

Abstract
The tempo and mode of protein evolution have been central questions in biology. Genomic data have shown a strong influence of the expression level of a protein on its rate of sequence evolution (E-R anticorrelation), which is currently explained by the protein misfolding avoidance hypothesis. Here, we show that this hypothesis does not fully explain the E-R anticorrelation, especially for protein surface residues. We propose that natural selection against protein–protein misinteraction, which wastes functional molecules and is potentially toxic, constrains the evolution of surface residues. Because highly expressed proteins are under stronger pressures to avoid misinteraction, surface residues are expected to show an E-R anticorrelation. Our molecular-level evolutionary simulation and yeast genomic analysis confirm multiple predictions of the hypothesis. These findings show a pluralistic origin of the E-R anticorrelation and reveal the role of protein misinteraction, an inherent property of complex cellular systems, in constraining protein evolution.
Although molecular and evolutionary biologists unanimously agree that the key determinant of the evolutionary rate of a protein is its functional constraint, the exact nature of the functional constraint on a protein has remained largely mysterious. In the last decade, the advent of functional genomics has allowed empirical examinations of correlations between the evolutionary rate of a protein sequence and various properties of the protein such as its expression level, expression breadth across tissues, subcellular localization, gene structure, number of protein interaction partners, and KO fitness effect (1–17). Unexpectedly, the strongest determinant of the rate of protein sequence evolution was found to be its expression level, at least in unicellular organisms such as bacteria and yeast (2, 3, 13, 15). The reason why highly expressed proteins evolve slowly, however, is not well-understood. The prevailing explanation of the negative correlation between the expression level of a protein and its evolutionary rate (E-R anticorrelation) is the protein misfolding avoidance hypothesis, which asserts that natural selection against cytotoxic protein misfolding (18) is stronger for more highly expressed proteins and constrains the evolution of these proteins (13, 19, 20). The misfolding avoidance hypothesis has been supported by a molecular-level evolutionary simulation as well as multiple lines of empirical evidence (13, 20), and therefore, it has been well-established. What is unclear, however, is whether this hypothesis can fully explain the E-R anticorrelation. We pose this question, because misfolding avoidance is achieved primarily by the enhancement of protein stability (20), which is mainly determined by the selective use of residues located in the protein core; however, the E-R anticorrelation is not limited to the protein core. In this work, we first show that the E-R anticorrelation persists, especially on the protein surface, even when residues constrained for misfolding avoidance are removed. We then propose a mechanism for the E-R anticorrelation on protein surfaces, termed the protein misinteraction avoidance hypothesis. Finally, we provide evidence for this hypothesis using both computer simulation and empirical genomic analysis.
Results
Misfolding Avoidance Cannot Fully Explain the E-R Anticorrelation.
To assess whether the E-R anticorrelation is fully explainable by the misfolding avoidance hypothesis, we first removed sites in a protein that are constrained by misfolding avoidance and then examined whether the anticorrelation disappears. In a recent study (20), we derived an approximate formula for the probability of protein misfolding (pmisfold) of a mutant gene relative to its WT version. At each codon position of each protein coding gene in the budding yeast Saccharomyces cerevisiae, we determined the rank of the WT codon among the 61 possible sense codons in terms of pmisfold. For example, if the WT codon has the lowest pmisfold among the 61 possible codons, the WT codon has a rank of one. In a gene, such top-ranked codons are expected to be under stronger constraint for misfolding avoidance than not top-ranked codons. Consequently, the E-R anticorrelation should be weakened when top-ranked codons are eliminated. Evaluating the E-R anticorrelation requires an accurate estimation of the substitution rate. We estimated the substitution rate of each amino acid position by using sequence alignments from six yeast species that diverged after the whole-genome duplication (WGD) that occurred ∼100 Mya (21) (Materials and Methods).
We first removed all amino acid positions where the WT codons are ranked one by pmisfold; these sites were previously referred to as matching sites (20). For comparison, we randomly removed the same number of amino acid sites as the number of matching sites from each yeast protein. We then calculated the correlation between the mRNA expression level of an S. cerevisiae gene and its amino acid substitution rate estimated from the mean of the remaining amino acid sites of the protein. Consistent with the misfolding avoidance hypothesis, removing the top-ranked codons weakens the E-R anticorrelation significantly more than any of the 104 random removals of the same number of sites (P < 10−4) (Fig. 1A). However, the amount of decrease in E-R anticorrelation is small (from ρ = −0.549 to −0.545), and the anticorrelation remains very strong after the removal of the top-ranked codons (P < 10−292, Spearman's rank correlation test), which constitute 15.4% of all codons.
Fig. 1.

The protein misfolding avoidance hypothesis cannot fully explain the E-R anticorrelation in yeast, especially for sites on protein surfaces. (A–C) Spearman's rank correlation (ρ) between the mRNA expression level of a gene and the mean amino acid substitution rate of the gene. We used all residues (dashed line) and removed sites with pmisfold rank = 1 (A), pmisfold rank ≤ 6 (B), and pmisfold rank ≤ 30 (C). To compare with the three specific removals (indicated by arrows), we randomly removed the same numbers of sites from each gene and repeated the random removal 1,000 times (frequency distribution indicated by open bars). The statistical significance in the difference of ρ between the specific removals and random removals is indicated above the horizontal solid bar. (D) Fraction of stabilizing sites in buried regions is greater than the fraction on protein surfaces for most proteins. Each dot represents a gene. Numbers of genes below and above the diagonal line are indicated as well as the P value of the null hypothesis that these two numbers are equal. (E) Spearman's rank correlation (ρ) between mRNA levels and amino acid substitution rates for surface and buried residues separately after sites with pmisfold rank less than or equal to certain cutoffs are removed.
The genome-wide median pmisfold rank for WT yeast protein sequences is six. To further reduce the evolutionary constraint imposed by misfolding avoidance, we eliminated all codons with pmisfold rank ≤ 6. As expected, this removal is more effective than the random removal of the same number of codons in weakening the E-R anticorrelation (Fig. 1B); however, the anticorrelation remains strong (ρ = −0.538, P < 10−284). We then took a third and even more dramatic action by removing all codons with pmisfold ranks ≤ 30 (Fig. 1C). Because each codon has 61 available choices, the remaining sites all have pmisfold ranks ≥ 31. These sites should be equally or less stabilizing than the chance expectation and thus, are unlikely to be subject to selection for misfolding avoidance. Furthermore, this removal eliminated 88% of codons, resulting in a dataset that is substantially smaller than the original one. Surprisingly, the E-R anticorrelation remains strong (ρ = −0.496, P < 10−234). These results suggest that the protein misfolding avoidance hypothesis does not fully account for the E-R anticorrelation.
Misfolding Avoidance Is Especially Poor in Explaining the E-R Anticorrelation for Protein Surfaces.
Protein misfolding avoidance is accomplished by the reduction of both translational error-induced and -free misfolding (20) through the use of optimal synonymous codons, which likely increases translational accuracy (13), and the use of amino acid residues at key positions, which increases protein stability (20). It has been reported that optimal codons are preferentially used at buried sites of proteins and that this preference intensifies with rising expression level (22). Buried sites are also known to be more important than surface sites in determining protein folding stability (23). Thus, natural selection against misfolding is expected to act primarily on the buried residues in a protein. To confirm this prediction, we examined the distribution of residues with top-ranked pmisfold among exposed and buried sites. Buried sites are defined as those sites that are accessible by fewer than five water molecules simultaneously (Materials and Methods). Because the genome-wide median pmisfold rank is six for WT yeast protein sequences, we consider residues with pmisfold rank ≤ 6 as stabilizing sites and compare the fraction of stabilizing sites in buried and surface regions of yeast proteins. Indeed, in >97% of yeast proteins examined, the fraction of buried sites that are stabilizing is greater than the fraction of surface sites that are stabilizing (Fig. 1D). The enrichment of stabilizing sites in buried regions predicts that the removal of stabilizing sites would weaken the E-R anticorrelation in buried regions more than in surface regions. This prediction is indeed correct (Fig. 1E). Thus, protein misfolding avoidance is especially poor in explaining the E-R anticorrelation for protein surfaces. However, surface residues are not completely irrelevant to misfolding avoidance (24), which is evident from Fig. 1E. Furthermore, a positive correlation exists between protein abundance and the fraction of matching sites for protein surfaces (ρ = 0.243, P < 10−18), although the corresponding correlation for protein cores is much stronger (ρ = 0.415, P < 10−55).
Estimation of pmisfold using protein structure information is expected to be more accurate than using protein sequence information (20, 25). However, the above analyses were based on protein sequence information, because most yeast proteins do not have structure information. Nevertheless, qualitatively similar results were obtained when only those proteins with structure information were examined. For instance, in Fig. 1D, all 26 proteins with structure information are located below the diagonal line. In Fig. 1E, the remaining E-R anticorrelation after the removal of buried residues with pmisfold ranks ≤ 30 (−0.12) is much weaker than the remaining E-R anticorrelation after the removal of surface residues with pmisfold ranks ≤ 30 (−0.27).
Protein Misinteraction Avoidance Could Constrain the Evolution of Protein Surfaces.
What might constrain the evolution of protein surfaces in a protein concentration-dependent manner? Protein misinteraction could be the answer. Protein misinteraction refers to nonfunctional and typically nonspecific protein–protein interactions that occur upon random encounters between protein molecules. For two reasons, protein misinteraction is quite frequent in a cell. First, many proteins coexist at any given time in any cellular compartment, providing ample opportunities for misinteraction. For example, ∼1,800 proteins are coexpressed and colocalized to the yeast cytoplasm in standard laboratory conditions (26, 27). Because an average protein has only a few specific partners (28), the total concentration of nonspecific partners of a protein is much greater than the concentration of its specific partners. Second, although functional and specific protein interactions are usually stronger than misinteractions, the difference in binding energy is moderate (29, 30). Considering these factors, Zhang et al. (30) recently estimated that ∼22% of protein molecules that are not engaged in specific protein interactions are bound with nonspecific partners in yeast. Similar estimates of 23–28% were obtained for other model organisms, including the nematode worm, fruit fly, and human (30).
Protein misinteraction can be deleterious to an organism, because it (i) potentially leads to a higher demand for protein synthesis that wastes energy, (ii) interferes with functional interactions, and (iii) initiates nonphysiological and potentially damaging cellular processes. The notion that misinteraction could lead to gains of deleterious functions is exemplified by a mutant version of the tumor suppressor p53 that misinteracts with vitamin D3 (VD3) receptor. As a result of this misinteraction, the mutant p53 enhances VD3-induced transcription, compromises VD3-mediated repression, and converts VD3 into a harmful antiapoptotic agent (31). A recent study showed that the deleterious effect of gross gene overexpression observed in yeast is largely caused by increased protein misinteraction (32). Theoretical modeling has repeatedly shown that, because of its deleterious effect, misinteraction constrains the proteome size, affects optimal protein concentrations, and shapes the functional interaction network (30, 33, 34).
Because protein misinteraction is generally abundant and deleterious and involves protein surface residues, we hypothesize that selection against protein misinteraction constrains the evolution of protein surface residues. Specifically, highly expressed proteins are under stronger selective pressures than lowly expressed ones to avoid misinteraction (Fig. 2), because a misinteraction-enhancing mutation is more harmful when it occurs in a highly expressed gene than in a lowly expressed gene because of the presence of a greater number of misinteracting molecules from a highly expressed protein than from a lowly expressed protein (Fig. 2). Consequently, highly expressed proteins are less sticky on surfaces and more constrained in surface sequence evolution than lowly expressed ones (Fig. 2). Hence, at least in principle, protein misinteraction avoidance can generate an E-R anticorrelation for protein surfaces.
Fig. 2.

A schematic diagram explaining the protein misinteraction avoidance hypothesis. Functional interactions between proteins are shown with lock and key matched pairs of jigsaws, whereas misinteractions are shown with unmatched jigsaw pairs that are also boxed.
Although protein misinteraction may occasionally lead to protein aggregation, they differ in several aspects. First, although protein misinteraction usually occurs between correctly folded molecules, protein aggregation more often happens to misfolded/unfolded proteins. Second, protein misinteraction often (but not always) involves two different proteins, whereas protein aggregation normally involves multiple molecules of the same protein. Third, both protein misinteraction and aggregation can interfere with normal protein–protein interaction, but only misinteraction can induce potentially deleterious cellular signals that are passed on from the involved proteins.
Misinteraction Avoidance Generates an E-R Anticorrelation: Computer Simulation.
To show that protein misinteraction avoidance can generate an E-R anticorrelation for protein surfaces, we conducted a molecular-level evolutionary simulation using a 3D protein lattice model (Fig. 3A). In this simulation, we designed 100 pairs of proteins with specific and functional interactions. Each of these 200 proteins consists of 27 amino acid residues that fold into a 3 × 3 × 3 lattice and maintains at least baseline folding stability during evolution (Materials and Methods). Using the information in a previous study (35), for each pair of the specifically interacting proteins, we optimized their sequences such that the specific interaction is significantly stronger than any misinteraction. We randomly assigned expression levels to each pair of specific interacting partners using a power law distribution, because cellular protein concentrations are known to follow this distribution (36). We then calculated the probability that each protein is bound to any other protein in the cell. For each binary interaction, we considered all 6 × 6 × 4 = 144 possible orientations when calculating the interaction energy. For simplicity, we did not allow simultaneous interactions of three or more molecules, which are expected to be rarer than binary interactions. The fitness of a cell is calculated by considering two factors: (i) the reduction in the concentrations of functional interactions because of misinteractions and (ii) the toxicity of misinteractions (Materials and Methods).
Fig. 3.

A molecular-level evolutionary simulation shows that misinteraction avoidance can create an E-R anticorrelation. (A) The general scheme of the simulation (details in Materials and Methods). (B) The average contact energy of misinteractions involving a particular protein decreases with rising expression level of the protein. (C) The proportion of surface residues that are hydrophobic decreases with the rise of the protein's expression level. (D) The probability that a protein molecule involved in misinteraction decreases with the expression level of the protein. (E) Highly expressed proteins have high concentrations of misinteracting molecules. (F) The number of amino acid (AA) substitutions per surface residue in 100 generations of simulation declines with rising protein expression level. (G) The number of amino acid substitutions per buried residue in 100 generations of simulation does not decline with rising protein expression level. In B–G, each dot represents one gene, and the averaged results from 100 simulation replications are presented. The red lines are estimated using locally weighted scatterplot smoothing. B–E are based on the observations in the 20,000th generation of the simulation, whereas F and G are based on the period from the 19,900th generation to the 20,000th generation of simulation.
We constituted a population of 100 cells that evolves at the mutation rate of 0.0005 amino acid changes per residue per generation. After 19,900 generations of evolution, mutation selection balance is reached. We then evolved the population for another 100 generations and estimated the substitution rate during the last 100 generations by counting only fixed amino acid mutations. We repeated the entire simulation 100 times with fixed expression levels but variable protein sequences.
Selection against protein misinteraction should result in lower stickiness (37) for more abundant proteins. Indeed, our simulation shows that, as the expression level of a protein increases, the average contact energy of its misinteractions decreases (i.e., more positive) (Fig. 3B), which was also observed in the recent simulation by Heo et al. (34). Hydrophobic residues are more likely than hydrophilic residues to mediate protein misinteraction (30), because the contact energy is greater (i.e., more negative) for hydrophobic interactions than hydrophilic interactions (38). Consistent with this prediction, we observed a reduced fraction of hydrophobic residues on the entire protein surface as the protein expression level increases (Fig. 3C). As expected, the probability for a protein to engage in misinteraction at any time decreases with rising protein expression level (Fig. 3D). However, this decrease in probability is slower than the rise in expression level (Fig. 3D). Consequently, the number of molecules involved in misinteraction is still greater for more abundant proteins (Fig. 3E). In direct support of our hypothesis, highly expressed proteins show lower rates of amino acid substitution on the surface (Fig. 3F) but not in the core (Fig. 3G).
It is interesting to note that all of the above results still hold qualitatively even when misinteractions only reduce the concentrations of functional interactions but are not toxic (Fig. S1). The reason is that, when a highly expressed protein increases its stickiness, the concentrations of many functional protein complexes are reduced, because highly expressed proteins misinteract with many proteins. The same will not happen when a lowly expressed protein increases its stickiness by the same degree, because it misinteracts with only a small number of proteins. Thus, selection against stickiness is stronger in highly expressed proteins than in lowly expressed ones, generating an E-R anticorrelation. However, the simulation shows that the E-R anticorrelation created by misinteraction avoidance is much weaker when misinteraction is nontoxic (Fig. S1).
Yeast Genomic Data Support the Misinteraction Avoidance Hypothesis.
With the above simulation showing the sufficiency of misinteraction avoidance in generating an E-R anticorrelation on protein surfaces, we now turn to empirical evidence for the hypothesis. Our hypothesis makes two key predictions. First, because of stronger selection against misinteraction on more highly expressed proteins, the probability for each molecule to engage in misinteraction should decrease with its concentration (34). In other words, highly expressed proteins should be less sticky than lowly expressed ones. Second, because of the constraint imposed by misinteraction avoidance, nonsticky residues on protein surfaces are prohibited from changing to sticky residues, whereas no such constraints are imposed on sticky surface residues. Because the pressure to avoid misinteraction increases with protein abundance, we predict that the substitution rate of surface nonsticky residues, relative to the substitution rate of surface sticky residues, decreases with protein abundance.
Below, we provide evidence for the first prediction using information from protein sequences and protein misinteractions. As aforementioned, the fraction of surface residues that are hydrophobic can be used as a proxy for protein stickiness. Consistent with our prediction, this fraction decreases with rising protein abundance (Fig. 4A). We also used quantitative measures of amino acid hydrophobicity (39) and observed a negative correlation between the mean hydrophobicity of surface residues of a protein and the abundance of the protein (Fig. 4B). By contrast, these patterns were not observed for buried residues (Fig. S2). The two proxies of protein stickiness remain significantly correlated with protein abundance after we control the fraction of matching sites (i.e., pmisfold rank = 1) on protein surfaces (ρ = −0.105, P < 0.05 and ρ = −0.123, P < 0.03, respectively), suggesting that the lower stickiness of abundant proteins is not explainable by protein misfolding avoidance. Because different amino acids have different biosynthetic costs, it has been shown that amino acid frequencies vary among proteins of different expression levels (40). Nonetheless, the above two proxies of protein stickiness remain significantly negatively correlated with protein abundance even after we control the amino acid synthetic costs under either fermentative or respiratory conditions (Fig. 4 A and B legend). Another proxy for protein stickiness is the fraction of amino acid residues located in intrinsically unstructured or disordered regions of a protein, because these regions tend to mediate protein misinteraction (32). Again, we found this proxy of stickiness to decrease with rising protein abundance (Fig. 4C). We also confirmed that these patterns remain qualitatively unchanged even when proteins of the same gene ontology (41) functional categories (e.g., enzymes or ligands/receptors) were compared (Fig. S3). Thus, three lines of evidence from protein sequences support the first prediction of our hypothesis.
Fig. 4.

Yeast proteins with higher abundance (number of molecules per cell) are less sticky. (A) The fraction of surface residues that are hydrophobic decreases with rising protein abundance. The correlation becomes ρ = −0.134 (P < 10−6) and −0.098 (P < 10−3) after control for amino acid synthetic costs under fermentative and respiratory conditions, respectively. (B) The mean hydrophobicity on the surface decreases with rising protein abundance. Note that a more positive hydrophobicity score indicates higher hydrophobicity. The correlation becomes ρ = −0.188 (P < 10−11) and −0.156 (P < 10−8), respectively, after control for amino acid synthetic costs under fermentative and respiratory conditions, respectively. (C) The fraction of residues within disordered regions decreases with rising protein abundance. (D) The number of interaction partners of a protein determined by Y2H assays, representing both specific and nonspecific partners, decreases with rising protein abundance. (E) The number of interaction partners of a protein determined by affinity-based assays, representing specific partners, increases with rising protein abundance. (F) The number of Y2H partners that are not affinity partners, representing nonspecific partners only, decreases with rising protein abundance. Genes are grouped into 10 bins of equal size based on expression levels, and each bin contains 376 genes. The error bar represents 1 SE. The protein abundance data are from ref. 27. All correlation coefficients and P values are determined from the original data rather than the binned data.
Protein–protein interactions have been probed experimentally by several different methods. Using the information in an earlier study (30), we consider interactions detected by yeast two-hybrid (Y2H) assays to include both functional interactions and misinteractions, because the interacting proteins are highly overexpressed in this assay (42). We found that the number of Y2H interactions that a protein has is negatively correlated with its native expression level (Fig. 4D). We consider interactions detected by affinity-based methods as largely functional and specific interactions, because in this method, proteins are expressed at their natural levels in their natural subcellular locations (43). Consistent with a recent report (34), the interaction number from affinity-based methods shows a strong positive correlation with protein abundance (Fig. 4E). A weaker positive correlation was found when we guarded against potential false positives in affinity data by requiring each functional interaction to have been identified at least three times (Fig. S4A). Interactions detected by protein fragment complementation assays also reflect functional interactions (44), and they similarly show a positive correlation between the abundance of a protein and its number of interactions (Fig. S5A). We then infer the number of misinteractions that a protein has by the number of Y2H interactions that are not found by affinity-based methods (or protein fragment complementation assays). As predicted by our hypothesis, the number of inferred misinteractions decreases with protein abundance (Fig. 4F and Figs. S4B and S5B). Note that the inferred number of misinteractions can be compared among different proteins, because all proteins are overexpressed to a similar level in Y2H that is even higher than the expression of the most highly expressed gene in yeast. This overexpression also ensures that false positives and false negatives in high-throughput Y2H experiments do not differentially affect proteins of different natural concentrations. In affinity-based methods and protein fragment complementation assays, high-concentration proteins may have higher detectabilities than low-concentration proteins. However, our conclusion is not dependent on the positive correlations observed in Fig. 4E (Figs. S4A and S5A). That is, even when the numbers of functional interactions are comparable among proteins of different concentrations, the Y2H data still suggest that misinteractions are fewer for proteins of higher concentrations. Thus, the first prediction of the misinteraction avoidance hypothesis is also supported by protein misinteraction data.
To test the second prediction of our hypothesis, we calculated the ratio between the substitution rate of surface hydrophilic (i.e., nonsticky) residues and the substitution rate of surface hydrophobic (i.e., sticky) residues in S. cerevisiae proteins using the alignment of orthologous proteins from six post-WGD species. Because of the large sampling error of the ratio calculated from individual proteins, we calculated this ratio for groups of proteins with similar levels of abundance. To increase sensitivity, we focused on strongly hydrophobic (hydrophobicity score > 2) and strongly hydrophilic (hydrophobicity score < −2) amino acids (39). As predicted, this ratio decreases significantly with rising protein abundance (Fig. 5A). As a control, we also examined the same substitution rate ratio using protein cores, but we observed no significant relationship between the ratio and protein abundance (Fig. 5B).
Fig. 5.

Misinteraction avoidance constrains amino acid substitutions on protein surfaces but not cores. (A) The ratio between the substitution rate of surface hydrophilic residues and the substitution rate of surface hydrophobic residues decreases with rising protein abundance. (B) The ratio between the substitution rate of buried hydrophilic residues and the substitution rate of buried hydrophobic residues does not decrease with rising protein abundance. Each dot represents ∼4,700 aa from ∼40 proteins with similar abundances (number of protein molecules per cells). The protein abundance data were from an earlier study (27).
Misinteraction Avoidance Explains the Protein Surface E-R Anticorrelation Better than Misfolding Avoidance.
To assess the relative importance of misinteraction avoidance and misfolding avoidance in generating the E-R anticorrelation for protein surfaces, we separately removed sites under each constraint. Specifically, we progressively removed surface sites constrained for misinteraction avoidance from those sites with low hydrophobicity to those sites with high hydrophobicity. When two sites have the same hydrophobicity score, we first removed the one with the larger solvent accessibility (i.e., more exposed). As a comparison, in each protein, we separately removed the same number of surface sites constrained most for misfolding avoidance according to the pmisfold rank. We found that removing sites by hydrophobicity is more effective than removing sites by the pmisfold rank in weakening the E-R anticorrelation on protein surfaces (Fig. 6A). To evaluate the robustness of this result, we bootstrapped all yeast proteins 1,000 times and found that the above result is true in a vast majority of bootstrap samples (Fig. 6B). The pmisfold rank at a specific site explicitly measures the misfolding probability of the WT protein relative to the probability of the 60 possible codon replacements at the site (20), whereas hydrophobicity is only one of multiple determinants of misinteraction and is only amino acid-specific and not site-specific; therefore, the pmisfold rank likely measures the misfolding probability more accurately than hydrophobicity measures the misinteraction probability. Thus, the result in Fig. 6 is expected to be conservative.
Fig. 6.

Misinteraction avoidance explains the E-R anticorrelation for protein surfaces better than misfolding avoidance. (A) E-R anticorrelation for protein surfaces after progressive removals of surface hydrophilic residues (step size = 5%). For comparison, the same number of surface sites is removed from each protein based on the pmisfold rank. (B) Fraction of 1,000 bootstrap replications in which removing sites constrained by misinteraction avoidance is more effective than removing the same number of sites constrained by misfolding avoidance in weakening the E-R anticorrelation on protein surfaces.
Discussion
In this work, we showed that the protein misfolding avoidance hypothesis cannot fully explain the E-R anticorrelation, especially for protein surface residues. Instead, we propose and show that protein misinteraction avoidance explains the E-R anticorrelation for protein surfaces better than misfolding avoidance. The two hypotheses have several similarities that are worth commenting on. First, the deleterious effects from protein misfolding and misinteraction are both protein concentration-dependent, a requisite for any explanation of the E-R anticorrelation. Second, protein misfolding and misinteraction both reduce the amount of proteins available for performing physiological functions. Third, both misfolding and misinteraction can lead to protein aggregation, although the causes of the aggregation may differ. Fourth, both hypotheses can explain, at least in part, the phenomenon of biased synonymous codon use. It has been shown that misfolding avoidance is partially achieved by a reduction in mistranslation through the use of optimal codons that have high translational accuracies (13, 20, 22). In principle, the pressure to minimize misinteraction can also result in a reduction in mistranslation through the use of accurately translated codons. In this work, we have chosen to focus on protein sequence evolution only, and we will analyze the impact of misinteraction avoidance on synonymous codon use in a separate study.
Apart from the four similarities, the two hypotheses have three major differences. First, selection against misfolding acts primarily, albeit not exclusively, on the buried residues of a protein, which are most important for protein stability, whereas selection against misinteraction acts on protein surfaces, which determine protein–protein interaction. Hence, they complement each other in generating the E-R anticorrelation for entire protein molecules. Second, protein misinteraction can generate a gain of function effect, inducing erroneous cellular processes, which has been documented in some mutants of p53 (31, 45). By contrast, protein misfolding does not have such effects. Third, although misfolding affects only the misfolded protein itself, misinteraction affects multiple proteins. Hence, when a highly abundant protein is sticky, it could form misinteractions with many other proteins and affect multiple cellular processes. Thus, although the deleterious effect of misfolding is localized and predictable, the effect of misinteraction can be global and unpredictable.
In addition to the evidence documented here for the protein misinteraction avoidance hypothesis of E-R anticorrelation, there are additional observations in the literature that are consistent with this hypothesis. First, Plata et al. (46) found a positive correlation between protein abundance and the fraction of charged (i.e., hydrophilic) residues on solvent accessible sites in Escherichia coli, which is highly consistent with our yeast observation in Fig. 4A, suggesting the applicability of the protein misinteraction avoidance hypothesis in prokaryotes as well. Second, it was reported that the difference in sequence conservation between surface residues involved in functional protein interactions (i.e., functional interfaces) and other surface residues decreases with rising expression level (47). This observation is likely because of an increasing constraint on these nonfunctional interfaces with rising expression level caused by misinteraction avoidance compared with the constraint on functional interfaces. Third, as mentioned, Zhang et al. (30) and Heo et al. (34) studied the biophysical properties of protein misinteraction. Their results, from both simulation and empirical studies, strongly support our hypothesis.
We showed that removing surface hydrophilic residues, which are likely constrained by misinteraction avoidance, weakens the E-R anticorrelation for protein surfaces (Fig. 6A). Nevertheless, even when 50% of surface residues are removed, the E-R anticorrelation is still strong (Fig. 6A). This observation has at least two explanations. First, although hydrophobicity affects the stickiness of a residue, it is by no means the sole determinant. Stickiness is likely influenced by additional factors (e.g., disorder in structure). Thus, removing hydrophilic sites may be rather ineffective in eliminating residues constrained by misinteraction avoidance. Second, it is possible that misfolding avoidance and misinteraction avoidance are but two of potentially many mechanisms underlying the E-R anticorrelation. For example, Gout et al. (48) and Cherry (49) recently proposed a hypothesis of selection for protein function that, in principle, can also explain the E-R anticorrelation, although it has yet to be empirically verified. Regardless of whether their hypothesis is correct, the E-R anticorrelation is the result of at least two factors: misfolding avoidance and misinteraction avoidance. In the future, it would be interesting to identify sites that would most effectively weaken the E-R anticorrelation when deleted and then study the properties of these sites to find the potential causes of the E-R anticorrelation.
Although our computer simulation focused on the role of misinteraction avoidance in constraining the evolution of proteins with presumably unchanged functions, the same constraint can also hinder neofunctionalization in protein evolution; therefore, a mutation conferring a new function may be unacceptable, because it compromises misinteraction avoidance (50). It is possible that the E-R anticorrelation reflects reductions of both neutral substitution rates and advantageous substitution rates in highly expressed proteins. Our misinteraction avoidance hypothesis may also be extended to include misinteractions between proteins and nonprotein molecules such as DNA and RNA. Future work is needed to evaluate the impact of such events on protein evolution. Because misinteraction may result in a gain of function, it could occasionally be beneficial under certain conditions. Thus, new functional protein interactions could originate from initial misinteractions through mutation and selection (29). Because the smaller the effective population size, the weaker the selection against protein stickiness, one may predict that protein interactions and protein complexes are more prone to evolve in species with smaller populations, which has been recently confirmed (51). Misinteraction, an inevitable phenomenon in any complex system, may, thus, both constrain and channel the evolution of the system.
Materials and Methods
Yeast Genomic Data and Comparative Analysis.
The cDNA and protein sequences of S. cerevisiae were downloaded from the Saccharomyces Genome Database (52). Protein sequences of five other post-WGD fungi (S. paradoxus, S. mikatae, S. bayanus, Candida glabrata, and S. castellii) and their orthologous relationships with S. cerevisiae proteins were extracted from the Fungal Orthogroups Repository (53). Only those genes that have one to one orthologs in each of the six species were used. Orthologous protein sequences from the six species were aligned using ClustalW (54), and the substitution rate at each amino acid position of an alignment was estimated by GAMMA (55). We used microarray-based measurements of S. cerevisiae mRNA expression levels (56) and immunodetection-based measurements of protein expression levels (27). Amino acid hydrophobicity scores were previously published (39). Qualitatively, amino acids A, M, C, F, L, V, and I were considered hydrophobic because of their positive hydrophobicity scores (39), and the other 13 amino acids were considered hydrophilic because of their negative hydrophobicity scores. Protein–protein interaction data of S. cerevisiae were downloaded from BioGRID v3.1.82 (57).
Estimation of pmisfold.
We used a previously derived equation (20) to calculate pmisfold, the probability of protein misfolding of a mutant gene relative to that of the WT gene. Here, each examined mutant differs from the WT gene by one codon replacement, and all 60 possible codon replacements are examined at each codon position of every gene. The calculation of pmisfold considers both translational error-free and -induced misfolding and involves the use of a computationally predicted change of protein stability (ΔΔG) due to a codon replacement (25) and the probability of translational error (20).
Protein Structures.
To determine whether a residue lies on the surface of a protein molecule, we BLASTed yeast proteins against all protein sequences from the Protein Data Bank (PDB) (58) using an E-value cutoff of 10−6. A yeast protein was considered to have sufficient matches in PDB only when, in total, over 50% of its residues were aligned to the significant hits. For each yeast protein with sufficient PDB matches, the matched PDB entries were analyzed by the program DSSP to obtain a solvent accessibility score for each residue (59). Because sequence similarity usually coincides with structural similarity, this score was used as the solvent accessibility score for the aligned yeast protein residue. Sometimes, a multidomain yeast protein was matched to multiple PDB entries. Because the conformations of different domains in the same protein are relatively independent from one another and linkers between domains rarely cover surfaces, we accepted accessibility scores from different PDB entries for different parts of a protein based on the best match of each domain. Such a strategy was supported by the observation that use of the best PDB hit or second best hit for solvent accessibility determination yielded similar results: 84.3% of residues were identically categorized into surface and buried residues. Amino acids with solvent accessibility scores larger than 50, meaning that the residue is simultaneously accessible by at least five water molecules (59), were considered as surface residues; otherwise, they were considered buried. Potential errors in solvent accessibility determination make our findings of differences between surface and buried residues conservative.
We used RONN to estimate the probability that a residue is natively disordered for every residue of every yeast protein, and those residues with the probability > 0.5 were considered as disordered residues (60).
Computer Simulation of the Interactome.
We built a molecular-level biophysical model with baseline selective constraints on protein folding to investigate the impact of protein misinteraction avoidance on protein sequence evolution. First, 200 protein sequences, each with a fixed length of 27 aa, were generated randomly. Given the sequence of a protein, we calculated its folding energy for each possible structure in a 3 × 3 × 3 lattice by the sum of the contact energies of spatially adjacent residues (61). A folding Z score for structure i of a protein sequence was defined as (Eq. 1)
 |
where Ei is the folding energy of structure i and μE and σE are the mean and SD of the folding energies of all possible structures of the protein, respectively. For each protein sequence, we randomly chose a structure with Fi < −7 as its native structure, which ensured fast and stable folding to the native structure (35). The native structure of a protein was fixed during the simulation of evolution. Second, we need to define the interaction energies for 40,000 possible pairs of folded protein cubes. To simplify the problem, we considered only interactions mediated by the whole surface on one side of a cube (that is, by nine intermolecule pairs of amino acids). For any two cubes and an interaction orientation, the contact energies of the nine intermolecule pairs of interacting amino acids were summed up as the interaction energy between the two proteins for the specific orientation. Third, we randomly divided the 200 proteins into 100 pairs of specific interaction partners. We optimized the specific interaction for each protein, and therefore, its binding Z score, defined as (Eq. 2)
 |
was as small as possible (35). Here, protein i and protein j are specific interaction partners with a specific orientation, with the interaction energy being Eij. Additionally,  and
and  are the mean and SD of interaction energies of all other interactions involving protein i in any possible orientation, respectively (including with the specific partner in nonspecific orientations). The specific interaction between proteins i and j was required to satisfy Fi < −6, Fj < −6, and Bij + Bji < −14 (35). All interactions except those interactions between specific partners in specific orientations are considered as misinteractions.
are the mean and SD of interaction energies of all other interactions involving protein i in any possible orientation, respectively (including with the specific partner in nonspecific orientations). The specific interaction between proteins i and j was required to satisfy Fi < −6, Fj < −6, and Bij + Bji < −14 (35). All interactions except those interactions between specific partners in specific orientations are considered as misinteractions.
The genome of the progenitor cell in the in silico evolution consisted of these 200 genes. We randomly generated 100 expression levels that follow a power law distribution (36) and assigned them to each pair of specific interacting partners. In other words, specific interaction partners had exactly the same expression levels, whereas nonspecific interaction partners could have different expression levels. We required all of the expression levels to be integers no less than 1 μM, and the largest expression should be at least 50 μM. To have a gradient of expression levels among genes, we required that the expression difference between two adjacent genes when ranked by expression level should be less than 5 μM.
With the expression levels and interaction energies determined and thermodynamic equilibrium assumed, we estimated the probability that protein i is in a complex with protein j by solving the following quadratic system (Eq. 3):
 |
 |
Here, Ci is the concentration of free molecules of protein i (unbound to any molecule), Cij is the concentration of the protein complex composed of a protein i and a protein j, Di is the total concentration of protein i in the cell (i.e., the expression level), R is the Boltzmann constant of 1.986 cal/mol per K, T is the absolute temperature, ∀ means for any, and ∃ means there exists. In Eq. 3, Eij is the overall binding energy between proteins i and j in all 144 orientations, and is calculated by
 |
where Eijk is the binding energy between i and j in the kth orientation, calculated from the contact energy between the nine amino acid pairs of i and j that are in contact.
There are 20,300 equations with 20,300 variables to be solved in this quadratic system. We used an iterative method to approach the solution of this quadratic system. Specifically, we started with an arbitrary set of Ci values and calculated Cij values based on the interaction energies using Eq. 3. We then adjusted Ci to be 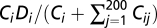 , where Di is the assigned expression level of the protein i. We repeated this process many times until the absolute value of the fractional adjustment in the sum of Ci between two consecutive iterations was smaller than 10−5. We tried multiple different sets of initial values of Ci and found no difference in final results.
, where Di is the assigned expression level of the protein i. We repeated this process many times until the absolute value of the fractional adjustment in the sum of Ci between two consecutive iterations was smaller than 10−5. We tried multiple different sets of initial values of Ci and found no difference in final results.
We defined the fitness of a cell by 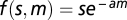 , where s is the product of the fractions of molecules engaged in specific interactions across 100 specific complexes, m is the total concentration (in micromolar) of misinteraction complexes, and a is a constant that determines the toxicity of an average misinteraction. Without loss of generality, we assigned a = 1. The above fitness function ensures that the relative fitness cost of each additional misinteraction is the same. We also repeated the simulation using a = 0 to examine the outcome when misinteraction is not toxic (Fig. S1).
, where s is the product of the fractions of molecules engaged in specific interactions across 100 specific complexes, m is the total concentration (in micromolar) of misinteraction complexes, and a is a constant that determines the toxicity of an average misinteraction. Without loss of generality, we assigned a = 1. The above fitness function ensures that the relative fitness cost of each additional misinteraction is the same. We also repeated the simulation using a = 0 to examine the outcome when misinteraction is not toxic (Fig. S1).
At the beginning of the simulated evolution, the population contained 100 identical cells. Random mutations were introduced at the rate of 0.0005 per residue per generation, with the requirement that the folding Z score of any protein must be lower than −2. Fitness was calculated for each cell, and the next generation of cells was generated by considering each cell's fitness and genetic drift. This process of mutation, selection, and drift was repeated 19,900 generations to reach the equilibrium. We then evolved the population for 100 additional generations and counted the number of fixed amino acid changes from the 19,900th to the 20,000th generation. We repeated the whole simulation 100 times with different protein sequences but the same set of expression levels.
Supplementary Material
Acknowledgments
We thank Meg Bakewell, Chungoo Park, Wenfeng Qian, the editor, and three anonymous reviewers for constructive comments. This work was supported by a research grant from the US National Institutes of Health (to J.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 5158 (volume 109, number 14).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1117408109/-/DCSupplemental.
References
- 1.Hurst LD, Smith NG. Do essential genes evolve slowly? Curr Biol. 1999;9:747–750. doi: 10.1016/s0960-9822(99)80334-0. [DOI] [PubMed] [Google Scholar]
- 2.Pál C, Papp B, Hurst LD. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang J, He X. Significant impact of protein dispensability on the instantaneous rate of protein evolution. Mol Biol Evol. 2005;22:1147–1155. doi: 10.1093/molbev/msi101. [DOI] [PubMed] [Google Scholar]
- 4.Wall DP, et al. Functional genomic analysis of the rates of protein evolution. Proc Natl Acad Sci USA. 2005;102:5483–5488. doi: 10.1073/pnas.0501761102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- 6.Zhang L, Li WH. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol. 2004;21:236–239. doi: 10.1093/molbev/msh010. [DOI] [PubMed] [Google Scholar]
- 7.Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- 8.Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2001;411:1046–1049. doi: 10.1038/35082561. [DOI] [PubMed] [Google Scholar]
- 9.Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW. Evolutionary rate in the protein interaction network. Science. 2002;296:750–752. doi: 10.1126/science.1068696. [DOI] [PubMed] [Google Scholar]
- 10.Jordan IK, Wolf YI, Koonin EV. No simple dependence between protein evolution rate and the number of protein-protein interactions: Only the most prolific interactors tend to evolve slowly. BMC Evol Biol. 2003;3:1. doi: 10.1186/1471-2148-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolf MY, Wolf YI, Koonin EV. Comparable contributions of structural-functional constraints and expression level to the rate of protein sequence evolution. Biol Direct. 2008;3:40. doi: 10.1186/1745-6150-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolf YI, Carmel L, Koonin EV. Unifying measures of gene function and evolution. Proc Biol Sci. 2006;273:1507–1515. doi: 10.1098/rspb.2006.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–352. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Subramanian S, Kumar S. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics. 2004;168:373–381. doi: 10.1534/genetics.104.028944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rocha EP, Danchin A. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 2004;21:108–116. doi: 10.1093/molbev/msh004. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Zhang J. Why is the correlation between gene importance and gene evolutionary rate so weak? PLoS Genet. 2009;5:e1000329. doi: 10.1371/journal.pgen.1000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao BY, Weng MP, Zhang J. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2010;2:39–43. doi: 10.1093/gbe/evp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geiler-Samerotte KA, et al. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci USA. 2011;108:680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci USA. 2005;102:14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JR, Zhuang SM, Zhang J. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol Syst Biol. 2010;6:421. doi: 10.1038/msb.2010.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 22.Zhou T, Weems M, Wilke CO. Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol Evol. 2009;26:1571–1580. doi: 10.1093/molbev/msp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaramillo A, Wernisch L, Héry S, Wodak SJ. Folding free energy function selects native-like protein sequences in the core but not on the surface. Proc Natl Acad Sci USA. 2002;99:13554–13559. doi: 10.1073/pnas.212068599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berezovsky IN, Zeldovich KB, Shakhnovich EI. Positive and negative design in stability and thermal adaptation of natural proteins. PLoS Comput Biol. 2007;3:e52. doi: 10.1371/journal.pcbi.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capriotti E, Fariselli P, Casadio R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huh WK, et al. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 27.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 28.Qian W, He X, Chan E, Xu H, Zhang J. Measuring the evolutionary rate of protein-protein interaction. Proc Natl Acad Sci USA. 2011;108:8725–8730. doi: 10.1073/pnas.1104695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuriyan J, Eisenberg D. The origin of protein interactions and allostery in colocalization. Nature. 2007;450:983–990. doi: 10.1038/nature06524. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Maslov S, Shakhnovich EI. Constraints imposed by non-functional protein-protein interactions on gene expression and proteome size. Mol Syst Biol. 2008;4:210. doi: 10.1038/msb.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stambolsky P, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 2010;17:273–285. doi: 10.1016/j.ccr.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell. 2009;138:198–208. doi: 10.1016/j.cell.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Johnson ME, Hummer G. Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks. Proc Natl Acad Sci USA. 2011;108:603–608. doi: 10.1073/pnas.1010954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heo M, Maslov S, Shakhnovich E. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc Natl Acad Sci USA. 2011;108:4258–4263. doi: 10.1073/pnas.1009392108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deeds EJ, Ashenberg O, Gerardin J, Shakhnovich EI. Robust protein protein interactions in crowded cellular environments. Proc Natl Acad Sci USA. 2007;104:14952–14957. doi: 10.1073/pnas.0702766104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ueda HR, et al. Universality and flexibility in gene expression from bacteria to human. Proc Natl Acad Sci USA. 2004;101:3765–3769. doi: 10.1073/pnas.0306244101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deeds EJ, Ashenberg O, Shakhnovich EI. A simple physical model for scaling in protein-protein interaction networks. Proc Natl Acad Sci USA. 2006;103:311–316. doi: 10.1073/pnas.0509715102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miyazawa S, Jernigan R. Estimation of effective interresidue contact energies from protein crystal structures: Quasi-chemical approximation. Macromolecules. 1985;18:534–552. [Google Scholar]
- 39.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 40.Akashi H, Gojobori T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci USA. 2002;99:3695–3700. doi: 10.1073/pnas.062526999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashburner M, et al. Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 43.Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 44.Tarassov K, et al. An in vivo map of the yeast protein interactome. Science. 2008;320:1465–1470. doi: 10.1126/science.1153878. [DOI] [PubMed] [Google Scholar]
- 45.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plata G, Gottesman ME, Vitkup D. The rate of the molecular clock and the cost of gratuitous protein synthesis. Genome Biol. 2010;11:R98. doi: 10.1186/gb-2010-11-9-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eames M, Kortemme T. Structural mapping of protein interactions reveals differences in evolutionary pressures correlated to mRNA level and protein abundance. Structure. 2007;15:1442–1451. doi: 10.1016/j.str.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gout JF, Kahn D, Duret L. The relationship among gene expression, the evolution of gene dosage, and the rate of protein evolution. PLoS Genet. 2010;6:e1000944. doi: 10.1371/journal.pgen.1000944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cherry JL. Expression level, evolutionary rate, and the cost of expression. Genome Biol Evol. 2010;2:757–769. doi: 10.1093/gbe/evq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liberles DA, Tisdell MD, Grahnen JA. Binding constraints on the evolution of enzymes and signalling proteins: The important role of negative pleiotropy. Proc Biol Sci. 2011;278:1930–1935. doi: 10.1098/rspb.2010.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernández A, Lynch M. Non-adaptive origins of interactome complexity. Nature. 2011;474:502–505. doi: 10.1038/nature09992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Engel SR, et al. Saccharomyces Genome Database provides mutant phenotype data. Nucleic Acids Res. 2010;38:D433–D436. doi: 10.1093/nar/gkp917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wapinski I, Pfeffer A, Friedman N, Regev A. Natural history and evolutionary principles of gene duplication in fungi. Nature. 2007;449:54–61. doi: 10.1038/nature06107. [DOI] [PubMed] [Google Scholar]
- 54.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 55.Gu X, Zhang J. A simple method for estimating the parameter of substitution rate variation among sites. Mol Biol Evol. 1997;14:1106–1113. doi: 10.1093/oxfordjournals.molbev.a025720. [DOI] [PubMed] [Google Scholar]
- 56.Holstege FC, et al. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 57.Stark C, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berman HM, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 60.Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- 61.Mirny LA, Shakhnovich EI. How to derive a protein folding potential? A new approach to an old problem. J Mol Biol. 1996;264:1164–1179. doi: 10.1006/jmbi.1996.0704. [DOI] [PubMed] [Google Scholar]
