.
reveals a high degree of plasticity in the binding pocket (Fig. 2
C and D). The chloro-substituent at the six-position of DCAI anchors the ligand into the hydrophobic pocket. DCAI is larger than BZDN and BZIM, and the 4-chloro group of DCAI is accommodated in the binding pocket through reorienting its indole core by 65° relative to BZIM. Additionally, the side chain of D54 of KRasm moves away from the binding site by 2.5 Å (Fig. 2C). D54 normally forms a salt bridge with R41 in the absence of compound binding. This compound-induced rotamer conformation of D54 effects an outward flipping (7.5 Å) of R41 and preserves the salt bridge (Fig. 2C). Overall, the pocket surface retreats by about 3.5 Å upon DCAI binding (Fig. 2D), rendering the ligand binding site opening to a size of 7 × 10.5 Å, although the extended region is relatively shallow.
DCAI Inhibits SOS-Mediated Nucleotide Exchange for Ras.
Comparison of the Ras-DCAI structure with the structure of the Ras-SOS1 complex (11) indicates that the small-molecule binding pocket described above is located adjacent to the Ras–SOS interaction surface (Fig. 3A), suggesting that ligand binding may affect SOS-mediated nucleotide exchange for KRas. To investigate the nucleotide exchange reaction, a nucleotide exchange and a nucleotide release assay were employed using fluorescent N-methylanthraniloyl (MANT) derivatives of guanine nucleotides, recombinant wild-type KRas, and a catalytically active form of SOS1 (SOScat) consisting of its Ras exchanger motif and Cdc25 domains (11, 12) (Fig. S3). The kinetics of these reactions were monitored by changes in fluorescence intensity of MANT-nucleotide in its Ras-bound versus free state. Our nucleotide exchange assay detects inhibitors against any step in the reaction mechanism, whereas the nucleotide release assay specifically identifies inhibitors against the first phase of the exchange reaction, nucleotide release (Fig. S3).
Fig. 3.

DCAI specifically inhibits the SOScat-catalyzed nucleotide release and exchange from KRas and KRasm. (A) Ras/SOS contact surface was calculated using the HRas–SOS1 cocrystal structure (11). Ras residues within 4.5 Å of any SOS1 atom in the HRas–SOS1 complex are colored blue; Ras residues that fall outside the HRas–SOS1 interface are colored in white. DCAI (carbon in yellow color) was modeled into the HRas–SOS1 complex by superimposing the nonswitch regions of the KRasm-DCAI structure onto the apo-HRas structure in the HRas–SOS1 complex. (B) SOScat-catalyzed nucleotide release assays were performed in duplicate with 125 nM SOScat in the presence of DCAI (red), BZIM (black), or INDL (gray), and the plot shows the rates normalized to that of the DMSO control. Average IC50 for DCAI over three independent experiments under identical conditions was 155 ± 36 μM. (C) SOScat-catalyzed nucleotide release assays were performed, in triplicate, as described in C for wild-type KRas (black) and mutant KRasm (red), except that a higher SOScat concentration, 500 nM, was used here to provide an optimal assay window for both KRasm and KRas. (D)SOScat-catalyzed nucleotide exchange reactions were performed in duplicate with KRasGDP/SOScat (black), RhoAGDP/Dbs (cyan), or Cdc42GDP/Dbs (magenta) in the presence of DCAI. The plot shows the rates normalized to that of DMSO. Error bars in B–D are the SEM.
We analyzed representative hits from the fragment screen in these two assays and found that DCAI effectively inhibited both reactions (Fig. S4A). Titration experiments indicated that DCAI blocked both nucleotide exchange and release reactions with an IC50 of 342 ± 22 μM and 155 ± 36 μM, respectively, whereas BZIM and indole (INDL) had no or minimal effects (Fig. 3B and Fig. S4B). We conclude that DCAI blocks the first phase of the exchange reaction by preventing nucleotide release from KRas. This inhibitory effect is not restricted to DCAI. We found that 2-(4,6-dichloro-2,3-dimethyl-1H-indol-1-yl)ethanamine, a close structural analog of DCAI, also binds to KRas and inhibits SOS-mediated nucleotide release from KRas (Fig. S4C).
In addition to wild-type KRas, DCAI also inhibits SOS-catalyzed nucleotide release from mutant KRas (KRasm) (Fig. 3C). In fact, DCAI exhibits a lower IC50 against KRasm than wild-type KRas, consistent with the different kinetics of the SOS-catalyzed release reaction for KRas vs. KRasm. At saturating SOS concentrations, the nucleotide release rate is fivefold slower for KRasm than for wild-type KRas (Fig. S4D), indicating that SOScat is less effective at releasing nucleotide from KRasm compared to wild-type KRas.
DCAI inhibits nucleotide release from Ras in an SOS-dependent manner only. EDTA chelates Mg2+ ions and promotes nucleotide release from KRas independent of SOS; DCAI had no effect on the rate of EDTA-stimulated nucleotide release from KRas or KRasm (Fig. S5A). In addition, we found that DCAI has no effect on the intrinsic ability of KRas to release nucleotide in the absence of EDTA and SOS (Fig. S5 B–C).
DCAI specifically inhibits the SOS-mediated nucleotide exchange on Ras, but not the Dbs-mediated nucleotide exchange on RhoA and Cdc42 (Fig. 3D), consistent with the fact that bulky residues, Trp58 in RhoA and Phe56 in Cdc42, occupy the space equivalent to the DCAI-binding site in RhoA and Cdc42, thereby potentially preventing the binding of DCAI to RhoA and Cdc42 (Fig. S6).
DCAI Inhibits Nucleotide Exchange by Blocking the Interactions Between Ras and SOS.
Next, we investigated the mechanism of the DCAI-mediated inhibition of nucleotide release. Based on the overlap of the small-molecule binding pocket with the Ras-SOS interaction surface (Fig. 3A), we predicted that DCAI binds to KRas and competes with SOS association. Further structural analysis of the apoRas-SOS complex suggests two mechanisms by which binding of DCAI to KRas interferes with the formation of the exchange reaction intermediates and thereby inhibits SOS-mediated catalysis (Fig. 4A and Fig. S7A). First, the 2-methyl and 3-aminoethyl side chains of DCAI sterically hinder the binding of SOScat to Ras. Second, binding of DCAI to KRas results in a rotation of both D54 and R41 to enlarge the binding pocket (Figs. 2 C and D, 4A, and Fig. S7A); reorientation of these side chains will prevent them from participating in two salt bridges that stabilize the Ras–SOS interaction (to SOS H911 and SOS D910, respectively; Fig. 4A and Fig. S7A) (11). Consistent with this model, the KRas R41S mutant reduces the rate of the SOS-mediated nucleotide release reaction by 2.5-fold (Fig. 4B), even though this mutation does not affect the binding of DCAI to KRas (Fig. S2G). We conclude that R41 is important for SOS-catalyzed nucleotide exchange on Ras, but dispensable for recognition by DCAI.
Fig. 4.

DCAI is a competitive inhibitor that blocks the Ras-SOScat interaction. (A) Ras coordinates from the DCAI-KRasm and BZIM-KRasm complexes were superimposed onto those from the HRas–SOS complex (1NVV; HRas bound to the substrate site of SOS; ref. 19) to highlight the locations of the DCAI and BZIM binding sites. The carbon atoms of Ras R41 and D54 are colored in green in the Ras–SOS complex and in yellow in the Ras–DCAI complex. (B) Nucleotide release assays were performed with wild-type RasGDP (black) or Ras-R41SGDP (red) across a range of SOScat concentrations. SOScat is less effective in catalyzing nucleotide release from Ras-R41Smant-GDP. (C, Top) Nucleotide-free biotinylated KRas was generated by incubation with EDTA. Streptavidin beads were incubated with SOScat alone (lane 1; negative control) or with SOScat plus nucleotide-free biotinylated KRas in the presence of increasing concentrations of DCAI (lanes 3–7). Bead-associated SOScat was purified, washed, and detected by Western blotting. (Bottom) Identical reactions as above, with the exception that EDTA was omitted in the reaction. Results shown are representative of three independent experiments. (D) Nucleotide release assays were performed, in duplicates, with a dose titration of SOScat in the presence of DMSO or of either 125 or 250 μM DCAI. The relative reaction rates were determined by normalizing to those of DMSO control, and the percentage of inhibition by DCAI was plotted for each concentration of SOScat. Error bars in B and D are the SEM.
In contrast, structural analysis suggests that the DCAI-related compound, BZIM, would not sterically hinder SOS binding (Fig. 4A and Fig. S7A) and would not alter the ability of D54 and R41 to participate in the interactions (Figs. 2B, 4A, and Fig. S7A) at the apoRas–SOS interface. Consistent with this structural prediction, BZIM does not inhibit the nucleotide release or exchange (Fig. 3B and Fig. S4 A–C). Interestingly, both DCAI and BZIM bind to KRas with similar affinity as measured by compound-induced chemical shift perturbations of Ras in NMR experiments (Kd of 1.1 ± 0.5 mM and 1.5 ± 0.3 mM for DCAI and BZIM, respectively), suggesting that binding to Ras itself is not sufficient to inhibit exchange. Our data indicate that the steric hindrance and/or disruption of salt bridges contribute to the inhibition of the SOScat-catalyzed nucleotide exchange by DCAI.
To further elucidate the mechanism of DCAI function, we developed biochemical assays to directly analyze the interaction between KRas and SOS. Biotinylated wild-type KRasGDP was incubated with SOScat in the absence of free nucleotide with or without EDTA. The KRas-associated SOScat was purified on streptavidin beads and assayed by Western blotting (Fig. 4C). In the absence of EDTA, KRasGDP and SOScat formed a complex that represents the first intermediate of the nucleotide release reaction, which is then converted into the apoRas–SOS complex upon nucleotide release (Fig. S3A). Addition of EDTA to the reaction chelates Mg2+ from the nucleotide-binding site in KRas and promotes the formation of the apoRas–SOS complex, a later intermediate in the exchange reaction (Fig. S3A). We found that DCAI, but not BZIM, compromised the formation of the Ras–SOS complex independent of the presence of EDTA (Fig. 4C and Fig. S7 B and C). We conclude that DCAI inhibits the nucleotide release reaction by blocking the association of KRas with SOS.
DCAI may block the formation of the KRasGDP-SOScat complex (step I) and/or inhibit the conversion of this complex to apoRas–SOS (step II) in the nucleotide release assay (Fig. S3A). If DCAI blocks step I in the reaction, we predict that the inhibitory effect of DCAI at a given compound concentration should be reduced as the SOS concentration increases. On the other hand, if DCAI blocks step II in the reaction, the inhibitory effect of DCAI should be constant independent of the SOS concentration. In the nucleotide release assay, we titrated the SOS concentration in the presence of fixed DCAI concentrations (either 125 or 250 μM) and observed greater inhibition by DCAI at lower SOS concentrations (Fig. 4D), indicating that DCAI blocks the formation of the KRasGDP-SOScat complex. Thus, DCAI acts as a competitive inhibitor of nucleotide release via its association with RasGDP. This mechanism of action for DCAI also predicts that the IC50 of DCAI in the nucleotide release assay should be highly dependent on the concentration of SOScat with lower SOScat concentrations resulting in lower IC50 values. Titration of SOScat in the nucleotide release assay confirmed this prediction (Fig. S7D).
DCAI Inhibits Ras Activation in Cells.
To measure the intracellular RasGTP level, we developed a biosensor system in HEK-293T cells inducibly expressing Venus fluorescence protein fused to the Ras-binding domain and cysteine-rich domain (RBD-CRD) domain of cRaf (13). In live cell imaging experiments, we demonstrated that endogenous plasma membrane-associated RasGTP recruited Venus-RBD-CRD to the membrane (13) (Fig. S8A; time 0). DCAI disrupted this recruitment with EC50 values of 15.8 ± 0.4 μM (Fig. S8 A–C). Neither BZIM nor INDL, which are inactive in in vitro exchange assays, had any effect on RBD-CRD localization (Fig. S8 A and C).
This inhibition of membrane targeting of the RBD-CRD reporter is not due to loss of Ras at the membrane, as DCAI had no effect on the localization of fluorescently tagged Ras in HEK-293T cells (Fig. S8D). In addition, the inhibition by DCAI does not result from a general disruption of protein interactions at the plasma membrane, as the recruitment of Smad-7 by Smurf-1 at the plasma membrane was not affected by DCAI (Fig. S8E).
We also examined the effect of DCAI on activation of Ras by EGF, a response critically dependent on SOS. Addition of EGF increased the RasGTP level in HEK-293T cells, whereas DCAI attenuated this EGF-stimulated activation of Ras (Fig. S8 F and G).
Discussion
This paper describes our efforts to identify and characterize small molecules that inhibit Ras activity. Extensive efforts have been invested in the past two decades in the development of an effective therapy against Ras-driven tumors. Initial work was focused on farnesyl transferase inhibitors (FTIs) and inhibitors of Ras converting enzme 1 and isoprenylcysteine methyltransferase, which disrupt the processing and localization of Ras. However, these strategies failed for the KRas mutant tumors because these enzymes have many substrates besides Ras and because KRas can be modified by geranylgeranylation in the presence of FTIs.
More recent efforts have focused on directly targeting the Ras oncoprotein by (i) restoring the GTPase activity, which is greatly reduced due to Ras mutations, (ii) blocking the formation of RasGTP (i.e., inhibiting Ras-GEFs), or (iii) blocking Ras signaling to downstream effectors. The first approach failed despite extensive efforts; retrospective analysis of the mutant Ras-GAP structure indicates a lack of any small-molecule binding space at the GTPase active site (14). Targeting Ras signaling to effectors and inhibition of Ras nucleotide exchange both require disruption of protein–protein interactions, which is challenging.
FBLD represents an effective approach to probe protein surface using low molecular weight but highly soluble compounds. Through FBLD, we identified a group of compounds that bind to a site adjacent to the functionally important switch I/II regions of KRas. This binding site exists on part of the Ras surface commonly involved in Ras-effector, Ras-GEFs, and Ras-GAP interactions (11, 14, 15). The fact that all 25 confirmed hits from the NMR screen of 3,300 fragments bind to the same site suggests that this site is likely to be the primary compound-binding site on Ras. High-resolution crystal structures elucidate the specific mode of molecular interactions and delineate a well-defined site responsible for binding.
Although several previous studies have reported potential small-molecule association with Ras, none have characterized the binding site by a high-resolution structure analysis and none reported a defined binding pocket (3–7). The best-characterized Ras-binding compounds reported so far are SCH54292 (16) and its more water-soluble derivative (7). In silico calculation based on NMR mapping suggests that these compounds bind to a region between switch II and helix 4 of RasGDP, although other binding areas cannot be ruled out. This hypothesized SCH54292-binding site is distinct from the DCAI-binding pocket that we determined.
An intriguing feature of the DCAI-binding pocket is its ability to expand upon compound binding and the functional importance of such compound-induced conformational change. Altering rotamer conformation of KRas R41 and D54 removes two hydrogen bonds at the interface between Ras and SOS, which likely contributes to the DCAI-mediated inhibitory activity. ADP-ribosylation on R41 has been reported to interfere with SOS-catalyzed nucleotide exchange on Ras (17), and we speculate that ADP-ribosylation on R41 also compromises the salt bridge with SOS. Interestingly, sequence and structure homology between SOS1 and RasGRF1 (18), another exchange factor, suggests that these hydrogen bonds are likely conserved in the complex of Ras and RasGRF1 as well, and therefore DCAI may inhibit other Ras-GEFs.
DCAI not only binds to Ras, but also blocks the interaction of Ras as a substrate with SOS (Fig. S7E). RasGTP also binds to an allosteric site on SOS1 and up-regulates its nucleotide exchange activity (19). DCAI does not appear to affect the binding of Ras to the allosteric site (Fig. S9A). Functionally, DCAI blocks the SOS-mediated nucleotide release and inhibits the activation of Ras. Consistent with this specific mechanism of action, the inhibitory effect of DCAI is only toward SOS-catalyzed nucleotide exchange and does not affect the intrinsic nucleotide exchange by Ras. This mode of action is in sharp contrast to that of a Rac exchange inhibitor, EHT 1864, which inhibits both intrinsic and GEF-mediated nucleotide exchange for Rac (20). Patgiri et al. recently described an SOS1-derived peptide that inhibited Ras signaling (21). Although this peptide and DCAI interact with different regions of Ras, both inhibit Ras nucleotide release in vitro and Ras activation in vivo.
DCAI exhibits cellular activity in HEK-293T cells; it attenuates the EGF-stimulated activation of Ras and blocks the recruitment of the cRaf RBD-CRD domain to the cytoplasmic membrane. Given that DCAI binds to Ras and acts as a competitive inhibitor against association of SOS with Ras, it is expected that its potency in cellular assays is in part determined by the cellular concentrations of SOS and Ras. Quantitative analysis determined the cellular SOS1 and Ras concentrations at 5 and 350 nM in HEK-293T cells, respectively. The EC50 of DCAI in the RBD-CRD redistribution assay is at 15.8 ± 0.4 μM (Fig. S8C), which is consistent with the inhibitory potency of DCAI in the in vitro nucleotide release reaction (Fig. S7D). Although these studies suggest that DCAI inhibits the nucleotide exchange via binding on Ras in cells, we cannot exclude the possibility of off-target driven effects, given its low biochemical affinity to Ras. In addition, our cellular assays in Fig. S8 measure RasGTP level via interaction with RBD or RBD-CRD of cRaf, and it is possible that the cellular activity we observed results from the inhibition of both Ras nucleotide exchange and Ras–Raf interaction. Superimposing the Ras-DCAI and the Ras-effector complexes suggests that DCAI binding does not alter the protein–protein interface and that DCAI should exert minimal effects on the Ras/effector interactions (Fig. S9 B and C).
Through an integrated approach of fragment-based small-molecule screening, high-resolution structural analyses and functional studies, we have identified a unique small-molecule binding pocket and elucidated its functional role in inhibiting Ras activation. The identified fragment hits have low affinity to Ras, and our efforts represent only the very first step toward developing a lead compound. Although further medicinal chemistry efforts are needed to enhance potency while controlling selectivity, the current results may reinvigorate the search for therapeutics of this challenging and yet important oncology target (see SI Text for discussion). Interestingly, additional small G proteins share similar structural features. Thus, the FBLD approach represents a potential route to target these small G proteins with small-molecule inhibitors, thereby opening up therapeutic opportunities for this class of biologically important regulatory proteins.
Materials and Methods
Fragment Screening and Hit Validation.
STD experiments were performed at 280 K in pools of up to six compounds each at a concentration of 250 μM per compound. The pools of compounds were incubated with a 1∶1 mixture of 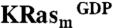 and
and 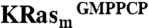 (5 μM each) in 20 mM d11-tris (hydroxymethyl) aminomethane hydrochloride pH 8.0, 200 mM NaCl, 2 mM MgCl2, 99%
(5 μM each) in 20 mM d11-tris (hydroxymethyl) aminomethane hydrochloride pH 8.0, 200 mM NaCl, 2 mM MgCl2, 99% 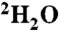 , and 50 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid. Compounds with STD signal-to-noise ratio greater than five were remeasured as individual compounds for binding to
, and 50 μM 4,4-dimethyl-4-silapentane-1-sulfonic acid. Compounds with STD signal-to-noise ratio greater than five were remeasured as individual compounds for binding to 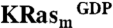 or
or 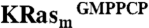 separately.
separately.
Hits were further tested for specific binding to KRasm by measurement of 1H15N HSQC protein spectra. Positive hits were defined as compounds that exhibit cross-peak perturbation by more than 15 Hz (combined 15N and 1H chemical shift change) when compared to the spectrum recorded in the absence of compounds.
Crystallization, Structure Determination, and Biochemical and Cellular Assays.
See SI Materials and Methods for details.
Supplementary Material
Acknowledgments.
We thank Dr. Sharon Campbell (University of North Carolina, NC) for helpful discussions on Ras biochemistry. We are grateful to Ken Dong, Yvonne Franke, William Forrest, Imola Fodor, Janet Jin, Julia Kong, Hong Li, Maria Lorenzo, Kyle Mortara, Olivier René, Qinghua Song, and Christine Tam for technical contributions. We thank Reciprocal Space Consulting (Berkeley, CA) for collecting diffraction data and the Stanford Synchrotron Facility and the Advanced Light Source for radiation light sources.
Footnotes
Conflict of interest statement: All authors are employees of Genentech, Inc.
*This Direct Submission article had a prearranged editor.
Data deposition: The KRas structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4DSO, 4DST, and 4DSU).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1116510109/-/DCSupplemental.
References
- 1.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 2.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 3.Taveras AG, et al. Ras oncoprotein inhibitors: The discovery of potent, ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg Med Chem. 1997;5:125–133. doi: 10.1016/s0968-0896(96)00202-7. [DOI] [PubMed] [Google Scholar]
- 4.Waldmann H, et al. Sulindac-derived Ras pathway inhibitors target the Ras-Raf interaction and downstream effectors in the Ras pathway. Angew Chem Int Ed Engl. 2004;43:454–458. doi: 10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- 5.Kato-Stankiewicz J, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci USA. 2002;99:14398–14403. doi: 10.1073/pnas.222222699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosnizeck IC, et al. Stabilizing a weak binding state for effectors in the human ras protein by cyclen complexes. Angew Chem Int Ed Engl. 2010;49:3830–3833. doi: 10.1002/anie.200907002. [DOI] [PubMed] [Google Scholar]
- 7.Palmioli A, et al. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorg Med Chem Lett. 2009;19:4217–4222. doi: 10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- 8.Chessari G, Woodhead AJ. From fragment to clinical candidate—a historical perspective. Drug Discov Today. 2009;14:668–675. doi: 10.1016/j.drudis.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 10.Milburn MV, et al. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 11.Boriack-Sjodin PA, Margarit SM, Bar-Sagi D, Kuriyan J. The structural basis of the activation of Ras by Sos. Nature. 1998;394:337–343. doi: 10.1038/28548. [DOI] [PubMed] [Google Scholar]
- 12.Lenzen C, Cool RH, Wittinghofer A. Analysis of intrinsic and CDC25-stimulated guanine nucleotide exchange of p21ras-nucleotide complexes by fluorescence measurements. Methods Enzymol. 1995;255:95–109. doi: 10.1016/s0076-6879(95)55012-7. [DOI] [PubMed] [Google Scholar]
- 13.Anderson DJ, et al. Live-cell microscopy reveals small molecule inhibitor effects on MAPK pathway dynamics. PloS One. 2011;6:e22607. doi: 10.1371/journal.pone.0022607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheffzek K, et al. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 15.Nassar N, et al. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 16.Ganguly AK, et al. Detection and structural characterization of ras oncoprotein-inhibitors complexes by electrospray mass spectrometry. Bioorg Med Chem. 1997;5:817–820. doi: 10.1016/s0968-0896(97)00021-7. [DOI] [PubMed] [Google Scholar]
- 17.Ganesan AK, Frank DW, Misra RP, Schmidt G, Barbieri JT. Pseudomonas aeruginosa exoenzyme S ADP-ribosylates Ras at multiple sites. J Biol Chem. 1998;273:7332–7337. doi: 10.1074/jbc.273.13.7332. [DOI] [PubMed] [Google Scholar]
- 18.Freedman TS, et al. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci USA. 2006;103:16692–16697. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margarit SM, et al. Structural evidence for feedback activation by Ras. GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 20.Shutes A, et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 21.Patgiri A, Yadav KK, Arora PS, Bar-Sagi D. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585–587. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.