

Abstract
Alzheimer's disease (AD), the most common neurodegenerative disorder, is a growing public health problem and still lacks effective treatments. Recent evidence suggests that microtubule-associated protein tau may mediate amyloid-β peptide (Aβ) toxicity by modulating the tyrosine kinase Fyn. We showed previously that tau reduction prevents, and Fyn overexpression exacerbates, cognitive deficits in human amyloid precursor protein (hAPP) transgenic mice overexpressing Aβ. However, the mechanisms by which Aβ, tau, and Fyn cooperate in AD-related pathogenesis remain to be fully elucidated. Here we examined the synaptic and network effects of this pathogenic triad. Tau reduction prevented cognitive decline induced by synergistic effects of Aβ and Fyn. Tau reduction also prevented synaptic transmission and plasticity deficits in hAPP mice. Using electroencephalography to examine network effects, we found that tau reduction prevented spontaneous epileptiform activity in multiple lines of hAPP mice. Tau reduction also reduced the severity of spontaneous and chemically induced seizures in mice overexpressing both Aβ and Fyn. To better understand these protective effects, we recorded whole-cell currents in acute hippocampal slices from hAPP mice with and without tau. hAPP mice with tau had increased spontaneous and evoked excitatory currents, reduced inhibitory currents, and NMDA receptor dysfunction. Tau reduction increased inhibitory currents and normalized excitation/inhibition balance and NMDA receptor-mediated currents in hAPP mice. Our results indicate that Aβ, tau, and Fyn jointly impair synaptic and network function and suggest that disrupting the copathogenic relationship between these factors could be of therapeutic benefit.
Introduction
Current treatments for Alzheimer's disease (AD) are only modestly effective and do not appear to alter the underlying disease progression. For good reasons, the search for better AD therapies has focused on amyloid-β peptides (Aβ), generally thought to drive AD pathogenesis (Hardy and Selkoe, 2002; Tanzi and Bertram, 2005). However, disappointing outcomes of initial drug trials targeting Aβ (Gilman et al., 2005; Green et al., 2009) have broadened interest in other, downstream targets, including the microtubule-associated protein tau.
Considerable evidence suggests that tau mediates or enables the pathogenic effects of Aβ (Götz et al., 2001; Lewis et al., 2001; Oddo et al., 2003; Tanzi and Bertram, 2005; Roberson et al., 2007; Ittner et al., 2010; Vossel et al., 2010). We showed that reducing or eliminating tau expression had striking beneficial effects in human amyloid precursor protein (hAPP) transgenic mice with high levels of Aβ in the brain (line hAPPJ20), preventing their learning and memory deficits, other behavioral abnormalities, and early mortality (Roberson et al., 2007). Similar protective effects of tau reduction were observed in an independent hAPP mouse model (line APP23) (Ittner et al., 2010). The mechanisms of these beneficial effects are unclear.
Fyn tyrosine kinase has also been implicated in Aβ/tau-dependent neuronal dysfunction (Haass and Mandelkow, 2010; Ittner et al., 2010). Increasing neuronal expression of Fyn worsens Aβ-dependent cognitive impairments, neuronal abnormalities, and premature mortality in hAPP mice, whereas ablation of Fyn protects against Aβ toxicity (Lambert et al., 1998; Chin et al., 2004, 2005). Fyn also modulates glutamatergic signaling pathways affected by Aβ (Salter and Kalia, 2004), phosphorylates tau at Tyr18 (Lee et al., 2004), and is crucial in tau-dependent Aβ toxicity at postsynaptic sites (Ittner et al., 2010).
Regulation of synaptic and network function is a potential mechanistic point of convergence for Aβ, tau, and Fyn. The synapse is a primary target of Aβ (Selkoe, 2002; Palop and Mucke, 2010), and AD mouse models have abnormal synaptic transmission and plasticity (Chapman et al., 1999; Hsia et al., 1999; Walsh et al., 2002; Kamenetz et al., 2003). Dysfunction at the circuit and network levels in these models may relate to neural network dysrhythmias in AD (Palop and Mucke, 2009). Pathologically elevated Aβ levels cause aberrant neuronal activity, as demonstrated by electroencephalographic (EEG) recordings (Palop et al., 2007; Minkeviciene et al., 2009; Vogt et al., 2009) and in vivo imaging (Busche et al., 2008). Increasing Fyn levels also reduces seizure thresholds (Cain et al., 1995; Kojima et al., 1998). Tau reduction increases resistance of mice to pharmacologically induced convulsive seizures without changing Aβ levels (Roberson et al., 2007; Ittner et al., 2010).
To elucidate the role of the Aβ–tau–Fyn triad in AD-related neuronal dysfunction, we examined the effects of tau reduction on EEGs and synaptic function in several lines of mice expressing hAPP/Aβ in the absence or presence of a transgene that increases neuronal Fyn levels. Our results support the hypothesis that, together, Aβ, tau, and Fyn dysregulate synaptic activity, causing aberrant neuronal synchrony.
Materials and Methods
Animals.
hAPPJ9 and hAPPJ20 mice express an hAPP minigene with the Swedish (K670N, M671L) and Indiana (V717F) mutations under the control of the PDGF β-chain promoter; hAPP and Aβ levels in hAPPJ20 mice are about double those in hAPPJ9 mice (Mucke et al., 2000). hAPPJ20 mice were crossed with tau-deficient mice (Dawson et al., 2001) as described (Roberson et al., 2007) to produce offspring with two (Tau+/+), one (Tau+/−), or no (Tau−/−) functional Tau alleles in the presence or absence of hAPP. hAPPJ9 mice were crossed with Tau−/− mice for two generations to produce hAPPJ9/Tau−/− mice. hAPPJ9/Tau+/+ mice do not have robust behavioral abnormalities (Chin et al., 2005) unless challenged with additional AD-related insults, such as reduction of the Aβ-degrading enzyme neprilysin (Farris et al., 2007) or overexpression of the tyrosine kinase Fyn (Chin et al., 2004, 2005). Doubly transgenic hAPPJ9/Fyn mice exhibit prominent AD-related abnormalities (Chin et al., 2004, 2005). To examine the role of tau in these mice, we separately crossed hAPPJ9 mice and mice overexpressing wild-type murine Fyn (line N8) (Kojima et al., 1997, 1998) onto the Tau−/− background and then bred hAPPJ9/Tau−/− mice with Fyn/Tau−/− mice.
TASD41 mice express hAPP with the Swedish and London (V717I) mutations from the Thy1 promoter (Rockenstein et al., 2001). TASD41 mice were bred with the same Fyn transgenic line to produce TASD41/Fyn/Tau+/+ mice. TASD41 mice were also crossed with Tau−/− mice to produce TASD41/Tau−/− mice, which were then bred with Fyn/Tau−/− mice to generate TASD41/Fyn/Tau−/− mice.
SOD1G93A mice from The Jackson Laboratory (stock number 004453) were crossed with Tau−/− mice (Dawson et al., 2001). SOD1G93A/Tau+/− offspring were bred with Tau+/− mice to produce SOD1G93A/Tau+/+, SOD1G93A/Tau+/−, and SOD1G93A/Tau−/− mice. SOD1G93A mice were weighed regularly, and age at disease onset was defined as the point at which weight peaked before beginning to decline. Survival calculation was based on the age at which mice either died spontaneously or were killed for being unable to right themselves within 30 s after being placed on their sides.
A second tau-deficient line, in which the tau locus is replaced by green fluorescent protein (GFP) (Tucker et al., 2001), was crossed with hAPPJ20 to produce hAPPJ20/Tau+/GFP mice, heterozygous for tau. These mice were crossed with nontransgenic (NTG)/Tau+/GFP mice to produce both hAPPJ20/Tau+/+ and hAPPJ20/TauGFP/GFP mice.
All lines had been backcrossed to a consistent strain background, C57BL/6J. Males and females were used for all experiments, and no sex-dependent effects were identified (data not shown). Animals were housed in a pathogen-free barrier facility with a 12 h light/dark cycle and ad libitum access to food and water. All experiments were approved by the Institutional Animal Care and Use Committees of the University of California, San Francisco or Baylor College of Medicine.
Water maze.
The water maze pool (122 cm diameter) contained opaque water with a 14 cm, square platform submerged 2 cm below the surface. For cued training sessions, a black-and-white striped mast was mounted above the platform. Mice were trained to locate the platform over six sessions (two per day, 4 h apart), each with two trials (15 min apart). The platform location was changed for each session. Hidden platform training began 3 d later, consisting of 10 sessions (two per day, 4 h apart), each with three trials (15 min apart). Platform location remained constant in hidden platform sessions, and entry points were changed semirandomly between trials. The day after hidden platform training was completed, a 60 s probe trial was performed. The platform was removed, and the entry point was in the quadrant opposite the original target quadrant. Performance was monitored with an EthoVision video-tracking system (Noldus Information Technology).
Elevated plus maze.
The elevated plus maze consisted of two open arms and two closed arms equipped with rows of infrared photocells and a computer interface (Hamilton-Kinder). Mice were placed individually into the center of the maze and allowed to explore for 10 min. Beam breaks were quantified to calculate the amount of time spent and distance moved in each arm. The apparatus was cleaned with 0.25% bleach between the testing of each mouse to standardize odors.
Novel object recognition.
Mice were transferred to the testing room and acclimated for at least 1 h before testing. The testing was performed in a 20 × 40 cm white plastic chamber under red light. On days 1 and 2, mice were habituated to the testing arena for 15 min. On day 3, each mouse was presented with two identical objects in the same chamber and allowed to explore freely for 10 min. Twenty-four hours after this training session, mice were placed back into the same arena for the test session, during which they were presented with an exact replica of one of the objects used during training along with a novel, unfamiliar object of different shape and texture. Object locations were kept constant during training and test sessions for any given mouse, but objects were changed semirandomly between mice. Arenas and objects were cleaned with 70% ethanol between each mouse. Behavior was recorded with a digital camcorder and time spent exploring each object was scored.
Pentylenetetrazole challenge.
Pentylenetetrazole (PTZ) (Sigma) dissolved in PBS was used at a concentration of 5 mg/ml. A dose of 40 mg/kg was administered intraperitoneally. Each mouse was placed in a cage and observed for 20 min after administration, with video recording. An investigator analyzed the videotapes to quantify the time course and severity of seizures according to published scales (Racine, 1972; Loscher et al., 1991). Seizure severity scores were as follows: 0, normal behavior; 1, immobility; 2, generalized spasm, tremble, or twitch; 3, tail extension; 4, forelimb clonus; 5, generalized clonic activity; 6, bouncing or running seizures; 7, full tonic extension; 8, death.
Video-EEG monitoring.
For placement of electrodes, mice were anesthetized with Avertin, and four pairs of cranial burr holes were made; the most anterior were used for reference and ground, and others were positioned for bilateral recordings over the temporal, parietal, and occipital cortices. Teflon-coated silver wire electrodes (0.005 inch diameter) were implanted in the subdural space and connected to a microminiature connector (Omnetics Connector). All recordings were performed at least 7 d after surgery on mice freely moving in the test cage. Digital EEG activity with simultaneous video was recorded with Harmonie software, version 6.1c (Stellate Systems). EEG activity was monitored for a median of 8 h total per mouse in three 2–3 h sessions during a 1 week period. The number of abnormal epileptiform spikes (sharp positive or negative deflections with amplitudes exceeding twice the baseline and lasting 25–100 ms) in each hour of recording and the latency to detection and the number of seizures were scored. The severity of spontaneous seizures in each mouse was scored on a scale as follows: 0, no seizures; 1, only nonconvulsive seizures; 2, mixed convulsive and nonconvulsive seizures; and 3, all convulsive seizures.
Immunohistochemistry.
Immunohistochemistry was performed on floating 30-μm-thick microtome sections (Palop et al., 2011). Primary antibodies were rabbit anti-calbindin D-28K (1:30,000; Swant) or rabbit anti-NPY (1:8000; Immunostar). Labeling was detected with a biotinylated secondary antibody and the Vectastain Elite avidin–biotin complex kit (Vector Laboratories). Images were acquired with a digital microscope (Axiocam; Carl Zeiss). Densitometric quantifications were performed with the Bioquant software package (Bioquant Image Analysis). Hippocampal remodeling was investigated in hAPPJ9/Fyn mice that had undergone PTZ challenge 20 min before being killed. Because PTZ does not induce changes in calbindin or NPY on this timescale (data not shown), the remodeling observed is attributable to previous spontaneous epileptiform activity, not to induced seizures.
Electrophysiology.
Four- to 6-month-old mice were anesthetized with Avertin and decapitated. The brain was quickly removed and placed in ice-cold slicing solution containing the following (in mm): 2.5 KCl, 1.25 NaPO4, 10 MgSO4, 0.5 CaCl2, 26 NaHCO3, 11 glucose, and 234 sucrose, pH ≈7.4 (305 mOsm). Horizontal slices were prepared on a Vibratome-3000 at either 450 μm (for field recordings) or 350 μm (for whole-cell patch clamp) in the solution above. Slices were incubated for 30 min in standard artificial CSF (ACSF) (30°C) containing the following (in mm): 2.5 KCl, 126 NaCl, 10 glucose, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, and 26 NaHCO3 (290 mOsmol; the pH was ∼7.4 when gassed with a mixture of 95% O2/5% CO2) and then at room temperature for at least another 30 min before recording. No recordings were made from slices >5 h after dissection. For recording, individual slices were transferred to a submerged chamber in which they were maintained at 30°C and perfused with ACSF at a rate of 2 ml/min.
Field EPSPs (fEPSPs) were evoked every 20 s with a parallel bipolar tungsten electrode (FHC) and recorded with glass electrodes (∼3 MΩ tip resistance) filled with 1 m NaCl and 25 mm HEPES, pH 7.3. Recordings were filtered at 2 kHz (−3 dB, eight-pole Bessel), digitally sampled at 20 kHz with a Multiclamp 700A amplifier (Molecular Devices), and acquired with a Digidata-1322A digitizer and pClamp 9.2 software. Data were analyzed offline with pClamp9 software and OriginPro 8.0 (OriginLab). For recordings in area CA1, the stimulating electrode was placed in the stratum radiatum at the border of CA3 and CA1, and the recording electrode was placed ∼150 μm away in CA1 stratum radiatum. Synaptic transmission strength in CA1 was assessed by generating input–output curves for the relationship between peak amplitude of the fiber volley and the initial slope of the resulting fEPSP. For each slice, the fiber volley amplitude and initial slope of the fEPSP responses were measured at stimulus strengths of 25–800 μA. For dentate gyrus recordings, the stimulating electrode was moved to the medial perforant path (MPP) in the dorsal blade; the recording electrode was also placed in the MPP ∼150 μm closer to CA3 than the recording electrode. To overcome inhibitory feedforward and feedback circuits that are activated during induction of long-term potentiation (LTP) and that normally prevent robust LTP induction in the dentate, inhibition by GABAA receptors was blocked by addition of picrotoxin (50 μm; Tocris Bioscience) for field recordings from the dentate. Input–output curves were generated as in CA1. Stimulus strength was then adjusted to ∼30% of the maximal fEPSP response for recordings that followed. Paired-pulse ratios were determined by evoking two fEPSPs 50 ms apart and dividing the initial slope of the second fEPSP by the initial slope of the first (fEPSP2/fEPSP1). After a 15 min stable baseline was established, LTP was induced in the dentate by theta burst stimulation (10 theta bursts were applied at 15 s intervals; each theta burst consisted of 10 bursts, at 200 ms intervals, of four 100 Hz pulses).
To evaluate epileptiform bursting, we adjusted the stimulus strength to evoke fEPSPs ∼30% of maximal size (∼150 μA), stimulated every 20 s in the stratum radiatum near the CA1/CA3 border, and recorded field responses ∼200 μm away in stratum pyramidale of area CA1. Epileptiform activity was induced by superfusing bicuculline into the bath (10 μm). We quantified the strength of discharges by calculating the coastline burst index (CBI) (Korn et al., 1987), which is a measure of the length of the outline of the burst waveform. The CBI is the summated distance between successive data points in a time window containing the entire burst using the formula, , where Vi is field voltage at time point i (ti), and Vi − 1 is field voltage at the previous time point (ti − 1). To correct for underlying baseline “noise,” the CBI for an equal duration of recording in the absence of bursts was subtracted from the burst CBI. This sensitive measure of burst intensity is useful for determining the effects of anti-epileptic interventions (Korn et al., 1987; Tallent and Siggins, 1999).
Whole-cell recordings were made from individual granule cells located within the dentate gyrus, identified under infrared differential interference contrast video microscopy with a Carl Zeiss Axioskop 2 FS plus microscope. Patch pipettes were pulled from borosilicate glass (WPI) and filled with the following (in mm): 120 Cs-gluconate, 10 HEPES, 0.1 EGTA, 15 CsCl2, 4 MgCl2, 4 Mg-ATP, and 0.3 Na2-GTP, pH 7.25 (adjusted with 1 m CsOH; 285–290 mOsm; patch electrode resistance, 3–6 MΩ). Electrophysiological data were obtained from granule cells that had a holding current between −100 and 50 pA at −60 mV and a membrane input resistance (Rm) >100 MΩ. Series resistance was monitored throughout recordings; if it increased by >15 MΩ or varied by >15%, recordings were discarded. Voltage-clamp data were acquired with a Digidata-1322A digitizer and pClamp 9.2 software (Molecular Devices), low-pass filtered at 6 kHz (−3 dB, eight-pole Bessel), and digitally sampled at 10 kHz with a Multiclamp 700A amplifier (Molecular Devices).
EPSCs were recorded at a holding voltage of −60 mV, at which there are no currents through GABAA receptors. IPSCs were isolated by recording at a holding potential of +15 mV, the reversal potential of AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated currents. To block GIRK (G-protein-coupled inwardly rectifying K+ channel) channels activated by GABAB receptor activation, Cs+ was included in the recording pipette. Evoked PSCs were generated by a bipolar glass stimulating electrode filled with ACSF and placed in the molecular layer of the dentate gyrus within 100 μm of the patched granule cell. Stimulus pulses were delivered through a stimulus isolation unit (IsoFLEX; A.M.P.I.). To generate input–output relationships, the minimal stimulus intensity that evoked a synaptic current was first determined for a 40 μs stimulus, which was typically 50–100 μA. Stimulus duration was then increased in 20 μs intervals to vary input strength. Evoked (e) EPSCs that were ∼80% of the maximal response were used for subsequent analysis. Spontaneous and miniature currents were analyzed with event detection software (wDetecta; J. R. Huguenard, Stanford University, Stanford, CA). The frequencies of spontaneous (s) and miniature (m) EPSCs were determined from the average frequency of 200 randomly selected sequential events for each cell. For amplitude, individual currents without other currents contaminating the decay phase were isolated. In each cell, eEPSCs and sEPSCs were first recorded at −60 mV, then eIPSCs and sIPSCs were recorded at +15 mV. Then, mEPSCs and mIPSCs were recorded after addition of tetrodotoxin. Calculations involving comparisons between various PSC types (e.g., sIPSC − mIPSC frequency) were made for individual cells in which both datasets were obtained.
To determine the NMDAR/AMPAR ratio, EPSCs were evoked in standard ACSF in the presence of 50 μm picrotoxin (picrotoxin was not used for other whole-cell recordings). Stimulus amplitude was adjusted to produce single-peaked responses with a short constant latency (2–3 ms, to ensure monosynaptic responses) and average amplitude of ∼60 pA. Evoked EPSCs were recorded at −80 and +35 mV, and in some cells, the complete current–voltage (I–V) relationship was determined from −80 to +35 mV in 5 mV steps. Because steps to +35 mV can activate voltage-dependent currents that decay very slowly (t1/2 > 5 s), EPSCs were evoked at low frequency (once every 40 s) to minimize their accumulation and the accumulation of slowly decaying NMDAR-mediated currents. Three to five traces were used to generate a single average trace at each holding potential for final analysis. Occasional traces were contaminated by stimulus-evoked epileptiform events (resulting from disinhibition by picrotoxin), preventing the determination of peak monosynaptically evoked currents; these traces were not included in the final average trace used for analysis. The peak current at −80 mV, considered to be fully mediated by AMPAR attributable to magnesium block of NMDAR, was used to establish AMPAR-mediated responses. The time for AMPAR currents to decay fully to baseline was determined from the recordings at −80 mV, and a time window after that point was selected for measurement of the NMDAR current in recordings at +35 mV. This current was designated as the NMDAR measurement, and INMDA at +35mV/IAMPA at −80 mV was taken as the NMDAR/AMPAR ratio.
Data analysis.
Investigators acquiring and scoring data were blinded to the genotype and treatment of mice. Data were analyzed with GraphPad Prism and SPSS 16.0. Unless specified otherwise in figure legends, statistical comparisons were made with the log-rank test for survival data, the exact test for categorical data, Kolmogorov–Smirnoff test for comparison of cumulative probability plots, and ANOVA with Bonferroni's post hoc tests for other data. A p value <0.05 was considered significant. Bar graphs show mean ± SEM.
Results
Tau reduction prevents behavioral abnormalities and early mortality caused by synergistic effects of Aβ and Fyn
We used the Morris water maze to assess the effect of tau reduction on spatial learning and memory deficits in hAPPJ9/Fyn mice, which express approximately half as much hAPP and Aβ in the brain as hAPPJ20 mice but are sensitized to pathogenic Aβ effects by neuronal overexpression of Fyn (Chin et al., 2004, 2005). Compared with Tau+/+ and Tau−/− mice lacking hAPP and Fyn transgenes (NTG/Tau+/+ and NTG/Tau−/−), hAPPJ9/Fyn/Tau+/+ mice showed a mild but significant delay in learning the spatial (hidden platform) component of the water maze (Fig. 1A). hAPPJ9/Fyn/Tau−/− mice, however, performed significantly better than hAPPJ9/Fyn/Tau+/+ mice (Fig. 1A). The benefits of tau reduction were more obvious in a probe trial conducted 24 h after completion of hidden platform training. Only hAPPJ9/Fyn/Tau+/+ mice were impaired, showing lower percentage dwell time in the target quadrant, fewer platform crossings, and a longer latency to the first crossing of the area where the platform had been located (Fig. 1B–D).
Figure 1.
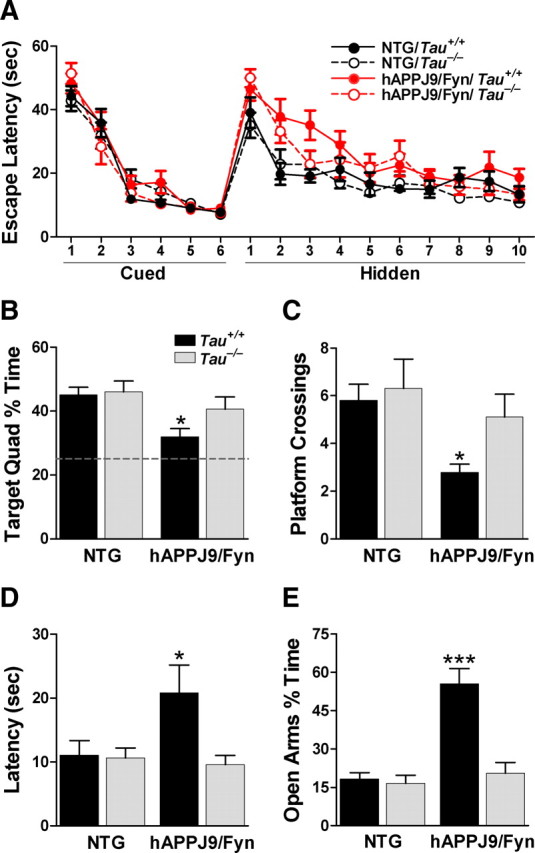
Tau reduction prevents behavioral deficits in hAPPJ9/Fyn mice. A, Morris water maze. There were no differences on the cued platform task. hAPPJ9/Fyn/Tau+/+ mice, but not hAPPJ9/Fyn/Tau−/− mice, were impaired on the hidden platform task (repeated-measures ANOVA, p < 0.005; on post hoc tests, only hAPPJ9/Fyn/Tau+/+ mice differed from controls; n = 9–10 mice per group; 4.5–8 months old). B–D, Morris water maze probe trial. Only hAPPJ9/Fyn/Tau+/+ mice were impaired, showing less time in the target quadrant (B), fewer platform crossings (C), and a longer latency to the first crossing of the location where the target platform had been (D). *p < 0.05 on post hoc tests. E, Elevated plus maze. hAPPJ9/Fyn/Tau+/+ mice showed abnormally high time in the open arms, but hAPPJ9/Fyn/Tau−/− mice did not (ANOVA, p < 0.0001; Tau × hAPPJ9/Fyn interaction, p < 0.0005; on post hoc tests, hAPPJ9/Fyn/Tau+/+ mice differed from other groups, ***p < 0.001; n = 9–10 mice per group; 3.5–7 months old).
hAPPJ9/Fyn/Tau+/+ mice also spent more time exploring the open arms of an elevated plus maze than NTG wild-type controls (Fig. 1E), consistent with previous results (Chin et al., 2005). In contrast, hAPPJ9/Fyn/Tau−/− mice showed no abnormalities in this test (Fig. 1E).
Like other hAPP transgenic lines, hAPPJ9/Fyn/Tau+/+ mice exhibit early mortality (Chin et al., 2004). We investigated whether tau reduction improved their survival. Approximately half of the hAPPJ9/Fyn/Tau+/+ mice died before 8 months of age (Fig. 2A). No early mortality was seen in hAPPJ9/Fyn/Tau−/− mice (Fig. 2B).
Figure 2.
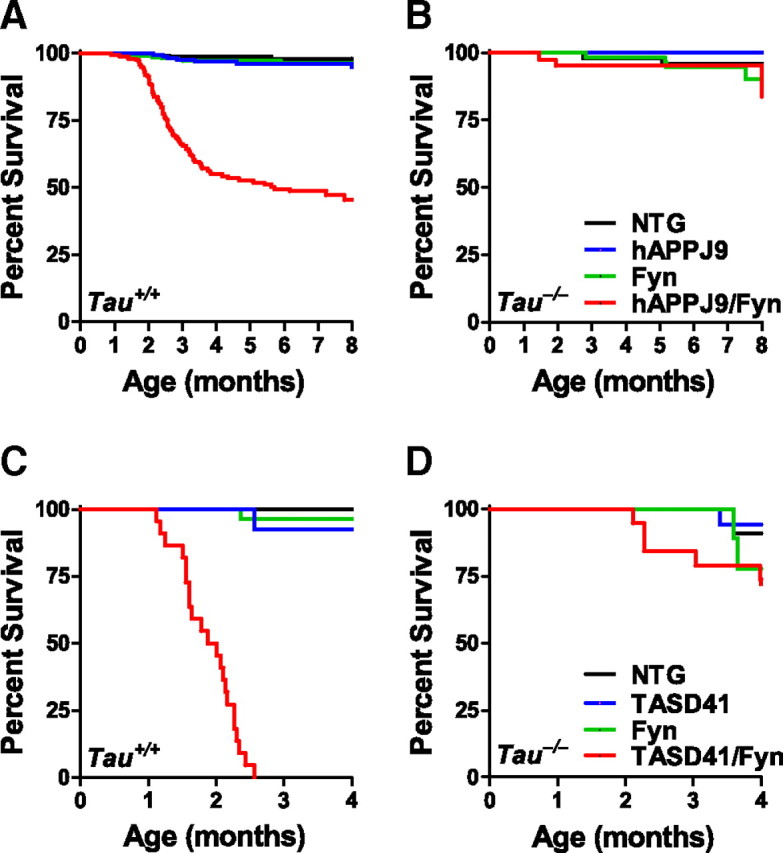
Tau reduction prevents early mortality in hAPPJ9/Fyn mice and TASD41/Fyn mice. A, B, Kaplan–Meier survival plots in NTG, hAPPJ9, Fyn, and hAPPJ9/Fyn mice on the Tau+/+ (A) or Tau−/− (B) background. There was high mortality in hAPPJ9/Fyn/Tau+/+ mice (log-rank test vs other Tau+/+ groups, p < 0.0001) but not in hAPPJ9/Fyn/Tau−/− mice (log-rank test vs hAPPJ9/Fyn/Tau+/+, p < 0.0001). C, D, Kaplan–Meier survival plots in NTG, TASD41, Fyn, and TASD41/Fyn mice on the Tau+/+ (C) or Tau−/− (D) background. There was high mortality in TASD41/Fyn/Tau+/+ mice (log-rank test vs other Tau+/+ groups, p < 0.0001), whereas TASD41/Fyn/Tau−/− mice had longer survival (log-rank test vs TASD/Fyn/Tau+/+, p < 0.0001).
We observed similar effects in a different hAPP line, TASD41, crossed with Fyn transgenic mice. Consistent with the higher Aβ levels and greater impairment of TASD41 mice than of hAPPJ9 mice, TASD41/Fyn/Tau+/+ mice showed even earlier mortality than hAPPJ9/Fyn/Tau+/+ mice; none survived longer than ∼2.5 months (Fig. 2C), which precluded analysis of behavioral deficits at later ages. A beneficial effect of tau reduction was apparent also in this line, as evidenced by significantly longer survival in TASD41/Fyn/Tau−/− mice than in TASD41/Fyn/Tau+/+ mice (Fig. 2D).
Thus, tau reduction prevents cognitive deficits and early mortality not only in hAPP singly transgenic mice (Roberson et al., 2007; Ittner et al., 2010) but also in hAPP mice that have been sensitized to Aβ-induced deficits through the overexpression of Fyn.
Tau reduction does not prevent early mortality in a mouse model of amyotrophic lateral sclerosis
Having observed protective effects of tau reduction in several AD-related models, and also after systemic kainate administration (Roberson et al., 2007), we wondered whether tau reduction is generally neuroprotective or targets a specific pathophysiological aspect shared by these models. To address this issue, we investigated the effects of tau reduction in a mouse model of amyotrophic lateral sclerosis (ALS). SOD1G93A mice, which express the human SOD1 gene containing a familial ALS mutation (Gurney et al., 1994), were crossed with Tau−/− mice to generate SOD1G93A/Tau+/+, SOD1G93A/Tau+/−, and SOD1G93A/Tau−/− mice. We examined both time to disease onset and time to death or terminal disease, because different manipulations influence these parameters differentially. Tau reduction did not improve either age at onset or survival to end-stage disease (Fig. 3). We conclude that tau reduction is not universally neuroprotective and hypothesize that it impinges on a mechanism shared by Aβ- and kainate-induced neuronal dysfunction.
Figure 3.

Tau reduction does not alter disease onset or survival in a mouse model of ALS. A, Age at onset in SOD1G93A mice, defined as the age at which weight started to decline, was not affected by tau reduction. B, Survival to end-stage disease was not altered by tau reduction.
Tau reduction reduces network excitability and ameliorates the seizure phenotype in hAPPJ9/Fyn mice
We next explored the basis of the protective effect of tau reduction in hAPPJ9/Fyn mice. High levels of Aβ elicit abnormal patterns of neuronal activity (Palop and Mucke, 2010), and multiple mouse models of AD exhibit epileptic activity and related hippocampal remodeling (Hsiao et al., 1995; LaFerla et al., 1995; Moechars et al., 1999; Kumar-Singh et al., 2000; Lalonde et al., 2005; Palop et al., 2007; Minkeviciene et al., 2009). Because tau reduction increases the resistance of mice to excitotoxin-induced seizures (Roberson et al., 2007), we hypothesized that tau reduction exerts its beneficial effects in hAPPJ9/Fyn mice by preventing aberrant neuronal activity.
We first examined network excitability by measuring susceptibility to PTZ-induced seizures. Consistent with previous findings in CRND8 (Del Vecchio et al., 2004) and hAPPJ20 (Palop et al., 2007) mice, hAPPJ9/Fyn mice had more severe seizures at shorter latencies than NTG mice (Fig. 4A). Tau reduction prevented increased susceptibility to PTZ-induced seizures in hAPPJ9/Fyn mice (Fig. 4A). Singly transgenic hAPPJ9 mice were also more susceptible to PTZ-induced seizures, an effect also blocked by tau reduction (Fig. 4A). In fact, tau reduction reduced susceptibility to PTZ-induced seizures across all genotypes, including NTG mice (Fig. 4A) (Roberson et al., 2007).
Figure 4.
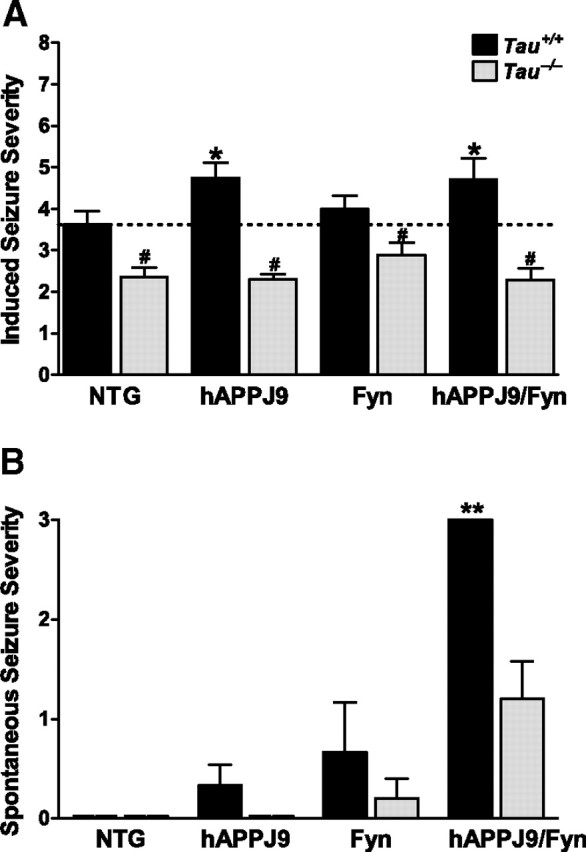
Effect of tau reduction on network excitability and spontaneous epileptiform activity in hAPP mice. A, Induced seizure severity was monitored after intraperitoneal injection of 40 mg/kg PTZ in NTG, hAPPJ9, Fyn, and hAPPJ9/Fyn mice (Tau effect, p < 0.001 by ANOVA; *p < 0.05 vs NTG and #p < 0.05 vs corresponding Tau+/+ group by post hoc testing; n = 16–20 mice per group; 5–8 months old). B, Severity of electrographically detected spontaneous seizures was scored (Tau effect, p < 0.005 by ANOVA; **p < 0.01 by post hoc tests; n = 5–6 mice in each group, median 8 h of EEG; 8–10 months old).
We also investigated whether tau reduction modulates spontaneous epileptiform activity. Monitoring spontaneous seizures in hAPPJ9/Fyn mice by video-EEG recording confirmed a protective effect of tau reduction. Every hAPPJ9/Fyn/Tau+/+ mouse we examined had spontaneous convulsive seizures. Tau reduction had a potent anticonvulsant effect in hAPPJ9/Fyn mice, shifting the type of epileptic activity from convulsive seizures to milder, nonconvulsive seizures (Fig. 4B).
Cognitive deficits and early mortality in hAPPJ9/Fyn mice are probably caused by synergistic actions of Aβ and Fyn, because singly transgenic hAPPJ9 or Fyn lines showed no major behavioral impairments or early mortality (Chin et al., 2004, 2005). We also looked for electrographic seizures in these singly transgenic lines. As expected, the severity of spontaneous seizures in hAPPJ9 and Fyn mice was much lower than in hAPPJ9/Fyn mice, and even these rare and mild events were ameliorated by tau reduction (Fig. 4B).
We conclude that Aβ and Fyn have synergistic pro-epileptic effects in hAPPJ9/Fyn mice and that tau reduction reduces the severity of the epilepsy phenotype in both singly transgenic lines and hAPPJ9/Fyn mice.
Tau reduction prevents seizure-induced hippocampal remodeling in hAPPJ9/Fyn mice
Epileptiform activity in hAPP mice induces inhibitory remodeling in the dentate gyrus (Palop et al., 2007; Minkeviciene et al., 2009), which likely represents adjustments in the hippocampal “gate” to chronic overexcitation (Heinemann et al., 1992; Lothman et al., 1992; Palop et al., 2007). Hallmarks of this remodeling include reduction of calbindin in dentate granule cells, GABAergic sprouting of NPY-positive axons in the molecular layer of the dentate gyrus, and ectopic expression of NPY in the mossy fiber axons of the granule cells. These changes are seen in many models of chronic overexcitation and can be induced subacutely by seizure activity (Vezzani et al., 1999; Palop et al., 2007). Because tau reduction reduced network excitability and spontaneous epileptiform activity, we tested whether it also prevented inhibitory hippocampal remodeling in hAPPJ9/Fyn mice.
Consistent with previous findings (Chin et al., 2005; Palop et al., 2007), hAPPJ9/Fyn mice had robust loss of calbindin in the dentate gyrus (Fig. 5A,B) and aberrant expression of NPY in the dentate molecular layer and mossy fibers (Fig. 5C–E). Tau reduction prevented these alterations in hAPPJ9/Fyn mice (Fig. 5A–E), consistent with the finding that it reduces the severity of the seizures that likely trigger this hippocampal remodeling.
Figure 5.
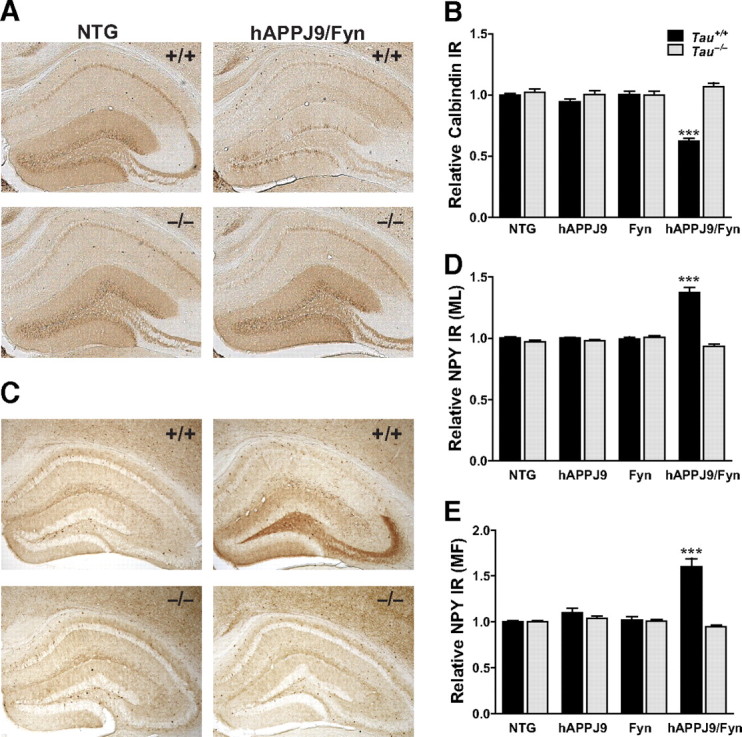
Tau reduction prevents hippocampal remodeling in hAPPJ9/Fyn mice. A, B, Calbindin levels in the molecular layer of the dentate gyrus were determined by immunohistochemistry (representative examples in A) and quantified by densitometry (B). Levels were significantly decreased in hAPPJ9/Fyn mice. This change was prevented by tau reduction (Tau × hAPPJ9/Fyn interaction, p < 0.001; ***p < 0.0001 by post hoc testing; n = 38–43 mice for Tau+/+ groups and 11–12 mice for Tau−/− groups; 6–9 months old). C–E, NPY levels were determined by immunohistochemistry (representative examples in C). Immunoreactivity in the molecular layer (D) and mossy fibers (E) was quantified by densitometry. Levels were not changed in hAPPJ9 or Fyn mice but were significantly increased in hAPPJ9/Fyn mice; the increase was prevented by tau reduction (Tau × hAPPJ9/Fyn interaction, p < 0.001; ***p < 0.0001 by post hoc testing; n = 43–53 mice for Tau+/+ groups and 18–20 mice for Tau−/− groups; 6–9 months old).
Tau reduction prevents EEG abnormalities and seizures in hAPPJ20 mice
We showed previously that tau reduction prevents behavioral abnormalities (Roberson et al., 2007) and hippocampal remodeling (Palop et al., 2007) in hAPPJ20 mice. Given our EEG findings in hAPPJ9/Fyn mice, we examined the effects of tau reduction on ictal and interictal EEG activity in hAPPJ20 mice.
We quantified the frequency of epileptiform spikes in freely moving hAPPJ20 mice. Consistent with previous findings (Palop et al., 2007), hAPPJ20/Tau+/+ mice had frequent spikes (Fig. 6A,B). Tau reduction effectively prevented this abnormal activity. In hAPPJ20/Tau−/− mice, cortical EEGs showed no spiking, and even in hAPPJ20/Tau+/− mice, spike frequency was not significantly higher than in controls (Fig. 6A,B). Tau reduction had no effect on EEG activity in NTG mice lacking hAPP transgenes (Fig. 6B and data not shown).
Figure 6.

Tau reduction prevents spontaneous epileptiform activity in hAPPJ20 mice. A, EEG tracings from hAPPJ20 mice with different levels of tau expression had a high frequency of generalized epileptiform spikes in the presence of normal tau levels but markedly fewer spikes in the presence of reduced tau levels. L, Left; R, right; T, temporal; P, parietal; O, occipital. Calibration bars: 2 s, 0.6 mV. B, Quantification of spontaneous epileptiform spiking in NTG and hAPPJ20 mice with different tau levels. Tau reduction blocked Aβ-induced spiking in a dose-dependent manner (hAPP × tau interaction, p < 0.001 by ANOVA; ***p < 0.001 vs all other groups by post hoc test; n = 20–32 h of EEG from 2–4 mice for each of NTG groups, 48–77 h from 5–10 mice for each of the hAPPJ20 groups; 7–14 months old). C, Kaplan–Meier analysis showing the duration of recording until an electrographic seizure was captured in hAPPJ20 mice with different tau levels. No seizures were observed in hAPPJ20/Tau+/− or hAPPJ20/Tau−/− mice (log-rank test, p = 0.05).
Tau reduction had a similar effect on seizures. hAPPJ20 mice did not exhibit convulsive seizures during routine husbandry or behavioral testing. During EEG recordings, however, some hAPPJ20/Tau+/+ mice had electrographic seizures. These seizures were always nonconvulsive, and the only behavioral correlate was motor arrest, sometimes accompanied by a subtle generalized myoclonic jerk (data not shown). Recording EEGs for a median of 8 h, we observed such seizures in 4 of 12 hAPPJ20/Tau+/+ mice but in 0 of 6 hAPPJ20/Tau+/− or 10 hAPPJ20/Tau−/− mice (exact test, p < 0.05). Kaplan–Meier analysis of the latency to electrographic seizure detection further demonstrated the protective effect of tau reduction in hAPPJ20 mice (Fig. 6C). These data indicate that tau reduction potently suppresses Aβ-induced epileptogenesis.
Tau reduction prevents epileptiform bursting in hippocampal slices
We further explored the anti-epileptic effect of tau reduction in a hippocampal slice model of epileptiform bursting induced by the GABAA receptor antagonist bicuculline (Gutnick et al., 1982). We applied bicuculline while recording in area CA1 and quantified the strength of resulting epileptiform discharges using coastline burst analysis, which measures the outline of the burst waveform and is useful for assessing burst intensity and determining the effects of anti-epileptic interventions (Korn et al., 1987; Tallent and Siggins, 1999). Bicuculline-induced epileptiform bursts were larger in slices from hAPPJ20/Tau+/+ mice than in slices from NTG/Tau+/+ controls (Fig. 7). Tau reduction markedly suppressed epileptiform bursting in slices from both NTG and hAPPJ20 mice, consistent with the increased resistance of NTG/Tau−/− mice and hAPPJ20/Tau−/− mice to chemically induced seizures in vivo (Fig. 4) (Roberson et al., 2007).
Figure 7.

Tau reduction prevents epileptiform bursting in acute hippocampal slices from NTG and hAPPJ20 mice. Bicuculline (10 μm) was superfused into the bath during recording in area CA1, and resulting epileptiform discharges were quantified by CBI. A, Representative traces 5 min before and 15 min after bicuculline. Note that traces after bicuculline are plotted on a longer timescale to accommodate the prolonged response. B, Time course. C, Average CBI during minutes 15–25. Slices from hAPPJ20/Tau+/+ mice had larger epileptiform bursts than slices from NTG/Tau+/+ mice, and tau reduction markedly reduced bursting in both NTG and hAPPJ20 slices (hAPP × tau interaction, p < 0.002 by two-way ANOVA; ***p < 0.001 vs all other groups; ###p < 0.001 vs Tau+/+ groups; NTG/Tau−/− and hAPPJ20/Tau−/− were not significantly different; n = 6–8 slices from 3–4 mice per genotype).
Tau reduction prevents synaptic impairments in hAPPJ20 mice
To better understand the cellular basis of this excitoprotective effect, we assessed the effects of tau reduction on synaptic transmission and plasticity in hAPPJ20 mice. We first examined miniature postsynaptic currents by whole-cell recording from dentate granule cells in the presence of tetrodotoxin. The frequency of mIPSCs onto granule cells was increased in hAPPJ20 mice (Fig. 8A,B), consistent with previous reports and with the sprouting of inhibitory axons seen histologically (Palop et al., 2007). In contrast, the frequency of mEPSCs was decreased (Fig. 8C,D), which is also consistent with other reports of the effects of hAPP/Aβ (Kamenetz et al., 2003; Shankar et al., 2007). There were no changes in mIPSC or mEPSC amplitude (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). These results suggest an increase in the number of effective inhibitory synaptic contacts and a decrease in the number of excitatory synaptic contacts onto dentate granule cells in hAPPJ20 mice. Tau reduction prevented both abnormalities (Fig. 8).
Figure 8.
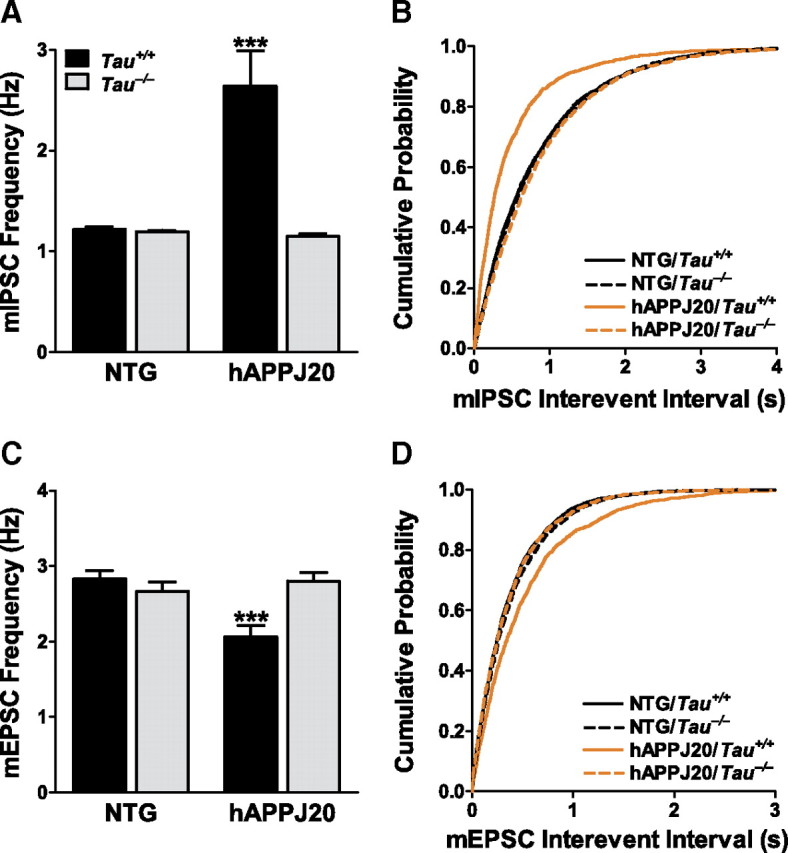
Tau reduction prevents abnormalities in miniature postsynaptic potentials in dentate granule cells of hAPPJ20 mice. Whole-cell recordings were made from acute hippocampal slices. A and C show mean frequency; B and D show cumulative probability plots for interevent intervals. A, B, mIPSC frequency was increased in hAPPJ20 granule cells, and the change was blocked by tau reduction (***p < 0.001 vs NTG/Tau+/+ by Kolmogorov–Smirnoff test). C, D, mEPSC frequency was reduced in hAPPJ20 granule cells, and the change was blocked by tau reduction (***p < 0.001 vs NTG/Tau+/+ by Kolmogorov–Smirnoff test). For both mIPSC and mEPSCs, n = 2000–2400 events from 10–12 cells (200 events from each) in 4 mice per genotype.
To examine how granule cells respond to spontaneous network activity in hippocampal slices, we also recorded spontaneous postsynaptic currents in dentate granule cells in the absence of tetrodotoxin. sIPSC frequency was reduced in hAPPJ20/Tau+/+ (Fig. 9A,B). In contrast to NTG controls, the frequency of sIPSCs in hAPPJ20/Tau+/+ mice was not much greater than the frequency of mIPSCs (Fig. 9C), suggesting that inhibitory neurons synapsing onto dentate granule cells had little spontaneous, action potential-driven activity. Thus, whereas inhibitory interneurons in hAPPJ20/Tau+/+ mice make an increased number of effective synapses onto granule cells (as reflected by the higher mIPSC frequency), they are less active than inhibitory interneurons in NTG mice. The decreased activity of inhibitory interneurons in hAPPJ20/Tau+/+ mice may be caused by reduced excitatory drive onto these cells, because blocking excitatory neurotransmission with 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) and APV reduced sIPSC frequencies less markedly in hAPPJ20/Tau+/+ mice than in NTG mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Reduced intrinsic excitability of the inhibitory interneurons may also contribute. sIPSCs recorded in the presence of NBQX and APV include both action potential-independent, stochastic release of vesicles (mIPSCs) and events caused by action potentials generated spontaneously in inhibitory cells. Although the sum of these two components was similar in NTG/Tau+/+ and hAPPJ20/Tau+/+ mice (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), the higher mIPSC frequency in hAPPJ20/Tau+/+ mice (Fig. 8A) indicates that the sIPSC component attributable to spontaneous action potentials in inhibitory interneurons was smaller in hAPPJ20/Tau+/+ mice, suggesting reduced intrinsic excitability of the inhibitory neurons.
Figure 9.

Tau reduction prevents excitation–inhibition imbalance in dentate granule cells of hAPPJ20 mice. Whole-cell recordings were made from acute hippocampal slices without pharmacological receptor blockers. A, B, sIPSC frequency was lower in hAPPJ20 granule cells and higher in Tau−/− groups than in NTG/Tau+/+ controls (***p < 0.001 and **p < 0.01 vs NTG/Tau+/+ by Kolmogorov–Smirnoff test; n = 2800–3000 events from 14–15 cells, 200 events from each, in 4 mice per genotype). C, Frequency of action potential-driven IPSCs. For each cell from which both sIPSC and mIPSC frequency data were obtained, we subtracted the frequency of mIPSCs (recorded in the presence of TTX; see Fig. 8A) from the frequency of sIPSCs; the difference reflects IPSCs driven by action potentials. hAPPJ20/Tau+/+ cells had almost no action potential-driven IPSCs, because the sIPSC frequency was essentially the same as the mIPSC frequency (hAPP × tau interaction, p < 0.001 by two-way ANOVA; ***p < 0.001 vs all other groups by post hoc test; n = 10–12 cells from 4 mice per genotype). D, hAPPJ20/Tau+/+ cells had an increased ratio of sEPSC to sIPSC frequency, and this abnormality was prevented by tau reduction (hAPP × tau interaction, p < 0.05 by two-way ANOVA; ***p < 0.001 vs all other groups by post hoc test; n = 14–15 cells from 4 mice per genotype). E, Amplitude of IPSCs onto granule cells evoked by stimulation in the molecular layer was reduced in hAPPJ20/Tau+/+ mice but not in hAPPJ20/Tau−/− mice (hAPP × tau interaction, p < 0.002 by two-way ANOVA; ***p < 0.001 vs all other groups by post hoc test; n = 10 cells from 4 mice per genotype). F, Amplitude of evoked EPSCs onto granule cells was mildly increased in hAPPJ20/Tau+/+ mice but not in hAPPJ20/Tau−/− mice (hAPP × tau interaction, p = 0.14; hAPP effect, p < 0.05 by two-way ANOVA; *p < 0.05 vs NTG/Tau−/− by post hoc test; n = 10 cells from 4 mice per genotype).
Tau reduction prevented the decrease in sIPSC frequency in hAPPJ20 mice; in fact, sIPSC frequencies were higher in NTG/Tau−/− and hAPPJ20/Tau−/− mice than in NTG/Tau+/+ controls (Fig. 9A,B). Thus, tau reduction may counteract Aβ, at least in part, by enhancing the activity of inhibitory interneurons.
hAPPJ20/Tau+/+ mice showed no alterations in the frequency of sEPSCs or in the amplitudes of either sIPSCs or sEPSCs (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). However, the ratios of sEPSC to sIPSC frequency provided evidence that excitation and inhibition are imbalanced in dentate granule cells of hAPPJ20/Tau+/+mice, with a shift toward more excitation (Fig. 9D). This change was also blocked by tau reduction (Fig. 9D). A similar pattern was observed when we recorded evoked postsynaptic currents. eIPSC amplitude was reduced in hAPPJ20/Tau+/+ slices (Fig. 9E), whereas eEPSC amplitude was mildly increased (Fig. 9F). This increase is likely attributable to the decrease in inhibition (Fig. 9A–C,E), because hAPPJ20/Tau+/+ mice showed no increase in AMPA receptor-mediated postsynaptic currents or input–output relationships in the presence of picrotoxin (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) (Palop et al., 2007; Harris et al., 2010). Tau reduction also blocked the changes in evoked IPSCs and EPSCs (Fig. 9E,F). We conclude that tau reduction blocks the excitation/inhibition imbalance induced by hAPP/Aβ in dentate granule cells.
We also investigated the effects of tau reduction on abnormalities of synaptic plasticity in the dentate gyrus. At medial perforant path synapses onto dentate granule cells, paired-pulse facilitation (PPF) and LTP are impaired in hAPPJ20 mice (Palop et al., 2007; Sun et al., 2008; Harris et al., 2010). Tau reduction blocked both of these abnormalities (Fig. 10A,B), preventing impairment of both short-term and long-term plasticity. The impairment of LTP in hAPPJ20 mice is unlikely to be attributable to changes in inhibition, because GABA receptors were blocked during the recordings. Thus, we investigated NMDA receptor function in dentate granule cells by whole-cell recordings of isolated NMDA- and AMPA-receptor-mediated currents. The NMDA/AMPA current ratio was depressed in hAPPJ20/Tau+/+ mice (Fig. 10C) because of impairments in NMDA receptor-mediated currents (Supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This effect, too, was blocked by tau reduction (Fig. 10C).
Figure 10.

Tau reduction prevents abnormalities in synaptic transmission and plasticity in hAPPJ20 mice. Field (A, B, D) and whole-cell (C) recordings were made from acute hippocampal slices. A, Paired-pulse ratio in the medial perforant pathway was reduced in hAPPJ20/Tau+/+ mice but normal in hAPPJ20/Tau−/− mice (hAPP × tau interaction, p < 0.01 by two-way ANOVA; **p < 0.01 vs all other groups by post hoc tests; n = 8 slices from 3–4 mice per genotype.) B, Theta burst stimulation-induced LTP at the medial perforant path synapse was impaired in hAPPJ20/Tau+/+ mice, and this deficit was blocked by tau reduction (repeated-measures ANOVA on data from minutes 51–60, p < 0.01; n = 10–12 slices from 3–4 mice per genotype). C, Evoked NMDA and AMPA currents were recorded by whole-cell patch clamp on dentate granule cells. NMDA/AMPA ratios were depressed in hAPPJ20/Tau+/+ mice, and this deficit was blocked by tau reduction (hAPP × tau interaction, p < 0.001 by two-way ANOVA; ***p < 0.001 vs all other groups by post hoc tests; n = 15–18 cells from 4 mice per genotype). D, Synaptic strength in area CA1 was significantly decreased in hAPPJ20/Tau+/+ mice (repeated-measures ANOVA, p < 0.01) but normal in hAPPJ20/Tau−/− mice (n = 10–12 slices from 3–4 mice per genotype).
Finally, we examined synaptic physiology in area CA1. PPF and LTP are unimpaired in area CA1, but synaptic transmission is decreased in hAPPJ20 mice, as evidenced by altered input–output relationships (Palop et al., 2007; Sun et al., 2008; Harris et al., 2010). We reproduced this finding in hAPPJ20/Tau+/+ mice and found that tau reduction normalized the input–output curves (Fig. 10D).
Thus, tau reduction prevents a wide range of abnormalities in synaptic transmission and plasticity in hippocampal subfields of hAPPJ20 mice.
Discussion
This study demonstrates that tau reduction suppresses spontaneous seizure activity and corrects synaptic deficits in hAPP mice with high Aβ levels and in hAPP/Fyn mice with lower Aβ levels that are sensitized to Aβ by coexpression of Fyn. Our data indicate that tau enables synaptic effects implicated in Aβ-induced neuronal dysfunction, because tau reduction prevented LTP impairment, NMDA receptor dysfunction, and excitation/inhibition imbalance in hAPP mice. At the network level, singly transgenic hAPP mice had interictal spikes and nonconvulsive seizures that were eliminated by tau reduction. Coexpression of hAPP/Aβ and Fyn caused overt convulsive seizures that were also ameliorated by tau reduction. Furthermore, tau reduction prevented cognitive impairment and early mortality in mice with deficits caused by synergistic effects of Aβ and Fyn. Our findings illuminate the synaptic and network mechanisms underlying copathogenic effects of Aβ, tau, and Fyn. Together with previous studies showing dual interactions between Fyn and Aβ or between Fyn and tau (Lambert et al., 1998; Lee et al., 1998; Williamson et al., 2002; Chin et al., 2004, 2005; Ittner et al., 2010), these findings suggest that the Aβ–Fyn–tau triad contributes to AD-related cognitive impairments, at least in part, by eliciting aberrant neural network activity and that disrupting their copathogenic relationship could be of therapeutic benefit.
Tau reduction is protective in several AD-related models but not in a model of ALS
The protective effects of tau reduction we identified in hAPPJ9, hAPPJ9/Fyn, and TASD41/Fyn mice add to a growing body of evidence that tau reduction protects against the pathogenic effects of Aβ in vivo. We showed previously that tau reduction prevented learning and memory deficits and early mortality in hAPPJ20 mice (Roberson et al., 2007), and similar results were recently obtained in APP23 mice (Ittner et al., 2010). Tau reduction also prevented learning and memory impairments in a mouse overexpressing glycogen synthase kinase 3 (Gómez de Barreda et al., 2010), which has been implicated in Aβ-induced pathogenesis (Hooper et al., 2008; Li et al., 2009).
In contrast, we found no beneficial effect of tau reduction in a mutant SOD1-transgenic mouse model of ALS. This selectivity is interesting, particularly because elimination of neurofilaments was protective in a related ALS model (Williamson et al., 1998), indicating that manipulation of different cytoskeletal gene products can block distinct pathogenic pathways. Crossing a mouse model of Niemann–Pick type C (NPC) disease with tau knock-out mice actually had adverse effects (Pacheco et al., 2009), further highlighting that tau reduction is beneficial in specific, AD-related circumstances. The detrimental effect of tau reduction in NPC mice is probably not attributable to the use of a different Tau−/− line (a GFP knock-in) (Tucker et al., 2001), because that line had beneficial effects in hAPPJ20 mice (supplemental Fig. 5, available at www.jneurosci.org as supplemental material) and APP23 mice (Ittner et al., 2010). Tau ablation was reported recently to change Aβ levels and plaque loads in opposite directions and to worsen behavioral deficits in hAPP mice from line Tg2576 at an age when behavioral deficits in this line tend to be somewhat variable (Dawson et al., 2010). Because these results are inconsistent with the findings from hAPPJ20, hAPPJ9, hAPPJ9/FYN, and TASD41/FYN mice described here and in our previous study (Roberson et al., 2007) as well as with findings in APP23 mice (Ittner et al., 2010), they may be specific to the Tg2576 model.
Tau reduction prevents aberrant neuronal activity
Given the differential effects of tau reduction on Aβ-related versus SOD1- or NPC-related impairments, we hypothesized that tau reduction impinges on a specific aspect of Aβ-induced pathogenesis. A likely candidate is Aβ-induced aberrant excitatory activity, which has been documented in multiple mouse models (Hsiao et al., 1995; LaFerla et al., 1995; Moechars et al., 1999; Kumar-Singh et al., 2000; Lalonde et al., 2005; Palop et al., 2007; Minkeviciene et al., 2009; Vogt et al., 2009). Because tau reduction increases resistance to pharmacologically induced seizures (Roberson et al., 2007), we studied its effects on the epilepsy phenotype in several AD-related mouse models and in hippocampal slices. Examination of EEG spiking, spontaneous and PTZ-induced seizures, and compensatory hippocampal remodeling all confirmed a robust antiepileptic effect of tau reduction. Tau reduction prevented both the mild seizure phenotype in hAPPJ9 mice and the more severe phenotype in hAPPJ20 mice. It also reduced the severity of seizures and prevented compensatory remodeling in hAPPJ9/Fyn mice. Finally, it prevented pharmacologically induced epileptiform bursting in hippocampal slices from both NTG and hAPPJ20 mice. Thus, tau has an important role in regulating neuronal activity and synchrony, and its presence in the brain contributes to seizure susceptibility.
Genetic background is an important factor to consider in these types of experiments because different strains vary in susceptibility to seizures and to hAPP/Aβ-induced neuronal dysfunction. Interestingly, strains that are relatively resistant to seizures, such as 129 strains (McKhann et al., 2003), are also relatively resistant to hAPP/Aβ-induced premature mortality and cognitive impairment (Krezowski et al., 2004), whereas strains that are relatively susceptible to seizures, such as FVB/N (Kasugai et al., 2007), are also more susceptible to hAPP/Aβ-induced premature mortality and cognitive impairment (Hsiao et al., 1995; Carlson et al., 1997). Importantly, we used a consistent genetic background, C57BL/6J, which has susceptibility intermediate between 129 and FVB/N. Because AD is associated with both premature mortality (Walsh et al., 1990; Roberson et al., 2005) and increased incidence of seizures (Palop and Mucke, 2009), mouse strains that also combine these characteristics when challenged with high cerebral Aβ levels may simulate the human condition more closely than those that do not. However, premature mortality is not a universal feature of AD mouse models, and additional research is needed to clarify the causal relationships between hAPP/Aβ, epileptiform activity, premature mortality, and cognitive deficits, all of which are ameliorated by tau reduction.
Synaptic effects of tau reduction
Our synaptic physiology data indicate that tau reduction corrects several abnormalities in multiple hippocampal subregions of hAPPJ20 mice. In the dentate gyrus, tau reduction corrected the increased mIPSC frequency and decreased mEPSC frequency, excitation/inhibition imbalance (decreased spontaneous/evoked inhibitory responses and increased spontaneous/evoked excitatory responses), NMDA receptor dysfunction, and deficits in PPF and LTP. In area CA1, tau reduction corrected the decline in evoked excitatory field potentials and blocked pharmacologically induced epileptiform bursting. Future experiments will need to determine which of these abnormalities reflect primary effects of Aβ and which are secondary, compensatory changes in the circuit.
NTG/Tau−/− mice are protected from epileptogenic agents other than Aβ, such as PTZ and kainic acid (Roberson et al., 2007). We saw a similar effect electrophysiologically, with much less bursting after GABA receptor blockade in NTG/Tau−/− slices than in NTG/Tau+/+ slices. NTG/Tau−/− mice also had a higher sIPSC frequency than NTG/Tau+/+ mice, which might contribute significantly to the protective effect of tau reduction. Inhibitory interneurons may be an important target of Aβ (Busche et al., 2008; Palop and Mucke, 2010), and it will be important to examine the effects of tau reduction on GABAergic transmission in more detail.
Mechanisms and implications
The findings presented here indicate that tau mediates the effects of Aβ and Fyn at both the synaptic and network levels. A variety of mechanisms have been proposed to explain how tau reduction might prevent Aβ toxicity, ranging from altered microtubule stability (Rapoport et al., 2002; Liu et al., 2004; King et al., 2006), elimination of a toxic tau fragment (Park and Ferreira, 2005), regulation of neuronal activity (Roberson et al., 2007), and changes in subcellular localization of Fyn (Ittner et al., 2010). In addition, although Tau−/− mice do not exhibit abnormal axonal transport in the absence of exogenous Aβ (Yuan et al., 2008), tau is required for Aβ to disrupt axonal transport in primary neurons (Vossel et al., 2010). These mechanisms are not mutually exclusive, and additional studies are needed to assess their relative importance and potential interdependence. Another important objective is to determine whether the protective effects of tau reduction against Aβ-induced aberrant neuronal activity and cognitive deficits depend on removal of a toxic tau species or reduction of a normal tau function that permits Aβ to elicit neuronal dysfunction.
Footnotes
The study was supported by the Stephen Bechtel Fund (E.D.R.) and by National Institutes of Health Grants NS054811 (E.D.R.), NS29709 (J.L.N.), AG022074 and NS041787 (L.M.), and RR018928 (J. David Gladstone Institutes). We thank Hana Dawson and Michael Vitek for tau knockout mice, Eliezer Masliah for TASD41 mice, Nobuhiko Kojima for Fyn mice, Tim Miller for advice on working with SOD1 mice, Hilda Solanoy, Xin Wang, and Kaitlyn Ho for technical support, the Gladstone Behavioral Core for assistance with behavioral testing, and Gary Howard and Stephen Ordway for editorial review.
References
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Cain DP, Grant SG, Saucier D, Hargreaves EL, Kandel ER. Fyn tyrosine kinase is required for normal amygdala kindling. Epilepsy Res. 1995;22:107–114. doi: 10.1016/0920-1211(95)00029-1. [DOI] [PubMed] [Google Scholar]
- Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK. Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet. 1997;6:1951–1959. doi: 10.1093/hmg/6.11.1951. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu GQ, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Dawson HN, Cantillana V, Jansen M, Wang H, Vitek MP, Wilcock DM, Lynch JR, Laskowitz DT. Loss of tau elicits axonal degeneration in a mouse model of Alzheimer's disease. Neuroscience. 2010;169:516–531. doi: 10.1016/j.neuroscience.2010.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett. 2004;367:164–167. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- Farris W, Schütz SG, Cirrito JR, Shankar GM, Sun X, George A, Leissring MA, Walsh DM, Qiu WQ, Holtzman DM, Selkoe DJ. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am J Pathol. 2007;171:241–251. doi: 10.2353/ajpath.2007.070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM AN1792(QS-21)-201 Study Team. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Gómez de Barreda E, Pérez M, Gómez Ramos P, de Cristobal J, Martín-Maestro P, Morán A, Dawson HN, Vitek MP, Lucas JJ, Hernández F, Avila J. Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol Dis. 2010;37:622–629. doi: 10.1016/j.nbd.2009.11.017. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH Tarenflurbil Phase 3 Study Group. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302:2557–2564. doi: 10.1001/jama.2009.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen WJ, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- Haass C, Mandelkow E. Fyn-tau-amyloid: a toxic triad. Cell. 2010;142:356–358. doi: 10.1016/j.cell.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harris JA, Devidze N, Halabisky B, Lo I, Thwin MT, Yu GQ, Bredesen DE, Masliah E, Mucke L. Many neuronal and behavioral impairments in transgenic mouse models of Alzheimer's disease are independent of caspase cleavage of the amyloid precursor protein. J Neurosci. 2010;30:372–381. doi: 10.1523/JNEUROSCI.5341-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Beck H, Dreier JP, Ficker E, Stabel J, Zhang CL. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res Suppl. 1992;7:273–280. [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kasugai M, Akaike K, Imamura S, Matsukubo H, Tojo H, Nakamura M, Tanaka S, Sano A. Differences in two mice strains on kainic acid-induced amygdalar seizures. Biochem Biophys Res Commun. 2007;357:1078–1083. doi: 10.1016/j.bbrc.2007.04.067. [DOI] [PubMed] [Google Scholar]
- King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SG, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci U S A. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Ishibashi H, Obata K, Kandel ER. Higher seizure susceptibility and enhanced tyrosine phosphorylation on N-methyl-d-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998;5:429–445. [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Krezowski J, Knudson D, Ebeling C, Pitstick R, Giri RK, Schenk D, Westaway D, Younkin L, Younkin SG, Ashe KH, Carlson GA. Identification of loci determining susceptibility to the lethal effects of amyloid precursor protein transgene overexpression. Hum Mol Genet. 2004;13:1989–1997. doi: 10.1093/hmg/ddh210. [DOI] [PubMed] [Google Scholar]
- Kumar-Singh S, Dewachter I, Moechars D, Lübke U, De Jonghe C, Ceuterick C, Checler F, Naidu A, Cordell B, Cras P, Van Broeckhoven C, Van Leuven F. Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol Dis. 2000;7:9–22. doi: 10.1006/nbdi.1999.0272. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's Aβ peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behav Brain Res. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, Panchamoorthy G. Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci. 1998;111:3167–3177. doi: 10.1242/jcs.111.21.3167. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H. Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci. 2004;24:2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Perry G, Chan HW, Verdile G, Martins RN, Smith MA, Atwood CS. Amyloid-β-induced toxicity of primary neurons is dependent upon differentiation-associated increases in tau and cyclin-dependent kinase 5 expression. J Neurochem. 2004;88:554–563. doi: 10.1046/j.1471-4159.2003.02196.x. [DOI] [PubMed] [Google Scholar]
- Löscher W, Hönack D, Fassbender CP, Nolting B. The role of technical, biological and pharmacological factors in the laboratory evaluation of anticonvulsant drugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 1991;8:171–189. doi: 10.1016/0920-1211(91)90062-k. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl. 1992;7:301–313. [PubMed] [Google Scholar]
- McKhann GM, 2nd, Wenzel HJ, Robbins CA, Sosunov AA, Schwartzkroin PA. Mouse strain differences in kainic acid sensitivity, seizure behavior, mortality, and hippocampal pathology. Neuroscience. 2003;122:551–561. doi: 10.1016/s0306-4522(03)00562-1. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reversé D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Pacheco CD, Elrick MJ, Lieberman AP. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum Mol Genet. 2009;18:956–965. doi: 10.1093/hmg/ddn423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L, Roberson ED. Quantifying biomarkers of cognitive dysfunction and neuronal network hyperexcitability in mouse models of Alzheimer's disease: depletion of calcium-dependent proteins and inhibitory hippocampal remodeling. Methods Mol Biol. 2011;670:245–262. doi: 10.1007/978-1-60761-744-0_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates β-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99:6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Hesse JH, Rose KD, Slama H, Johnson JK, Yaffe K, Forman MS, Miller CA, Trojanowski JQ, Kramer JH, Miller BL. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005;65:719–725. doi: 10.1212/01.wnl.0000173837.82820.9f. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E. Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ1–42. J Neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- Salter MW, Kalia LV. Src kinases: A hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Zhou Y, Halabisky B, Lo I, Cho SH, Mueller-Steiner S, Devidze N, Wang X, Grubb A, Gan L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol. 1999;81:1626–1635. doi: 10.1152/jn.1999.81.4.1626. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA. Neurotrophins are required for nerve growth during development. Nat Neurosci. 2001;4:29–37. doi: 10.1038/82868. [DOI] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF. Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci. 1999;22:25–30. doi: 10.1016/s0166-2236(98)01284-3. [DOI] [PubMed] [Google Scholar]
- Vogt DL, Thomas D, Galvan V, Bredesen DE, Lamb BT, Pimplikar SW. Abnormal neuronal networks and seizure susceptibility in mice overexpressing the APP intracellular domain. Neurobiol Aging. 2009 doi: 10.1016/j.neurobiolaging.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh JS, Welch HG, Larson EB. Survival of outpatients with Alzheimer-type dementia. Ann Intern Med. 1990;113:429–434. doi: 10.7326/0003-4819-113-6-429. [DOI] [PubMed] [Google Scholar]
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: Involvement of Src family protein kinases. J Neurosci. 2002;22:10–20. doi: 10.1523/JNEUROSCI.22-01-00010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Bruijn LI, Zhu Q, Anderson KL, Anderson SD, Julien JP, Cleveland DW. Absence of neurofilaments reduces the selective vulnerability of motor neurons and slows disease caused by a familial amyotrophic lateral sclerosis-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A. 1998;95:9631–9636. doi: 10.1073/pnas.95.16.9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008;28:1682–1687. doi: 10.1523/JNEUROSCI.5242-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]