

Abstract
Receptor-initiated phospholipase C activation and generation of IP3 and DAG are important common triggers for a diversity of signal transduction processes in many cell types. Contributing to this diversity is the existence and differential cellular and subcellular distribution of distinct phospholipase C isoforms with distinct regulatory prop- erties. The recently identified PLCε enzyme is an isoform that is uniquely regulated by multiple upstream signals including ras-family GTP binding proteins as well as heterotrimeric G-proteins. In this review we will consider the well documented biochemical regulation of this isoform in the context of cell and whole animal physiology and in the context of other G protein-regulated PLC isoforms. These studies together reveal a surprisingly wide range of unexpected functions for PLCε in cellular signaling, physiology and disease.
Keywords: Phospholipase C ε, G protein-coupled receptor, Ras family GTPase, Cardiovascular function, Diabetes, Inflammation
1. Introduction
1.1. PLC overview
Receptor-stimulated hydrolysis of phosphatidylinositol 4,5 bisphosphate (PIP2) to generate inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG) is a fundamental signaling process in the bi- ology of virtually all mammalian cells and in many lower eukaryotes. The proteins that catalyze this reaction are the phosphoinositide- specific phospholipase Cs (PLCs), which function at key control points for directing signaling responses to a variety of regulatory stimuli[26,27,59,75] (Fig. 1). The immediate consequences of this reaction are manifold and are initiated through two canonical signaling pathways downstream: regulation of Ca2+ release into the cytoplasm from intra- cellular endoplasmic reticulum stores, and activation of protein kinase C (PKC) cascades. Additionally, PIP2 is a signaling molecule in its own right, most notably as a stabilizer of ion channel activity, with PIP2 deple- tion serving as a mechanism for the regulation of channel activity[30,74,76]. All of these processes have the potential to dramatically alter cellular physiology and contribute to cellular pathophysiology. There are multiple PLC isoforms that are differentially localized and regulated, enabling them to participate in a variety of distinct physiolog- ical processes. This review will focus on emerging physiological and pathophysiological roles for one recently identified isoform of PLC, PLCε. PLCε is unique in relation to other phospholipase C isoforms in terms of its ability to be regulated by multiple signaling inputs from both Ras family GTPases and heterotrimeric G proteins. In addition, it contains a small GTPase nucleotide exchange factor domain that serves to function in activating Ras family GTPases. Thus, PLCε has the capacity to respond to, and integrate diverse cellular information as well as partic- ipate in more sustained signal generation. In this review, we will discuss how biochemically defined cellular signaling mechanisms both upstream and downstream of PLCε function to regulate specific physiological functions.
Fig. 1.

Phospholipase C isoforms. A) Reaction catalyzed by PI-PLC and overall structure. Structure of PLCβ2 (Protein Database ID: 2ZKM) taken from [29] and rendered with Molsoft ICM. The catalytic domain comprised the X and Y domains is in red, the Pleckstrin Homology (PH) domain is in blue, the EF hand domain is in yellow and the C2 domain is in purple. B) PLC isoforms with domains color coded as in panel A. C) Two splice variants of PLCε which include in addition to the aforementioned common domains, 2 ras association ho- mology (RA) domains and a CDC25 homology guanine nucleotide exchange factor (GEF) domain.
1.2. PLC isoforms and general regulatory properties
The family of phospholipase C enzymes includes 13 different isozymes grouped into 6 different classes based on sequence homology (Fig. 1B and C). All phospholipase C isoforms contain highly conserved X and Y domains that fold to form the catalytic core of the enzyme, a phos- pholipid binding C2 domain, and an EF hand domain [21,26,27,59,75] (Fig. 1A, B and C). Outside of the core conserved regions, there is diver- sity in protein structure that reflects the range of mechanisms utilized for regulation of these enzymes, and that underlies the basis for classifica- tion of the different groups. There are 4 members of the β class, 2 mem- bers of the γ class and 3 members of the δ class. These groupings also correspond to functional classifications. Members of the PLCγ class are regulated by receptors that are coupled to tyrosine kinases [9,51,59]. Members of the PLCβ class are activated by G protein subunits[8,69,70,79,82]. PLCδ activation mechanisms are less well defined, but its unusual sensitivity to Ca2+ puts it in a position to amplify Ca2+ re- sponses initiated by other PLC isoforms [58] or other mechanisms [80]. PLCζ, comprises a class of enzyme localized to sperm and it is involved in fertilization [77]. PLCη is a neuron-specific class whose function has yet to be defined, but it appears to be regulated by G protein βγ subunits [52,91].
2. PLCε structure and regulatory properties
2.1. PLCε discovery
The first identification of a PLCε homologue was in a yeast two- hybrid screen of a Caenorhabditis elegans library with a C. elegans Ras homologue LET-60 as the bait [65]. This 210 kDa protein, PLC210, was shown to bind Ras and possess PLC activity. Subsequently, mammalian isoforms of PLC210 were independently identified and cloned by three groups using homology based screening of human and rat EST databases [43,47,71]. The PLC210 and PLCε clones were highly homologous, and contained Ras association (RA) homology do- mains and, surprisingly, a putative small GTPase nucleotide exchange factor domain (Fig. 1C). Subsequently, two forms of PLCε were found to arise from alternative splicing at the amino terminus. These are des- ignated as PLCε1a and PLCε1b, and they differ in size by 25 kDa [73] (Fig. 1C). No functional differences between the two splice variants have as yet been identified.
Analysis of the PLCε mRNA content of various tissues/organs by either northern blot [43,47] or reverse transcriptase PCR [73] suggests a relatively widespread distribution of both spliced transcripts, although there are some differences in splice variant distributions. Analysis by immunoblotting has proven to be more challenging even though rela- tively strong antibodies for PLCε detection have been developed [85]. In many tissues PLCε is not readily detectable by direct immunoblotting, but rather requires an immunoprecipitation step from tissue lysates to enrich the PLCε protein prior to immunoblotting ([85,90], and unpub- lished observations). These observations have led to the suggestion that PLCε protein is present in low abundance in most tissues and cell types and at significantly lower levels than other PLC isoforms, although a systematic quantitative analysis supporting this idea is lacking.
2.2. PLC regulation in transfected cells
Since the initial demonstration that PLCε can be activated by Ras in transfected cells [43,71], it has become clear PLCε can be activated by a wide variety of protein partners (Table 1). These include members of the Ras family such as Rap1, Rap2, TC21, Ral, Rho and Rac as well as the heterotrimeric G protein subunits Gα12, Gα13, and βγ [36,44,47,63,88]. Thus receptors that either directly or indirectly regulate these G proteins have the potential to regulate PLCε (Fig. 2) (Table 2). For example, G protein-coupled receptors (GPCRs) that bind lysophosphatidic acid (LPA), sphingosine-1-phosphate (S1P), and thrombin (PAR) activate re- combinant PLCε heterologously expressed in COS-7 cells [24], or endog- enous PLCε in Rat1 fibroblasts [44]. Blocking Gα12/13, Gi, or Rho activation could inhibit activation of PLCε depending on the receptor being tested[24,44]. Thus, individual G-protein coupled receptors appear to use distinct signaling pathways to activate PLCε.
Table 1.
Phospholipase Cε activators.
| Direct binding to PLCε |
PLCε domain that binds |
Assays/tools | Reference |
|---|---|---|---|
| Rho | Y-domain insert required for Rho- dependent activation but does not bind Rho |
In vitro reconstitution purified Y domain deletion |
[63] |
| H-Ras | RA2 domain | GST-pulldown, ITC,a | [43] |
| cotransfection enzyme activation assayb |
[7] | ||
| K-Ras, N-Ras | RA2 domain | GST-pulldown | [7] |
| R-Ras | RA2 domain |
GST-pulldown, ITC | [7] |
| Rap1A&B | RA2 domain | GST-pulldown, ITC, | [44] |
| cotransfection enzyme activation assay |
[7] | ||
| Rap2A | RA2 domain | GST-pulldown, ITC, | [44] |
| cotransfection enzyme activation assay |
[7] | ||
| Ral | ?, not RA dependent | GST-pulldown, cotransfection enzyme activation assay |
[44] |
| Rac | ?, not RA dependent | GST-pulldown, cotransfection enzyme activation assay |
[44] |
| TC21 | RA2 domain | GST-pulldown, | [44] |
| cotransfection enzyme activation assay |
[7] | ||
| Indirect activation of PLCε | |||
| Gα12/13 | ? | Cotransfection enzyme | [44] |
| activation assay, RGS inhibition |
[47] | ||
| β γ | ? | Cotransfection enzyme activation assay, GRK2 ct |
[88] |
Isothermal titration calorimetry.
Activator and PLCe transfected into cells and total inositol phosphates were measured.
Fig. 2.

Common mechanisms for PLCε regulation of CICR in cardiac myocytes and pancreatic β cells downstream of Gs-coupled receptors. A) Common upstream components regulate of PLCε by Gs coupled receptors in adult cardiac myocytes and isolated pancreatic β cells [19,54]. B) Signaling downstream of PLCε. In red are components found to be involved in regulation of CICR in both cardiac myocytes and pancreatic β cells [19,54]. An alternative pathway has also been proposed and is depicted in black and may operate in β cells as well [57].
Table 2.
Ligands/receptors shown to couple to phospholipase Cε activation.
| Ligands/receptora | Cell type | Signaling mechanism | |
|---|---|---|---|
| GPCRs | |||
| LPA/Edg | Astrocytes | Gi/Gβγ | [13] |
| Cos-7 | G12/13/Rho | [44] | |
| Rat-1 fibroblasts | ? | [42] | |
| Thrombin/PAR | Astrocytes | G12/13/Rho | [13] |
| Cos-7 | G12/13/Rho | [44] | |
| Rat-1 fibroblasts | ? | [42] | |
| S1P/Edg | Astrocytes | Gi/Gβγ | [13] |
| Cos-7 | G12/13/Rho | [44] | |
| Endothelin/ET-1R | Rat-1 fibroblast | ? | [42] |
| NRVMb | ? | [90] | |
| Isoproterenol/β-adrenergic receptor |
NRVM | Gs/cAMP/Epac1/Rap | [90] |
| AVMc | Gs/cAMP/Epac1/Rap | [54] | |
| HEK293 | Gs/cAMP/Epac1/Rap | [61] | |
| Exendin-4/GLP-1R | Pancreatic β cells | Gs/cAMP/Epac2/Rap | [20] |
| Tyrosine kinases | |||
| EGF/EGF-R | COS-7 | Ras | [71] |
| [44] | |||
| PDGF/PDGF-R | BaF3 | [72] | |
| IGF-1/IGF-1-R | NRVM | ? | [90] |
In most cases the specific receptor subtypes have not been defined.
Neonatal rat ventricular myocytes.
Adult mouse ventricular myocytes.
Interestingly, in HEK293 cells, agonist stimulation of recombinant β2 adrenergic receptors activated PLCε in a cAMP, Epac (exchange protein directly activated by cAMP) and Rap GTPase-dependent manner [61]. Epac is a cAMP-regulated guanine nucleotide exchange factor that can activate Rap which in turn binds directly to PLCε, thereby stimulating the phospholipase. Growth factor-activated receptor tyrosine kinases were also shown to stimulate PLC activity in cell lines transfected with recombinant PLCε [71,72]. These studies in various biochemical systems and cell lines laid the groundwork for further elucidating how PLCε par- ticipates in signal transduction pathways downstream of multiple GPCR and growth factor receptors. This information is critical to understanding how PLCε participates in distinct physiological and pathophysiological responses.
2.3. Mechanisms of activation of PLCε
Direct interactions of upstream regulators with PLCε can be divided into 2 general classes: those that interact directly with the phospholipa- se’s second Ras association homology domain (RA2 domain) on PLCε, and those that bind elsewhere in the PLCε protein [44]. For example Ras and Rap bind directly to the RA2 domain while Rho, Ral and G12/13 do not. The site on PLCε required for Rho-dependent activation is an in- sert in the catalytic domain unique to PLCε [63]. Thus, similar to other PLCs, different regions of the enzyme are involved in interactions with different upstream activators. It is not clear that regulators such as G12/13 and Gβγ bind directly to PLCε. Indeed, Gα12/13 activation of PLCε is not likely to be direct but rather appears to occur through p115 RhoGEF or other RhoGEFs catalyzing the activation of RhoA which in turn binds directly to PLCε to activate it [24].
How direct binding of various G protein regulators to any PLC isoform leads to increased PLC activity remains unclear despite the emergence of new structural data for complexes of PLCβ with G-proteins [29,49,83]. One mechanism could involve translocation of cytosolic PLCs to the membrane where they would gain access to the PIP2 substrate. Addition- ally, it has been proposed, based on structural information from PLCβ, that membrane association can remove an auto-inhibitory element, common to all PLC isoforms, from the PLC catalytic core and thereby lead to activation [27,29]. In COS-7 cells, transfection with activated Ras or Rap causes translocation of GFP-PLCε from the cytosol to mem- branes. Ras promotes plasma membrane association of PLCε [71] while Rap promotes translocation of PLCε to perinuclear regions such as the Golgi apparatus [36]. Epidermal growth factor (EGF) also promotes translocation to plasma or Golgi membranes, dependent on Ras or Rap1 activation respectively [36,71]. Together, these data suggest that translocation from cytosol to membrane may, in part, underlie the mechanism for activation of PLCε. However, membrane targeting is not the only factor driving activation. Directing overexpressed PLCε to the membrane by appending a membrane targeting CAAX sequence at the C terminus of PLCε leads increased activity relative to a non- membrane targeted PLCε, but expression of Ras or stimulation with EGF further increases the activity of the CAAX modified PLCε [7]. These data indicate that while membrane targeting may be part of the activation mechanism, additional physical alterations of the enzyme at the membrane surface occur upon interaction with Ras or other activators.
2.4. Feed forward regulation of PLCε activity by Rap GEF
In addition to its PIP2 hydrolytic activity, PLCε has a CDC25 homology domain suggesting a putative guanine nucleotide exchange factor (GEF) function (Fig. 1C). There are conflicting reports concerning the activity of this domain in PLCε. One study suggested that PLCε may be a GEF for Ras, but the evidence is indirect [47]. In a separate study, Rap1A, but not Ras was a substrate for PLCε GEF in an in vitro assay with purified compo- nents [36]. Similarly, recombinant PLCε stimulated formation of RapGTP but not RasGTP in transfected COS-7 cells. In astrocytes from PLCε KO mice, hormonal activation of Rap was diminished while that of Ras was not [13], as detailed below. The cell biological function of the CDC25 domain in PLCε has been investigated extensively by the Kataoka group. Expression of PLCε or the CDC25 domain led to activation of the Rap effector pathways B-Raf and ERK. Sustained, but not acute, EGF receptor-dependent activation of Rap was enhanced by PLCε but only if it contained an intact CDC25 domain. Similarly EGF and Rap-dependent translocation of PLCε to the Golgi apparatus were independent of the CDC25 domain at short times of stimulation but sustained Golgi associa- tion was shown to be CDC25 domain-dependent [36]. The model suggested by these data is that Rap localization at the Golgi drives PLCε to the Golgi where it has access to substrate. PLCε in turn activates Rap which serves to reinforce PLCε association with Golgi and maintain the signal at the membrane. Similarly, PLCε CDC25 activity is required for the sustained activation of PLCε activity in a stable cell line expressing the PDGF receptor [72]. These data reinforce the concept that activated Rap generated by the PLCε CDC25 domain feeds forward to PLCε by binding the RA2 domain to maintain PLC activation.
Further support for the idea that PLCε is a Rap GEF in a physiological setting comes from studies of Rap activation in primary cells or tissues isolated from PLCε knockout mice. In astrocytes isolated from PLCε knockout mice, thrombin-stimulated sustained Rap activation was sig- nificantly decreased compared to wild type mice, while thrombin- stimulated Ras activation was unaffected by PLCε deletion [13]. Similarly in isolated mouse hearts, perfusion with isoproterenol (Iso) stimulated Rap activation in a manner that was completely lost in PLCε null mice [53]. The bioactive lysophospholipid S1P also activates Rap1 in the isolated mouse heart and this effect is diminished in PLCε knockout mice (Xiang et al., in preparation). In astrocytes, PLCε knockout eliminates the sustained phase of ERK activation that can be rescued by re-expression of PLCε but not with re-expression of PLCε with a mutation that disables the GEF domain of PLC (ΔCDC25 PLCε) [13]. Finally, in cardiac myocytes isolated from PLCε knockout mice, isoproterenol regulation of cardiac calcium cycling is impaired, but can be rescued by expression of wild- type (wt) PLCε but not ΔCDC25 PLCε [53]. These data suggest that PLCε GEF activity is important for sustained Rap activity in maintaining calcium cycling. Since PLCε activity involved in the regulation of cardiac contraction (as discussed below) we propose that Rap also activation generates a feed-forward amplification signal that maintains sustained PLCε activity at specific sites in cardiac myocytes, such as at the sarcoplasmic reticulum. Rap activation may also serve to signal to other pathways such as observed for sustained ERK activation in isolated astrocytes [13], and activation of PKD in isolated astrocytes (Dusaban, in preparation).
2.5. Role of PLCε in IP3 and DAG formation
Given the multiple direct activators of PLCε it stands to reason that PLCε can be a central integrator of multiple upstream signals. As dis- cussed above, PLCε can be activated by both GPCRs and receptor tyrosine kinases [24,36,44]. This raises the question as to the role of PLCε in gen- erating signals relative to other PLCs such as PLCβ and PLCγ that are well established as major mediators of PIP2 hydrolysis downstream of GPCRs and tyrosine kinase signaling, respectively [59]. Studies knocking down PLCε in cultured cells or genetically deleting PLCε in higher organisms such as mice have begun to shed light on this. In an initial study in Rat1 fibroblasts, siRNA knockdown of endogenous PLCε vs. PLCβ3 had different temporal effects on IP production downstream of receptor ac- tivation [42]. In general, knockdown of PLCβ3 significantly reduced short term IP generation (<3 min) whereas knockdown of PLCε did not significantly affect this response. Conversely, knockdown of PLCε sig- nificantly reduced longer term IP accumulation (3-60 min) in a manner that was unaffected by PLCβ3 knockdown. Although these effects were somewhat dependent on the receptor being tested, they are understand- able if PLCε is responsible for sustained PIP2 hydrolysis, consistent with the model discussed above whereby Rap1 activation by the PLCε- CDC25 domain can sustain PLCε activity. It is important to note that these assays of IP3 production are indirect in that they measure levels of inositol-1P generated from the metabolism of inositol phosphates. The lifetime of IP3 in receptor-stimulated assays where IP3 is measured is generally very short with most of the IP3 generated in the first minute after receptor stimulation and back to baseline within 1 or 2 min, al- though this can be receptor dependent. Thus during the sustained phase of IP production the level of IP3 that accumulates is generally quite low. This suggests that a major function of PLCε is to control longer term processes requiring sustained phosphoinositide hydrolysis, but perhaps involving DAG rather than IP3 as a major product.
An understanding of the contributions of PLCε signaling downstream from various receptors, relative to other PLC isoforms, has also arisen from studies of PLCε knockout mice. For example, IP production down- stream of various receptors was examined in astrocytes isolated from PLCε+/+ and PLCε−/− mice [13]. These studies revealed a large percent-age of the total IPs generated by S1P, thrombin or LPA were dependent on the presence of PLCε while IP generation stimulated by carbachol, which acts on the Gq coupled muscarinic receptor, was unaffected. In the case of thrombin, longer term IP generation was significantly re- duced in the PLCε knockout but in the case of carbachol, IP generation was not attenuated at any time in the PLCε knockout cells. In considering the receptors involved, and the possible G proteins they activate it is im- portant to note that LPA, thrombin and S1P all activate GPCRs coupled to Gq as well as G12/13 and in some cases Gi. In contrast, the muscarinic receptors in these cells are thought to couple purely to Gq. In these studies, C3 exoenzyme and pertussis toxin were used to block RhoA and Gi signaling respectively and indicate that PLCε activation by S1P and LPA was strongly dependent on Gi while thrombin signals to PLCε through RhoA, likely in a G12/13-dependent manner. Thus each re- ceptor uses a unique pathway to couple PLCε activation to IP production. Finally, it is noteworthy that in neonatal rat ventricular cardiac myocytes, inositol phosphate generation by the Gq-coupled, hypertrophic receptors for ET-1 and norepinephrine were unaffected by PLCε knockdown, but hypertrophic responses to these agonists were ablated [90]. In contrast SIP, which does not produce cardiomyocyte hypertrophy, induced mod- est inositol phosphate production in these cells, through G12/13 coupling to RhoA and is inhibited by PLCε knockdown (Xiang et al., in prepara- tion). These findings suggest that distinct pools of phosphoinositides are under the control of PLCε with some required for the hypertrophic response and others serving other functions.
3. Physiological roles of PLCε revealed in PLCε deficient mice
While the data discussed above reveal detailed signaling mecha- nisms involved in PLCε regulation and cellular function, many of the physiological functions for PLCε have emerged from studies of PLCε knockout animals produced independently in two laboratories (known physiological functions are summarized in Table 3). The phenotypes of these two animals do not entirely overlap in part because the laborato- ries involved have taken their studies in different directions, but also be- cause the nature of the knockouts are not identical. Tohru Kataoka’s laboratory generated a mouse lacking a portion of the EF-hand and the catalytic domain (PLCεΔx/Δx) [78]. As a result, a shortened version of PLCε is produced that is catalytically inactive with respect to PIP2. The knockout generated in our laboratory deleted the first common exon 6 in the PLCε gene resulting in complete loss of detectable PLCε protein [85]. Thus it might be expected that the phenotypes of these animals would differ significantly.
Table 3.
Summary of physiological roles for PLCε signaling.
| System(s) | Signaling | Functional role | References | |
|---|---|---|---|---|
| Cardiac | ||||
| Contractility | Knockout mouse; | βAR/cAMP/Epac/Rap/ | Increases contractility | [85]; |
| AVM | PLCε/PKC/CaMKII/RyR2 | [54] | ||
| Hypertrophy | Knockout mouse; | Downstream of ET-1, β-AR, α1-AR, IGF1-R | Knockout animal suggests PLCε suppresses | [85]; |
| NRVM | hypertrophy but NRVM siRNA suggests PLCe mediates hypertrophy |
[90] | ||
| Valve development | Knockout mouse | EGF signaling | [78] | |
| Pancreas | ||||
| β-cell Ca2+ handling | Knockout mouse | GLP1R/cAMP/Epac/Rap/PLCe/PKC/CaMKII/RyR or IP3-R |
Enhances cAMP-dependent CICR | [19] |
| Insulin release | Knockout mouse | Presumably the same as for β-cell calcium handling |
Mediates cAMP-potentiation of insulin release | [20] |
| Cancer/inflammation | ||||
| Epidermal squamous cell tumors |
Knockout mouse | TPA induced Rap GEF activation/PLCε/?/ production of inflammatory mediators |
Pro-inflammatory action enhances tumor formation |
[3] |
| Allergic contact sensitivity |
Knockout mouse; PLCε transgenic |
?/PLCε/?/production of inflammatory mediators | Pro-inflammatory | [33] |
| Esophageal cancer | GWAS | ? | PLCε SNPs positively associated with Esophageal cancer and gastric adenocarcinoma |
[1,86] |
| Kidney disease | ||||
| Childhood nephrotic syndrome |
Human genetic analysis | Expressed highly in glomerular podocytes | May be involved in glomerular development but knockout mouse has no glomerular defect |
[31] |
| Brain | ||||
| Astrocyte proliferation | Knockout mouse | Thrombin/G12/13/rho/PLCε/Rap/B-RAF/ERK | Regulates DNA synthesis in response to thrombin | [13] |
3.1. PLCε in cardiac function
3.1.1. Semilunar valve development
Interestingly both the PLCεΔx/Δx and PLCε−/− mice have cardiac defects but the nature of these defects is different. PLCεΔx/Δx mice have enlarged hearts resulting from ventricular dilation that occurs over the course of development that does not result from cardiac myocyte hypertrophy [78]. PLCε−/− mice do not have enlarged hearts but have an increased susceptibility to hypertrophy development in response to adrenergic stress [85] (this phenotype will be discussed in greater depth below). In the PLCεΔx/Δx mice, the ventricular dila- tion results from aberrant development of the aortic and pulmonary semilunar valves manifest as thickening and stiffening of the valves [78]. This increases valvular regurgitation resulting in chronic volume overload which is likely the cause of the ventricular dilation. The defects in valvulogenesis observed in these mice are reminiscent of the pheno- type observed in mice with defects in HB-EGF or the EGF receptor. Since the EGF receptor could potentially signal to PLCε through the activation of Ras or Rap, the authors speculate that the phenotype is due to a defect in the ability of EGF to generate the appropriate signals from PLCε. Interestingly there is an increase in the levels of phospho-SMADs in the valves of the PLCεΔx/Δx mice and it has been suggested that SMADs play in important role in valve cell proliferation [62]. It is none- theless unclear what PLCε-dependent signals would lead to alterations in SMAD regulation. Furthermore, the pathway that underlies potential EGF-dependent regulation of PLCε activation remains to be elucidated.
3.1.2. Cardiomyocyte hypertrophy
PLCε−/− mice do not develop spontaneous hypertrophy. However, when exposed to a chronic adrenergic stimulation resulting from chronic delivery of isoproterenol for one week, PLCε−/− mice develop significantly greater hypertrophy than PLCε+/+ mice [85]. In addition, PLCε mRNA increases during aortic banding of the wt mice and is elevated in tissue samples from human heart failure patients. To inves- tigate the specific cellular signaling mechanism by which PLCε regulates cardiac hypertrophy we studied neonatal rat ventricular myocytes (NRVMs) in which PLCε was knocked down using siRNA. Hypertrophic responses to many neurohumoral agonists can be observed in this model [66-68], thus it was possible to examine stimulation of hypertro- phy by a variety of agonists that signal through different pathways [90]. Surprisingly, all of the stimuli tested required the presence of PLCε to elicit a hypertrophic response. These stimuli included: 1) Iso, which acts through β adrenergic stimulation of Gs and cAMP, 2) endothelin-1 through ET receptors coupled to Gq, G12/13 and/or Gi, 3) norepinephrine through α1-adrenergic receptors and Gq, and 4) IGF-1, through tyrosine kinase linked signaling likely activating either Ras or Rap1. The exact mechanisms by which these ligands signal through or require PLCε has not yet been determined, but the ability of PLCε to mediate the hypertro- phic response to such varied upstream regulatory molecules identifies PLCε as a nexus for responding to multiple upstream signals in cardiac cells.
While the data regarding the role of PLCε in the hypertrophic re- sponse of NRVMs is clear, several important issues remain unresolved. As discussed above, PLCε−/− mice are more susceptible to hypertrophy, but in NRVMs the opposite result is seen i.e. PLCε deletion protects against hypertrophy. One potential explanation for this discrepancy is that the PLCε−/− mice have PLCε deleted globally during development, thus compensatory changes and global effects from other cell types can impact the in vivo phenotype. Alternatively, this could be explained by a difference between a cell biological model of hypertrophy based on NRVMs and a whole animal model examining the adult mouse. Resolving this issue will require a more sophisticated gene deletion strategy in which PLCε expression is deleted conditionally, after development and specifically in cardiac myocytes. A second issue concerns the role of Gαq in hypertrophic signaling. Gαq overexpression stimulates hypertro- phy of NRVMs in vitro [2] and transgenic Gαq overexpression in the heart leads to development of hypertrophy and heart failure [14]. Mice with conditional cardiac myocyte specific deletion of both Gαq and Gα11 do not develop hypertrophy in response to pressure overload (transverse aortic constriction or “banding”) [87]. Thus, Gαq signaling is essential for the development of pathological forms of hypertrophy which transition to heart failure. Gq regulates PLCβ and does not directly regu- late PLCε. Furthermore, a splice variant of PLCβ1 was recently shown to be critical for hypertrophy downstream of Gαq in NRVMs [22]. Unpub- lished data from our own laboratory using PLCε siRNA in NRVMs shows that PLCε is essential for Gαq-dependent hypertrophy, as it is for responses to multiple agonists. A model that would reconcile these data would be that both PLCβ and PLCε are necessary for Gαq-dependent hypertrophy, with PLCβ being activated directly by Gαq, and PLCε being activated as an indirect consequence of Gαq signaling through an as yet unknown mechanism. Since these enzymes both have the same activi- ties, there must be spatiotemporal differences in their function that al- lows each to provide signals required for the hypertrophic response.
3.1.3. Cardiac myocyte contraction
In addition to showing alterations in hypertrophic responsiveness, PLCε−/− mice showed defective ionotropic responses to β-adrenergic stimulation [85]. At baseline, PLCε−/− and PLCε+/+ mice had similar heart rates and did not differ in contractile function (dP/dT, a measure of the force of contraction). Isoproterenol (Iso)-induced changes in heart rate were no different in the two sets of animals but the Iso- dependent increase in contractile force (dP/dT) was significantly blunted in the PLCε knockout mice. To investigate the nature of this de- fect, adult ventricular myocytes (AVM) were isolated from PLCε−/− mice and analyzed for alterations in Iso-dependent increases in depolarization induced Ca2+-induced Ca2+ release (CICR; note that the amplitude of CICR in cardiac myocytes determines in part the force of contraction by enhancing actin-myosin crossbridging). This analysis revealed that AVMs isolated from PLCε−/− mice had diminished Iso-dependent in- creases in CICR. Thus, PLCε in AVMS appears to play an important role to enable βAR-dependent regulation of cardiac CICR and contraction. Since β-adrenergic regulation of cardiac contractility has been widely shown to be controlled by cAMP-dependent regulation of PKA- mediated phosphorylation events [5], a role for PLC activity in this process was unexpected.
To understand the molecular basis for this contractility defect in the hearts of PLCε knockout mice, and to understand the function of PLCε in the regulation of CICR, many questions concerning the signaling mechanisms upstream and downstream of PLCε needed to be clarified. First, previous work had not demonstrated β-adrenergic stimulation of IP production in cardiac myocytes, and since β adrenergic receptors are coupled to Gs, a mechanism for PLC regulation by these receptors in the heart was not immediately clear. A clue came from studies of PLCε signaling in HEK293 cells showing that the cAMP-dependent Rap ex- change factor Epac could activate PLCε through Rap2B activation [61]. Surprisingly, direct stimulation of AVMs with an Epac activator, 8-(4- chloro-phenylthio)-2′-O-methyladenosine-3′-5′-cyclic monophosphate (cpTOME), caused an increase in CICR that was completely absent in the PLCε knockout mice [54]. Responsiveness to cpTOME could be restored by adenoviral re-expression of PLCε in AVMs isolated from PLCε−/− mice, but not by re-expression of a catalytically dead PLCε. These data indicated that a pathway downstream of the β-adrenergic re- ceptor involves cAMP-dependent activation of Epac which in turn, produces activated Rap which directly stimulates PLCε activity (Fig. 2A). Interestingly, expression of PLCε lacking a portion of the GEF domain did not rescue Epac-dependent regulation of CICR, suggesting that PLCε GEF activity is required for maintenance of Rap activation by Epac which is, in turn, required to sustain PLCε dependent regulation of CICR [53]. Under conditions of strong β-adrenergic receptor activa- tion, the Epac/PLCε pathway appeared to account for about half of the increase in CICR induced by activation of the β-adrenergic receptor. As discussed below, the reason that IP production is not observed upon β-adrenergic receptor stimulation may be because PLCε activity is restricted to discreet regions, or that relatively low but sustained PI hydrolysis that occurs upon PLCε activation in myocytes is not readily detected in assays for IP production.
A second question is how the products of PLCε activity regulate CICR and cardiac contraction. Prior to the above-mentioned studies, no mechanistic pathways for PLC regulation of CICR in the heart had yet been described. In cardiac myocytes the major determinants of CICR are the sarcolemmal L-type calcium channels and the type2 rya- nodine receptors (RyR2) in the sarcoplasmic reticulum [5]. The classi- cal effectors downstream of PLC activation are IP3 receptors and PKC. IP3 receptors are in very low abundance in the ventricular cardiac myocytes and are not thought to play a major role in ventricular CICR [17]. Treatment of AVMs with 2-APB, a blocker of IP3 receptors, did not affect Iso or Epac-dependent CICR in these studies [53]. A PKC inhibitor, on the other hand, partially blocked Iso-dependent regula- tion of CICR and completely blocked Epac-dependent enhancement of CICR [53]. Iso and Epac also induced activation of a single PKC isoform, PKCε, and PKCε siRNA inhibited Epac-dependent increases in CICR. PKC activity had not been directly implicated in the regulation of cardiac Ca2+ transients. However, a clue came from a study by Pereira et al. in- dicating that activation of Epac could stimulate CaMKII-dependent phos- phorylation of RyR2 [56]. We determined that PKC inhibition also blocked Epac-dependent CaMKII-autophosphorylation and phosphory- lation of downstream targets [53]. This data allowed us to more completely model a pathway in which the role for PLCε was in activation of PKC downstream of Iso stimulation and subsequent activation of CaM- KII and phosphorylation of RyR2 to regulate CICR [53] (Fig. 2B in red). Loss of this mechanism likely underlies, at least in part, the decreased ability of Iso to stimulate cardiac contraction in PLCε−/− mice. A later study showed that Epac stimulation leads to an increase in the sensitivity of cardiac myofilaments to Ca2+ [10]. This also appeared to require PKC and CaMKII activation, thus while not directly demonstrated, may also be mediated through activation of PLCε by Epac.
An alternative view for the mechanisms of Epac-dependent regula- tion of RyR2 in cardiac myocytes has been proposed in another recent study [57]. In this study, a role for PLC in Epac-dependent regulation of RyR2 was proposed based on the use of the PLC inhibitor, U73122. However, treatment with a PKC inhibitor did not influence Ca2 + release through RyR2 whereas the effect of Epac activation on Ca2 + release from RyR2 was blocked by 2-APB. Since IP3 receptors are localized to the nuclear envelope in ventricular cardiac myocytes the authors pro- pose that Ca2+ release through IP3 receptors near the nucleus locally activates CaMKII which in turn phosphorylates RyR2 in the SR leading to enhanced Ca2 + release (Fig. 2B in black). The complete lack of effect of a PKC inhibitor and full effect of an IP3 receptor antagonist on these responses is difficult to reconcile with the study by Oestreich et al. where PKC effects were rigorously demonstrated in mouse AVMs at multiple levels: a PKC inhibitor, measurements of Epac-dependent PKC activation, and isoform-specific PKCε siRNA. Also the IP3 receptor antagonist 2-APB had no effect on Epac stimulated CICR in the Oestreich study. These issues remain to be addressed but could have something to do with differences in methodological approaches.
3.1.4. Scaffolding of PLCε in the heart
From data discussed in the previous two sections, PLCε seems to be involved in two independent processes in the cardiac myocyte. One concerns regulation of hypertrophy and hypertrophic gene expression through an as yet undefined mechanism and a second concerns regulation of cardiac contraction through PKC and CaM- KII. This raises the question of whether these processes are directly connected or independent processes. To begin to address this question we began to examine the subcellular scaffolding of PLCε in the heart [90].
Immunoprecipitation experiments from heart tissue indicate that PLCε is in a complex with muscle specific A kinase anchoring protein (mAKAP) [90] where PLCε is part of a multiprotein complex assem- bled on mAKAP with PKA, adenylyl cyclase, PDE4D3, Epac and other proteins [16,90]. Cotransfection and purified protein binding experi- ments indicate that the PLCε directly interacts with mAKAP [90]. Thus the upstream activator Epac and the target PLCε are scaffolded together in the same complex. Since mAKAP is highly localized to the perinuclear region of myocytes [55] this serves to scaffold PLCε as well the other proteins of the macromolecular complex to the nu- clear membrane [41,90]. Scaffolding of PLCε at the nucleus suggested a role for mAKAP-bound, nuclear scaffolded PLCε in regulation of hypertro- phic gene expression and hypertrophy. To test for a role of PLCε/mAKAP scaffolding, the protein interaction surfaces between PLCε and mAKAP were mapped and small domains on either mAKAP or PLCε that could be used to disrupt PLCε/mAKAP binding were identified. Exogenous ex- pression of these domains in NRVMs disrupted endogenous PLCε- mAKAP complexes and suppressed ET-1-dependent hypertrophy [90]. Thus it appears that PLCε scaffolded to mAKAP at the nucleus is impor- tant for regulation of the development of hypertrophy and heart failure. This scaffolding could also be important for the Epac-dependent regulation of nuclear Ca2+ reported recently [57].
In addition to scaffolding to mAKAP, PLCε is found in a complex with RyR2 in PLCε immunoprecipitates from heart extracts. In con- trast to mAKAP, scaffolding of PLCε to RyR2 may be indirect since PLCε only binds weakly to RyR2 in cotransfected HEK293 cells (Malik, in preparation). mAKAP has previously been reported to scaf- fold to RyR2 and could possibly scaffold PLCε to RyR2 in the SR[50,60]. Arguing against this scaffolding mechanism for PLCε binding to RyR2, however, is that the amount of RyR2 in mAKAP complexes isolated from heart is much lower than the amount of RyR2 isolated from PLCε complexes. These data indicate that there is a significant pool of PLCε associated with RyR2 that is not associated with mAKAP. In addition, coexpression of mAKAP with RyR2 and PLCε does not en- hance interactions of PLCε with RyR2 suggesting that mAKAP binding to RyR2 does not mediate binding of PLCε to RyR2. Thus the protein- protein interactions that drive PLCε binding to RyR2 are undefined, but it is reasonable to speculate that this scaffolding interaction is im- portant for regulation of RyR2 function in the sarcoplasmic reticulum. Since RyR2 has been shown to scaffold multiple proteins including CaMKII and PKC, the scaffolding of PLCε to RyR2 would place many of the signaling partners in the correct place for regulation of RyR2 [50].
Overall, scaffolding of PLCε to discrete locations in the cardiac myocyte appears to be critical for its ability to regulate distinct func- tions (see Fig. 3). Thus, scaffolding at RyR2 is likely to be involved in CICR, whereas scaffolding at mAKAP located on the nuclear envelope may regulate hypertrophic gene expression. Thus far, scaffolding inter- actions in native cells have only been examined for cardiomyocytes, but it is likely that in other cell types PLCε can also scaffold, through different binding partners, to distinct locations in various differentiated cell types. Relative to other PLC isoforms, protein expression of PLCε is quite low, so scaffolding to specific sites may be important to allow specific signals to generate IP3 or DAG locally. In the case of SR Ca2+ release, the critical PLC product appears to be DAG since PKC activation is required for the response [53], although, as discussed earlier, one study debates this no- tion [57]. At the nucleus it is possible that either IP3 or DAG or both are needed locally to drive hypertrophy. In this regard PIP2 is thought to be localized primarily to the plasma membrane, while phosphatidylino- sitol 4-P (PI4P) is more broadly distributed in ER and Golgi [4]. Both PIP2 and PI4P are substrates for PLCε in vitro [63]. Since the ER is continuous with the nuclear envelope, PI4P may be the available substrate for PLCε at the nuclear envelope. This would suggest that the relevant product of PLCε at the nuclear envelope would be DAG since IP3 would not be produced from this reaction.
Fig. 3.
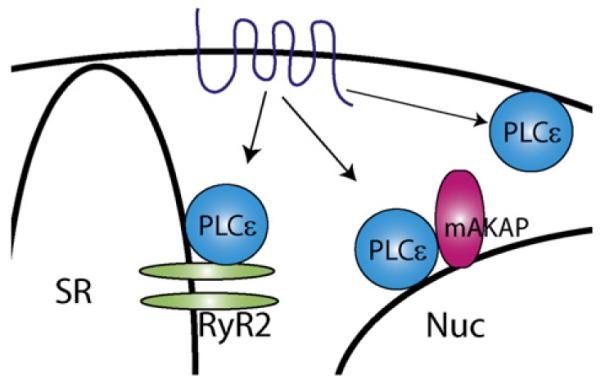
PLCε scaffolding in cardiac myocytes. PLCε is scaffolded at different subcellular loca- tions to perform distinct functions. PLCε scaffolded to RyR2 in the sarcoplasmic reticulum (SR) functions in CICR; PLCε scaffolded to mAKAP at the nuclear (Nuc) envelope is involved in hypertrophy. Additional roles for PLCε at the plasma membrane PM may also exist.
3.2. Role of PLCε in pancreatic β cell function
Glucagon-like peptide-1 (GLP-1), is a gastrointestinal “incretin” hormone that stimulates pancreatic β cell cAMP production. It lowers levels of blood glucose by stimulating the release of insulin from islet β cells. Thus, analogs of GLP-1 are now in use as novel blood glucose- lowering agents for treatment of patients with type 2 diabetes mellitus (T2DM) to increase insulin release [45]. GLP-1 binds to its GPCR expressed on β cells [81] resulting in cAMP production and a PKA- mediated potentiation of depolarization-induced CICR from endoplasmic reticulum Ca2+ stores. Based on these findings, it was proposed that under conditions in which Ca2 + influx is initiated by glucose- dependent depolarization, PKA-mediated phosphorylation of intracel- lular Ca2 + release channels results in a sensitization of these channels to the stimulatory action of Ca2 + so that CICR is facilitated [18,32]. However, evidence for a PKA-independent action of GLP-1 to raise the [Ca2 +]i was found in a study of mouse β cells [6]. Subsequently, it was demonstrated that this action of GLP-1 was mediated by Epac2 [37]. Consistent with such findings, cpTOME was shown to promote CICR in human β cells [40].
Given the role of PLCε in regulation of cAMP-dependent CICR in the heart, studies were then initiated to determine whether Epac- dependent activation of PLCε might explain the PKA-independent action of GLP-1 to regulate CICR in β cells. In these studies, the GLP-1 receptor agonist Exendin-4 activated PKA and Epac2 while also facilitating CICR triggered by the uncaging of Ca2 + [19]. The PKA-dependent action of Exendin-4 was antagonized by PKA inhibitor H-89, was mimicked by PKA activator 6-Bnz-cAMP-AM, and was still measurable in β cells of PLCε KO and Epac2 KO mice. In contrast, the Epac2-mediated action of Exendin-4 was resistant to H-89, was mimicked by cpTOME, and was absent in β cells of PLCε KO and Epac2 KO mice [19]. Furthermore, a rescue of CICR could be achieved after transduction of PLCε KO mouse β cells with wild-type PLCε, but not a catalytically dead PLCε [19]. The ability of cpTOME to facilitate CICR was abrogated in mouse β cells trans- duced with RapGAP, indicating that CICR was under the control of a signal transduction “module” comprised of Epac2, Rap, and PLCε (Fig. 2A).
These findings are remarkable in view of the fact that Epac1, Rap, and PLCε regulate CICR downstream of cAMP in mouse cardiomyo- cytes [53,54]. Just as remarkable is the finding that for both cell types, the ability of cpTOME to facilitate CICR was antagonized by in- hibitors of PKC and CaMKII [19,53]. However, the intracellular Ca2 + release channel that mediates Epac2/PLCε-dependent CICR in β cells has not yet been clearly defined. As has been discussed, in mouse heart the Epac/PLCε/CaMKII pathway modulates CICR through RyR2. In human β cells, CICR facilitated by GLP-1 resulted, at least in part, from the gating of RyR2 [32]. Furthermore, CICR stimulated by cpTOME was blocked by ryanodine [12,40]. However, in mouse β cells evidence exists that CICR is mediated not only by ryanodine re- ceptors, but also by IP3 receptors [18,38]. Thus, for human β cells and perhaps mouse β cells we envision three possible scenarios to explain available data (Fig. 2B): 1) PLCε directly modulates RyR2 in a PKC and CaMKII-dependent manner, analogous to what occurs in the cardiac myocyte, 2) PLCε indirectly modulates RyR2 by first activating IP3 recep- tors, which then release Ca2 + that serves as a stimulus for the gating of RyR2, or 3) some combination of both. Taken as a whole, such findings indicate the existence of an evolutionarily conserved mechanism of Ca2+ mobilization in β cells and cardiomyocytes, one that utilizes Epac proteins and Rap GTPases to activate PLCε, and results in activation of PKC and CaMKII and their sequealae.
PLCε is also important in the cAMP-dependent potentiation of glucose-stimulated insulin secretion (GSIS) from β cells [20]. GSIS from mouse β cells was potentiated by cpTOME, and this action of cpTOME was disrupted in β cells of PLCε KO mice. The defect in insulin secretion measured in the islets of PLCε KO mice was highly selective in that activators of PKA retained their abilities to potentiate GSIS. For β cells of wild-type mice, the insulin secretagogue action of cpTOME was associated with its ability to facilitate CICR, and was disrupted in β cells of PLCε KO mice [19]. Since CICR in β cells is known to be posi- tively coupled to Ca2 +-dependent exocytosis of insulin [38,39], these data together indicate that Epac2-mediated activation of PLCε regulates CICR in a PKA-independent manner to potentiate GSIS.
Based on these findings concerning β cells, we propose that the above-described signaling mechanism is of importance to the treat- ment of type 2 diabetes. β cells act as blood glucose sensors which match a rise of blood glucose concentration to an appropriate rate of β cell insulin secretion. Secreted insulin then acts at insulin-responsive tissues to stimulate glucose uptake, thereby lowering levels of blood glucose [64]. The ability of β cells to act as blood glucose sensors derives from the fact that an increase of blood glucose concentration leads to ac- celerated glucose metabolism within β cells. This glucose metabolism generates an increase of cytosolic ATP/ADP concentration ratio, and this metabolic signal is responsible for the closure of ATP-sensitive K+ channels with concomitant β cell depolarization. Ensuing influx of Ca2 + through voltage-dependent Ca2 + channels produces an in- crease of [Ca2 +]i that initiates exocytosis of insulin stored within the secretory vesicles [28]. In T2DM the blood glucose concentration is chronically elevated due to the inability of β cells to secrete sufficient quantities of insulin. This pathology is due, at least in part, to the failure of β cell glucose metabolism to generate an increase of [Ca2+]i that would normally initiate insulin exocytosis. One therapy for the treatment of T2DM involves the administration of agents that have the capacity to restore normal Ca2+ handling in β cells. GLP-1 analogs might have the ability to facilitate CICR in β cells of T2DM patients, thereby restoring Ca2+ handling and insulin exocytosis. In theory, strategies that target the Epac/PLCε pathway, such as Epac or PLCε activators, would also en- hance CICR and rescue insulin exocytosis perhaps under conditions where β-cells have become refractory to GLP-1 stimulation.
The observation that PLCε is a direct effector of the proto-oncogene Ras raised considerable excitement about the potential role of PLCε in cancer. Ras activates multiple effector pathways and in many cancers an important target of mutationally activated Ras in many cancers is the Raf/ERK pathway that drives cell division and transformation. To in- vestigate the role of PLCε in Ras-dependent transformation PLCεΔx/Δx and PLCε+/+ mice were exposed to a two-stage chemical carcinogenesis protocol. The first-stage of this treatment protocol utilizes 7,12- dimethylbenz(a)anthracene (DMBA) to induce Ras mutations; 12-O- tetradecanoylphorbol-13-acetate (TPA) treatment in a follow-up second stage facilitates clonal expansion of cells containing Ras mutations [3]. In this model Bai et al. demonstrated that both the number of squamous cell tumors and the size of the tumors were reduced in PLCεΔx/Δx animals. In the absence of an initiating DMBA stimulus, TPA also induced epidermal proliferation which was significantly reduced in PLCεΔx/Δx animals.
To understand the mechanisms for PLCε dependent regulation of tumor initiation and growth, epidermal keratinocytes and dermal fibroblasts were isolated from PLCεΔx/Δx and PLCε+/+ mice and tested for alteration in cell growth in response to growth factors in an organotypic cell culture system [35]. PLCε is expressed both in the fibroblasts and in keratinocytes but is much more abundant in the fibroblasts. This suggested a possible direct role for PLCε in the proliferation of these cells, but in contrast to the intact animal, no differences in growth were observed in the fibroblasts nor in the keratinocytes isolated from the wt vs. KO PLCε genotypes nor were differences in proliferative signals observed in response to TPA. This led to the hypothesis that PLCε was influencing dermal proliferation indirectly. It was noted that there was a reduction in skin inflammation in the skin of PLCεΔx/Δx mice exposed TPA treatment, suggesting that PLCε might indirectly promote tumor formation by local- ly regulating inflammation. In fact, it was shown that fibroblasts isolated from PLCεΔx/Δx mice had reduced production of several proinflammatory cytokines. In addition, fewer inflammatory leukocytes were recruited to the skin in response to TPA in the PLCεΔx/Δx mice. These findings support the hypothesis that PLCε can regulate inflammatory responses, and led to the proposal that it was the proinflammatory actions of PLCε in fibro- blasts that contribute to tumor growth in this model.
To test the notion that the role of PLCε in tumor cell proliferation in general might be due to regulation of the local extracellular envi- ronment, the Katoaka group examined another cancer model in which inflammation has been shown to play a major role. ApcMin/+ mice lacking one copy of the andenomatous polyposis coli (Apc) gene are a well established model for intestinal tumorigenesis where inflammatory processes play an important role in tumorigenesis and cancer progression. Spontaneous intestinal tumor formation was significantly suppressed in ApcMin/+ mice bred into a PLCεΔx/Δx background [1,46]. Additionally the conversion of low grade tumors into high grade adenocarcinomas was dramatically reduced by loss of PLCε activity. That two different stages of tumor development were influ- enced by PLCε suggested PLCε involvement in two distinct mechanisms. The development of low grade tumors is strongly dependent on angio- genesis, and VEGF production was shown to be significantly reduced in tumors isolated from PLCεΔx/Δx animals. The second stage is thought to be strongly dependent on inflammation, and COX2 expression was sig- nificantly reduced in PLCεΔx/Δx mice. Thus, for this model of intestinal tumorigenesis, PLCε regulates the expression of factors that regulate different stages of tumor progression and does so via distinct mecha- nisms, one involving angiogenesis and a second involving the production of inflammatory mediators. That both skin and intestinal cancer can be regulated in an inflammatory mediator-dependent manner through PLCε supports the idea that PLCε may regulate cancer progression for tu- mors that are modulated by inflammatory status. Recently, genome wide association (GWAS) studies have implicated PLCε in esophageal squamous cell carcinoma and gastric adenocarcinoma [1,86], conditions that may also depend on chronic inflammation, although whether the PLCε polymorphisms identified in this study may influence PLCε activity is unresolved.
The data described above suggest that PLCε may play a general role as a proinflammatory mediator in multiple tissues through actions that do not directly involve effects of PLCε actions in in- flammatory leukocytes. Follow up analyses have implicated PLCε in allergic contact hypersensitivity, and over expression of PLCε in the skin tissue of mice causes spontaneous skin inflammation [33]. Thus, the major role for PLCε in inflammation appears to be in reg- ulating the production of proinflammatory mediators. The molecular mechanism(s) underlying the regulation of the production of proinflam- matory mediators by PLCε remains uncertain. In the case of TPA- dependent skin hyperplasia, one proposed mechanism is through the regulation of two DAG dependent targets, Ras-GRP3 and PKC. RasGRP3 is a Rap1 GEF that is directly activated by DAG and TPA, and it is also de- pendent on PKC phosphorylation for activation [35]. Thus, TPA could stimulate PLCε activity in dermal fibroblasts through the production of activated Rap1 that can directly bind and regulate PLCε. Downstream of PLCε, one recent study in cell culture suggests that PLCε may cooperate with NfκB to modulate expression of certain cytokines [25]. Additionally, our recent work using astrocytes as a model of inflammatory cells in the brain demonstrates that GPCR agonists such as thrombin, LPA and SIP require PLCε to regulate NFκB and cyclooxygenase expression (Dusaban, in preparation).
PLC epsilon is not the only PLC isoform that has been linked to inflammation. In immune cells, PLCγ2 signals downstream of various tyrosine kinase-linked receptors to initiate pro-inflammatory responses[23,84]. PLCβ isoforms are involved in the innate immune response downstream of chemotactic peptide signaling, which is involved in the signaling cascades within leukocytes that direct immune cell migration and secretion of inflammatory factors [89]. PLCδ on the other hand, sup- presses production of inflammatory cytokine production in keratino- cytes, thereby suppressing activation and recruitment of immune cells [34]. These different enzymes elicit diverse reactions likely due, in part, to their expression in different cell types. In cells such as keratinocytes, both PLCε and PLCδ are expressed and they appear to perform opposing functions. The ability of Ras subfamily small G-proteins to regulate PLCε is unique, however, as is the aforementioned spatial and temporal activa- tion of this PLC isoform, thus it is likely that PLCε serves to regulate the expression of inflammatory genes in ways that other PLC isoforms do not.
4. Roles for PLCε in human disease
4.1. PLCε in the kidney
In a recent search for genes involved in childhood nephrotic syn- drome, PLCε mutations were identified as autosomal recessive deter- minants of this disease [31]. These mutations included missense mutations that resulted in truncation of the PLCε protein, or in single site polymorphisms. The disease course correlated with the severity of the PLCε mutation. Patients with homozygous truncation mutations had an earlier onset of proteinurea and earlier development of end-stage kidney disease than did patients with single amino acid substitutions in the gene.
PLCε is highly expressed in glomerular podocytes [31]. Podocytes are epithelial cells with foot-like processes that form connections through various adhesion proteins which form a key part of the glo- merular filtration barrier. This barrier functions in the glomerulus to allow the relatively free flow of liquid and aqueous solutes while retaining plasma proteins such as albumin to maintain blood protein levels. Various genetically inherited nephropathies result from inherited mutations in the proteins that form or maintain this filtration barrier, resulting in disruption of podocyte connections and leakage of proteins into the urine (proteinurea) [48]. The signaling role of PLCε in podocytes in the normal physiology of podocytes and in glomerular kidney disease is not understood. An intriguing possibility is that PLCε is involved in the regulation of a DAG sensitive Trp channel (TrpC6). TrpC6 plays an important role in regulating Ca2+ signaling in the podocyte, and mutations in TrpC6 are associated with glomerular kidney disease [15]. A connection between PLCε activity and TrpC6 function has not yet been supported by direct data. Some protein- protein interactions between PLCε and podocyte proteins such as IQGAP and B-Raf have been reported [11], but whether these interactions are involved in the ability of PLCε to regulate podocyte development or function remains unresolved.
Patients with PLCε mutations have impaired glomerular develop- ment, thus it has been suggested that PLCε signaling may be involved in glomerular development [31]. However, PLCε deletion in mice does not lead to increased proteinurea nor does it appear to affect glomer- ular development, a finding that seemingly rules out a direct role of PLCε signaling in podocyte or glomerular development (unpublished observations). On the other hand, PLCε deletion in zebrafish leads to a defect in glomerular filtration function and development of a disorga- nized filtration barrier characteristic of kidney disease [31]. It is likely that genetic background or environmental factors influence the ability of PLCε dysfunction to produce a glomerular phenotype.
4.2. PLCε in esophageal cancer and gastric adenocarcinoma
Recent studies using genomic based approaches have identified an association of the PLCε gene with various human cancers. Two separate genome-wide association (GWAS) studies identified PLCE1 single nucle- otide polymorphisms (SNPs) associated with esophageal squamous cell carcinoma (ESCC) [1,86]. These studies analyzed DNA SNPs from over 1000 patients with ESCC and compared them with DNA isolated from control individuals. Both studies identified polymorphisms in either the coding or non coding regions of the PLCE1 gene, and demonstrated that these polymorphisms were associated with ESCC. One of these studies found that the same PLCE1 SNPs associated with ESCC were also associated with gastric adenocarcinoma [1]. Two polymorphisms resulted in single amino acid substitutions in the PLCε coding sequence, while three other SNPs were silent in terms of the protein sequence. One substitution independently identified by both groups was R1927H which is located in the C2 domain, and the other was I177T which is in the Y domain portion of the catalytic domain (see Fig. 1). Whether these or the other non-coding alterations associated with the PLCE1 SNPs alter PLCE1 function or expression is unknown. Accordingly, while these SNPs are associated with the disease, it is unclear whether alterations in PLCε function or expression are causal or merely correlated with the ESCC or gastric adenocarcinoma.
5. Conclusion and outlook
One of the surprising results of these summarized studies is the dramatic impact of PLCε on cellular physiology despite the fact that it is a relatively low abundance protein expressed in the context of multiple other more abundant PLC isoforms in the same cell. This un- derscores an overall recognition that the functional importance of a protein in cellular physiology does not necessarily correlate with abundance. Of additional interest is the fact that PLCε activation oc- curs in response to the activation of GPCRs that have been tradition- ally thought to be coupled to inositol phosphate production via Gq and PLCβ . This is manifest in the role of PLCε in more sustained sig- nals emanating from multiple receptors. Another critical point is the apparent localization of PLCε to internal membrane structures, such as ER, Golgi and perinuclear membranes that are not thought to be rich in PIP2, but rather are enriched in PI4P; As discussed above, hy- drolysis of PI4P will generate DAG but not IP3 again suggesting a role in sustained processes perhaps involving DAG rather than IP3. Signals involving DAG formation and activation of downstream kinases such as PKC and PKD may be sustained and compartmentalized in this way. A final theme that emerges from these studies is that intracellular scaf- folding can specify cellular function. This is not a new concept overall, but the demonstration that PLC scaffolding has a significant impact on physiological functions is novel.
Several of the physiological functions discussed above indicate that PLCε may be a therapeutic target. In hypertrophy PLCε appears to be a central player that integrates signals from multiple stimuli. In this case inhibition of PLCε function could be a strategy for treatment of heart failure. Similarly, inhibition of PLCε could prevent inflammatory reactions associated with tumor development in some cancers or in other diseases in which inflammation plays a major role. Disrupting PLCε scaffolding in the cardiac myocyte inhibits development of hyper- trophy and could also be a useful strategy to inhibit development of heart failure in particular since the mAKAP scaffolding is likely to be unique to myocytes. In pancreatic β-cells increasing, rather than inter- fering with PLCε function has the potential to increase insulin secretion in response to natural changes in the levels of incretin hormones such as GLP1 and thus could be an approach to treatment of Type 2 diabetes.
It is likely that PLCε plays important roles in other physiologies that have yet to be investigated. PLCε is highly enriched in the lung suggesting that PLC signaling plays a role in asthma through mediating contraction of bronchial smooth muscle. PLCε signaling may be associat- ed with Epac in many cell types and could be a general regulator of CICR in response to cyclic AMP in multiple excitable cells. Answering these questions will require more detailed phenotype analysis of PLCε−/− mice. Thus, while the general basis for receptor stimulated phosphoinosi- tide hydrolysis is well established some unexpected roles for PLC isoform specific phosphoinositide signaling networks in specific physiological functions continue to emerge. Studies with PLCε knockout animals have been invaluable in revealing specific phenotypes and coupled with detailed tissue, cell and biochemical analysis, novel detailed physio- logical signaling mechanisms will be revealed.
Acknowledgments
This work was supported by National Institutes of Health grants GM R01 053536 to A. V. S., GM R01 036927 to J.H.B., and DK R01 045817 and DK R01 069575 to G.G.H.
Abbreviations
- PLC
phosphoinositide-specific phospholipase C
- PKC
protein kinase C
- LPA
lysophosphatidic acid
- S1P
sphingosine-1 phosphate
- Epac
Exchange protein direct- ly activated by cAMP
- GPCR
G protein-coupled receptor
- GEF
guanine nucleotide exchange factor
- EGF
epidermal growth factor
- PDGF
platelet-derived growth factor
- Iso
Isopro- terenol
- PKD
protein kinase D
- RA
Ras association domain
- IP
inositol phosphate
- DAG
diacylglycerol
- PIP2
phosphatidylinositol 4,5 bisphosphate
- IP3
inositol 1,4,5 triphosphate
- NRVM
neonatal rat ventricular myocyte
- AVM
adult ventricular myocyte
- IGF-1
insulin-like growth factor 1
- ET-1
endothelin-1
- CICR
calcium-induced calcium release
- PKA
pro- tein kinase A
- cpTOME
8-(4-chloro-phenylthio)-2′-O-methyladenosine-3′-5′-cyclic monophosphate
- AKAP
A kinase anchoring protein
- 2-APB
2-aminophenyl borate
- RyR
ryano- dine receptor
- CaMKII
calcium calmodulin-dependent kinase II
- SR
sarcoplasmic reticulum
- GSIS
glucose-stimulate insulin secretion
- T2DM
type 2 diabetes mellitus
- GLP-1
glucagon-like peptide-1
- TPA
12-O-tetradecanoylphorbol-13-acetate
- VEGF
vascular endothelial growth factor
- SNP
single nucleotide polymorphism
- ESCC
esophageal squamous cell carcinoma
References
- [1].Abnet CC, Freedman ND, Hu N, Wang Z, Yu K, Shu X-O, Yuan J-M, Zheng W, Dawsey SM, Dong LM, et al. Nature Genetics. 2010;42:764–767. doi: 10.1038/ng.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., II Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bai Y, Edamatsu H, Maeda S, Saito H, Suzuki N, Satoh T, Kataoka T. Cancer Research. 2004;64:8808–8810. doi: 10.1158/0008-5472.CAN-04-3143. [DOI] [PubMed] [Google Scholar]
- [4].Balla T, Szentpetery Z, Kim YJ. Physiology. 2009;24:231–244. doi: 10.1152/physiol.00014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bers DM. Annual Review of Physiology. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- [6].Bode HP, Moormann B, Dabew R, Göke B. Endocrinology. 1999;140:3919–3927. doi: 10.1210/endo.140.9.6947. [DOI] [PubMed] [Google Scholar]
- [7].Bunney TD, Harris R, Gandarillas N.L.o., Josephs MB, Roe SM, Sorli SC, Paterson HF, Rodrigues-Lima F, Esposito D, Ponting CP, et al. Molecular Cell. 2006;21:495–507. doi: 10.1016/j.molcel.2006.01.008. [DOI] [PubMed] [Google Scholar]
- [8].Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P. Nature. 1992;360:684–686. doi: 10.1038/360684a0. [DOI] [PubMed] [Google Scholar]
- [9].Carpenter G, Hernandez-Sotomayor T, Jones G. Advances in Second Messenger and Phosphoprotein Research. 1993;28:179–185. [PubMed] [Google Scholar]
- [10].Cazorla O, Lucas A, Poirier F, Lacampagne A, Lezoualc’h F. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14144–14149. doi: 10.1073/pnas.0812536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaib H, Hoskins BE, Ashraf S, Goyal M, Wiggins RC, Hildebrandt F. American Journal of Physiology. Renal Physiology. 2008;294:F93–F99. doi: 10.1152/ajprenal.00345.2007. [DOI] [PubMed] [Google Scholar]
- [12].Chepurny OG, Kelley GG, Dzhura I, Leech CA, Roe MW, Dzhura E, Li X, Schwede F, Genieser HG, Holz GG. American Journal of Physiology, Endocrinology and Metabolism. 2010;298:E622–E633. doi: 10.1152/ajpendo.00630.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Citro S, Malik S, Oestreich EA, Radeff-Huang J, Kelley GG, Smrcka AV, Brown JH. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15543–15548. doi: 10.1073/pnas.0702943104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., II Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dietrich A, Chubanov V, Gudermann T. Journal of the American Society of Nephrology. 2010;21:736–744. doi: 10.1681/ASN.2009090948. [DOI] [PubMed] [Google Scholar]
- [16].Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dorn GW, II, Brown JH. Trends in Cardiovascular Medicine. 1999;9:26–34. doi: 10.1016/s1050-1738(99)00004-3. [DOI] [PubMed] [Google Scholar]
- [18].Dyachok O, Gylfe E. Journal of Biological Chemistry. 2004;279:45455–45461. doi: 10.1074/jbc.M407673200. [DOI] [PubMed] [Google Scholar]
- [19].Dzhura I, Chepurny OG, Kelley GG, Leech CA, Roe MW, Dzhura E, Afshari P, Malik S, Rindler MJ, Xu X, et al. Journal de Physiologie. 2010;588:4871–4889. doi: 10.1113/jphysiol.2010.198424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dzhura I, Chepurny OG, Leech CA, Roe MW, Dzhura E, Xu X, Lu Y, Schwede F, Genieser H-G, Smrcka AV, et al. Islets. 2011;3:121–128. doi: 10.4161/isl.3.3.15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Essen LO, Perisic O, Cheung R, Katan M, Williams RL. Nature. 1996;380:595–602. doi: 10.1038/380595a0. [DOI] [PubMed] [Google Scholar]
- [22].Filtz TM, Grubb DR, McLeod-Dryden TJ, Luo J, Woodcock EA. The FASEB Journal. 2009;23:3564–3570. doi: 10.1096/fj.09-133983. [DOI] [PubMed] [Google Scholar]
- [23].Graham DB, Robertson CM, Bautista J, Mascarenhas F, Diacovo MJ, Montgrain V, Lam SK, Cremasco V, Dunne WM, Faccio R, et al. The Journal of Clinical Investigation. 2007;117:3445–3452. doi: 10.1172/JCI32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hains MD, Wing MR, Maddileti S, Siderovski DP, Harden TK. Molecular Pharmacology. 2006;69:2068–2075. doi: 10.1124/mol.105.017921. [DOI] [PubMed] [Google Scholar]
- [25].Harada Y, Edamatsu H, Kataoka T. Biochemical and Biophysical Research Communications. 2011;414:106–111. doi: 10.1016/j.bbrc.2011.09.032. [DOI] [PubMed] [Google Scholar]
- [26].Harden TK, Sondek J. Annual Review of Pharmacology and Toxicology. 2006;46:355–379. doi: 10.1146/annurev.pharmtox.46.120604.141223. [DOI] [PubMed] [Google Scholar]
- [27].Harden TK, Waldo GL, Hicks SN, Sondek J. Chemistry Review. 2011;111:6120–6129. doi: 10.1021/cr200209p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Henquin JC. Diabetes. 2000;49:1751–1760. doi: 10.2337/diabetes.49.11.1751. [DOI] [PubMed] [Google Scholar]
- [29].Hicks SN, Jezyk MR, Gershburg S, Seifert JP, Harden TK, Sondek J. Molecular Cell. 2008;31 doi: 10.1016/j.molcel.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hilgemann DW, Ball R. Science. 1996;273:956–959. doi: 10.1126/science.273.5277.956. [DOI] [PubMed] [Google Scholar]
- [31].Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, Garg P, Verma R, Chaib H, Hoskins BE, et al. Nature Genetics. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- [32].Holz GG, Leech CA, Heller RS, Castonguay M, Habener J. Journal of Biological Chemistry. 1999;274:14147–14156. doi: 10.1074/jbc.274.20.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu L, Edamatsu H, Takenaka N, Ikuta S, Kataoka T. Journal of Immunology. 2010;184:993–1002. doi: 10.4049/jimmunol.0901816. [DOI] [PubMed] [Google Scholar]
- [34].Ichinohe M, Nakamura Y, Sai K, Nakahara M, Yamaguchi H, Fukami K. Biochemical and Biophysical Research Communications. 2007;356:912–918. doi: 10.1016/j.bbrc.2007.03.082. [DOI] [PubMed] [Google Scholar]
- [35].Ikuta S, Edamatsu H, Li M, Hu L, Kataoka T. Cancer Research. 2008;68:64–72. doi: 10.1158/0008-5472.CAN-07-3245. [DOI] [PubMed] [Google Scholar]
- [36].Jin TG, Satoh T, Liao Y, Song C, Gao X, Kariya K, Hu CD, Kataoka T. Journal of Biological Chemistry. 2001;276:30301–30307. doi: 10.1074/jbc.M103530200. [DOI] [PubMed] [Google Scholar]
- [37].Kang G, Chepurny OG, Holz GG. Journal de Physiologie. 2001;536:375–385. doi: 10.1111/j.1469-7793.2001.0375c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kang G, Chepurny OG, Rindler MJ, Collis L, Chepurny Z, Li WH, Harbeck M, Roe MW, Holz GG. Journal de Physiologie. 2005;566:173–188. doi: 10.1113/jphysiol.2005.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kang G, Holz GG. Journal de Physiologie. 2003;546:175–189. doi: 10.1113/jphysiol.2002.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kang G, Joseph JW, Chepurny OG, Monaco M, Wheeler MB, Bos JL, Schwede F, Genieser HG, Holz GG. Journal of Biological Chemistry. 2003;278:8279–8285. doi: 10.1074/jbc.M211682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kapiloff MS, Jackson N, Airhart N. Journal of Cell Science. 2001;114:3167–3176. doi: 10.1242/jcs.114.17.3167. [DOI] [PubMed] [Google Scholar]
- [42].Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJH. Journal of Biological Chemistry. 2006;281:2639–2648. doi: 10.1074/jbc.M507681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kelley GG, Reks SE, Ondrako JM, Smrcka AV. EMBO Journal. 2001;20:743–754. doi: 10.1093/emboj/20.4.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kelley GG, Reks SE, Smrcka AV. Biochemical Journal. 2004;378:129–139. doi: 10.1042/BJ20031370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Leech CA, Dzhura I, Chepurny OG, Kang G, Schwede F, Genieser HG, Holz GG. Progress in Biophysics and Molecular Biology. 2011;107:236–247. doi: 10.1016/j.pbiomolbio.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li M, Edamatsu H, Kitazawa R, Kitazawa S, Kataoka T. Carcinogenesis. 2009;30:1424–1432. doi: 10.1093/carcin/bgp125. [DOI] [PubMed] [Google Scholar]
- [47].Lopez I, Mak EC, Ding J, Hamm HE, Lomasney JW. Journal of Biological Chemistry. 2001;276:2758–2765. doi: 10.1074/jbc.M008119200. [DOI] [PubMed] [Google Scholar]
- [48].Löwik M, Groenen P, Levtchenko E, Monnens L, van den Heuvel L. European Journal of Pediatrics. 2009;168:1291–1304. doi: 10.1007/s00431-009-1017-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lyon AM, Tesmer VM, Dhamsania VD, Thal DM, Gutierrez J, Chowdhury S, Suddala KC, Northup JK, Tesmer JJG. Nature Structural and Molecular Biology. 2011;18:999–1005. doi: 10.1038/nsmb.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- [51].Meisenhelder J, Suh PG, Rhee SG, Hunter T. Cell. 1989;57:1109–1122. doi: 10.1016/0092-8674(89)90048-2. [DOI] [PubMed] [Google Scholar]
- [52].Nakahara M, Shimozawa M, Nakamura Y, Irino Y, Morita M, Kudo Y, Fukami K. Journal of Biological Chemistry. 2005;280:29128–29134. doi: 10.1074/jbc.M503817200. [DOI] [PubMed] [Google Scholar]
- [53].Oestreich EA, Malik S, Goonasekera SA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Journal of Biological Chemistry. 2009;284:1514–1522. doi: 10.1074/jbc.M806994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Oestreich EA, Wang H, Malik S, Kaproth-Joslin KA, Blaxall BC, Kelley GG, Dirksen RT, Smrcka AV. Journal of Biological Chemistry. 2007;282:5488–5495. doi: 10.1074/jbc.M608495200. [DOI] [PubMed] [Google Scholar]
- [55].Pare GC, Easlick JL, Mislow JM, McNally EM, Kapiloff MS. Experimental Cell Research. 2005;303:388–399. doi: 10.1016/j.yexcr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- [56].Pereira L, Metrich M, Fernandez-Velasco M, Lucas A, Leroy J, Perrier R, Morel E, Fischmeister R, Richard S, Benitah JP, et al. Journal de Physiologie. 2007;583:685–694. doi: 10.1113/jphysiol.2007.133066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pereira L, Ruiz-Hurtado G, Morel E, Laurent A-C, Métrich M, Domínguez-Rodríguez A, Lauton-Santos S, Lucas A, Benitah J-P, Bers DM, et al. Journal of Mo-lecular and Cellular Cardiology. 2012;52:283–291. doi: 10.1016/j.yjmcc.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Rebecchi MJ, Pentyala SN. Physiological Reviews. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- [59].Rhee SG. Annual Review of Biochemistry. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Ruehr ML, Russell MA, Ferguson DG, Bhat M, Ma J, Damron DS, Scott JD, Bond M. Journal of Biological Chemistry. 2003;278:24831–24836. doi: 10.1074/jbc.M213279200. [DOI] [PubMed] [Google Scholar]
- [61].Schmidt M, Evellin S, Weernink P, von Dorp F, Rehmann H, Lomasney J, Jakobs K. Nature Cell Biology. 2001;3:1020–1024. doi: 10.1038/ncb1101-1020. [DOI] [PubMed] [Google Scholar]
- [62].Schroeder JA, Jackson LF, Lee DC, Camenisch TD. Journal of Molecular Medicine. 2003;81:392–403. doi: 10.1007/s00109-003-0456-5. [DOI] [PubMed] [Google Scholar]
- [63].Seifert JP, Wing MR, Snyder JT, Gershburg S, Sondek J, Harden TK. Journal of Biological Chemistry. 2004;279:47992–47997. doi: 10.1074/jbc.M407111200. [DOI] [PubMed] [Google Scholar]
- [64].Seino S, Shibasaki T, Minami K. The Journal of Clinical Investigation. 2011;121:2118–2125. doi: 10.1172/JCI45680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Shibatohge M, Kariya K, Liao Y, Hu CD, Watari Y, Goshima M, Shima F, Kataoka T. Journal of Biological Chemistry. 1998;273:6218–6222. doi: 10.1074/jbc.273.11.6218. [DOI] [PubMed] [Google Scholar]
- [66].Simpson P. Circulation Research. 1985;56:884–894. doi: 10.1161/01.res.56.6.884. [DOI] [PubMed] [Google Scholar]
- [67].Simpson P, McGrath A, Savion S. Circulation Research. 1982;51:787–801. doi: 10.1161/01.res.51.6.787. [DOI] [PubMed] [Google Scholar]
- [68].Simpson P, Savion S. Circulation Research. 1982;50:101–116. doi: 10.1161/01.res.50.1.101. [DOI] [PubMed] [Google Scholar]
- [69].Smrcka AV, Hepler JR, Brown KO, Sternweis PC. Science. 1991;251:804–807. doi: 10.1126/science.1846707. [DOI] [PubMed] [Google Scholar]
- [70].Smrcka AV, Sternweis PC. Journal of Biological Chemistry. 1993;268:9667–9674. [PubMed] [Google Scholar]
- [71].Song C, Hu CD, Masago M, Kariya K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T. Journal of Biological Chemistry. 2001;276:2752–2757. doi: 10.1074/jbc.M008324200. [DOI] [PubMed] [Google Scholar]
- [72].Song C, Satoh T, Edamatsu H, Wu D, Tadano M, Gao X, Kataoka T. Oncogene. 2002;21:8105–8113. doi: 10.1038/sj.onc.1206003. [DOI] [PubMed] [Google Scholar]
- [73].Sorli SC, Bunney TD, Sugden PH, Paterson HF, Katan M. Oncogene. 2004;64:90–100. doi: 10.1038/sj.onc.1208168. [DOI] [PubMed] [Google Scholar]
- [74].Suh B-C, Hille B. Annual Review of Biophysics. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, Katan M, Fukami K, Kataoka T, Yun S, Ryu SH. BMB Reports. 2008;41:415–434. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- [76].Sui JL, Petit-Jacques J, Logothetis DE. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1307–1312. doi: 10.1073/pnas.95.3.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Swann K, Larman MG, Saunders CM, Lai FA. Reproduction. 2004;127:431–439. doi: 10.1530/rep.1.00169. [DOI] [PubMed] [Google Scholar]
- [78].Tadano M, Edamatsu H, Minamisawa S, Yokoyama U, Ishikawa Y, Suzuki N, Saito H, Wu D, Masago-Toda M, Yamawaki-Kataoka Y, et al. Molecular and Cellular Biology. 2005;25:2191–2199. doi: 10.1128/MCB.25.6.2191-2199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Taylor SJ, Chae HZ, Rhee SG, Exton JH. Nature. 1991;350:516–518. doi: 10.1038/350516a0. [DOI] [PubMed] [Google Scholar]
- [80].Thompson JL, Shuttleworth TJ. Journal de Physiologie. 2011;589:5057–5069. doi: 10.1113/jphysiol.2011.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Thorens B. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:8641–8645. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Waldo GL, Boyer JL, Morris AJ, Harden TK. Journal of Biological Chemistry. 1991;266:14217–14225. [PubMed] [Google Scholar]
- [83].Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, et al. Science. 2010;330:974–980. doi: 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wang D, Feng J, Wen R, Marine J-C, Sangster MY, Parganas E, Hoffmeyer A, Jackson CW, Cleveland JL, Murray PJ, et al. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
- [85].Wang H, Oestreich EA, Maekawa N, Bullard TA, Vikstrom KL, Dirksen RT, Kelley GG, Blaxall BC, Smrcka AV. Circulation Research. 2005;97:1305–1313. doi: 10.1161/01.RES.0000196578.15385.bb. [DOI] [PubMed] [Google Scholar]
- [86].Wang L-D, Zhou F-Y, Li X-M, Sun L-D, Song X, Jin Y, Li J-M, Kong G-Q, Qi H, Cui J, et al. Nature Genetics. 2010;42:759–763. doi: 10.1038/ng.648. [DOI] [PubMed] [Google Scholar]
- [87].Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, Offermanns S. Nature Medicine. 2001;7:1236–1240. doi: 10.1038/nm1101-1236. [DOI] [PubMed] [Google Scholar]
- [88].Wing MR, Houston D, Kelley GG, Der CJ, Siderovski DP, Harden TK. Journal of Biological Chemistry. 2001;276:48257–48261. doi: 10.1074/jbc.C100574200. [DOI] [PubMed] [Google Scholar]
- [89].Wu D, Huang CK, Jiang H. Journal of Cell Science. 2000;113:2935–2940. doi: 10.1242/jcs.113.17.2935. [DOI] [PubMed] [Google Scholar]
- [90].Zhang L, Malik S, Kelley GG, Kapiloff MS, Smrcka AV. Journal of Biological Chemistry. 2011;286:23012–23021. doi: 10.1074/jbc.M111.231993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Zhou Y, Wing MR, Sondek J, Harden TK. Biochemical Journal. 2005;391:667–676. doi: 10.1042/BJ20050839. [DOI] [PMC free article] [PubMed] [Google Scholar]