

Abstract
p13suc1 has two native states, a monomer and a domain-swapped dimer. We show that their folding pathways are connected by the denatured state, which introduces a kinetic barrier between monomer and dimer under native conditions. The barrier is lowered under conditions that speed up unfolding, thereby allowing, to our knowledge for the first time, a quantitative dissection of the energetics of domain swapping. The monomer–dimer equilibrium is controlled by two conserved prolines in the hinge loop that connects the exchanging domains. These two residues exploit backbone strain to specifically direct dimer formation while preventing higher-order oligomerization. Thus, the loop acts as a loaded molecular spring that releases tension in the monomer by adopting its alternative conformation in the dimer. There is an excellent correlation between domain swapping and aggregation, suggesting they share a common mechanism. These insights have allowed us to redesign the domain-swapping propensity of suc1 from a fully monomeric to a fully dimeric protein.
Domain swapping was first described at the atomic level by Eisenberg and coworkers in 1994 in the crystal structure of diphtheria toxin (1). It refers to the process by which one protein molecule exchanges a structural domain with an identical partner (2–4). The domain can be a single element of secondary structure or an entire globular domain. The subunits of the resulting oligomer have the same structure as the monomer except in the “hinge loop”, which connects the exchanging subunit with the rest of the protein. Domain swapping has been proposed as a possible mechanism for protein aggregation (5). Further, it may be a novel regulatory motif for protein function§ (6–8) as well as a simple evolutionary strategy for evolving oligomers from monomers (9–11).
Monomeric and domain-swapped oligomeric forms of a protein have identical interactions, with the exception of the hinge loop, and thus comparable free energies per molecule. The small energetic difference between a monomer and its domain-swapped form has its origins in three different effects. First, the greater translational and rotational entropy favors the monomer. Second, the change in conformation of the hinge leads to a difference in free energy. Third, new intermolecular interfaces may form in the domain-swapped form, resulting in a free energy change.
There are now many examples of domain-swapped structures but still little experimental investigation of the mechanistic and thermodynamic details of the phenomenon (12, 13). Here we look at domain swapping in the cell cycle regulatory protein, p13suc1 (referred to subsequently as suc1). No new intermolecular interface is introduced in domain-swapped dimeric suc1, so the presence of this form must be explained solely by conformational changes originating in the hinge loop. We show that partitioning between monomer and dimer is controlled solely by the two proline residues located in this region. The proline side chain's unique characteristics introduce strain into the backbone, and we are able to manipulate this strain to redesign the assembly of suc1. Our results rationalize the earlier observation by Bergdoll et al. (14) of the occurrence of proline repeats in the hinge regions of domain-swapped proteins. Finally, we propose that domain swapping occurs via the denatured state in suc1 and that the protein aggregates by the same mechanism.
Materials and Methods
Materials.
High-purity urea was obtained from Rose Chemicals Ltd. (U.K.). All other chemicals were from Sigma or BDH. Site-directed mutagenesis was performed by using the Quick-Change site-directed mutagenesis kit (Stratagene).
Mutants were constructed and proteins expressed and purified as described previously (15, 16). The samples were pure as judged by SDS/PAGE and mass spectrometry. The buffer used for subsequent experiments was 50 mM Tris, pH 7.5/1 mM EDTA, unless stated otherwise. EDTA was used to prevent the formation of zinc-mediated nondomain-swapped dimers. Protein concentration was measured spectrophotometrically by using an extinction coefficient of ɛ280 = 19,940 cm−1 M−1, calculated by the method of Gill and Von Hippel (17). Samples were concentrated as required by using Centriprep 3 concentrators (Amicon).
Separation of Monomeric and Dimeric suc1.
Monomeric and dimeric suc1 were separated by high-resolution analytical size-exclusion chromatography by using an Amersham Pharmacia Superdex 75 h 10/30 column connected to an Amersham Pharmacia Äkta system and equilibrated with 50 mM Tris buffer, pH 7.5/300 mM NaCl/1 mM EDTA at 25°C. Monomeric suc1 has an elution volume of 12.4 ml, whereas dimeric suc1 elutes at 10.5 ml (Fig. 2A). The two peaks were well separated with the absorption signal returning to the baseline level between the two. The proportions of monomer and dimer were determined by integration of area of the elution peaks by using the Amersham Pharmacia Unicorn Evaluation package.
Figure 2.

Measurement of the stability of monomeric and dimeric suc1. (A) Elution profile of wild-type suc1 on a Superdex 75 h 10/30 column. (B) Plot of the square of the concentration of monomer vs. the concentration of dimer for wild-type suc1. The data are fitted to a straight line (r = 0.9977). (C) Urea-induced denaturation curve of monomeric wild-type suc1 monitored by fluorescence intensity. The best fits of the data are shown.
Determination of the Dissociation Constant of Wild-Type suc1.
Monomeric suc1 was concentrated to ≈4 mM, and a set of 10 dilutions was made to concentrations ranging from 75 μM to 2.5 mM in a sample volume of 5 μl. The dilution series was incubated at 50°C for a fixed length of time and then transferred to ice for 5 min. The amounts of monomer and dimer present were then determined by size-exclusion chromatography in 50 mM Tris, pH 7.5/300 mM NaCl/1 mM EDTA at 25°C. Because of the presence of a kinetic barrier between monomer and dimer at lower temperatures (see Results and Discussion), the equilibrium reached at 50°C is “frozen” instantly when the sample is transferred onto ice. PCR tubes that are designed to have a very low heat capacity were used to accelerate the rate of heat transfer. Thus, although the amounts of monomer and dimer are measured at 25°C, they reflect the equilibrium at 50°C. There were two reasons for using this “temperature jump” procedure. The first is that the column cannot be run at 50°C because the monomer and dimer peaks merge at this temperature (the rate of interconversion is of the same order of magnitude as the elution time). The second is that the concentrated protein could not be left for more than a few minutes at 50°C because aggregation occurred.
The procedure was then repeated using longer incubations at the 50°C equilibration temperature until no further change in the monomer and dimer ratios was observed by gel filtration. The system can then be assumed to be at equilibrium. A time of 5 min was found to be sufficient at the highest protein concentration in the dilution series, whereas the lower protein concentrations were incubated for up to 25 times longer (because monomer-to-dimer formation is slower for these). A plot of the square of the monomer vs. the dimer concentration could be fitted to a straight line through the origin, indicating that the incubation times used were long enough for the system to reach equilibrium. The slope of the linear fit gives the dissociation constant of the dimer. For all samples, only monomer and dimer species were observed, and there were no higher-order oligomers or soluble aggregates, as demonstrated by the absence of elution peaks other than those corresponding to monomer and dimer. Further, the sum of the monomer and dimer peak areas always corresponded to the amount of protein that was in the sample aliquot before incubation, showing that no irreversible aggregation had occurred.
Determination of the Dissociation Constant of suc1 Mutants.
The same procedure was used as that described for the wild type. Some mutants needed to be incubated at 50°C for a longer time than the wild type, because they equilibrated more slowly. The protein concentration range used depended on the mutant: those with high Kds needed very high concentrations (up to 20 mM for PA90), and the lowest protein concentration used was 40 μM (for PA92). As for the wild-type measurement, the incubation time was tested to ensure that the system had reached equilibrium and the absence of higher order oligomers and aggregation was also confirmed.
Measurement of the Kinetic Barrier for Interconversion Between Monomeric and Dimeric p13suc1.
To determine the effect of pH, temperature, and chemical denaturant on the kinetic barrier for monomer–dimer interconversion, samples of monomeric wild-type suc1 at a concentration of 2 mM were incubated in various conditions. The amounts of monomer and dimer present were then measured at regular time intervals by size-exclusion chromatography. The same experiment was performed with samples of dimeric wild-type suc1 at a concentration of 2 mM.
Determination of the Free Energy of Unfolding of Monomeric p13suc1.
The free energies of unfolding of wild type and the mutant monomers were determined at 25°C by urea-induced equilibrium denaturation experiments monitoring intrinsic fluorescence intensity, as described previously (16). A protein concentration of 2 μM was used. No oligomerization occurred under these conditions, as demonstrated by an absence of dependence of the stability on protein concentration in the range 1–10 μM.
The free energy of unfolding of monomeric suc1 in the presence of urea,
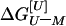 , is assumed to be linearly
proportional to the concentration of urea, [U] (18).
, is assumed to be linearly
proportional to the concentration of urea, [U] (18).
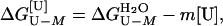 |
1 |
where 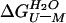 is the
free energy of unfolding in water, and m is the constant of
proportionality. At [U]50%, the concentration
of urea at which 50% of the protein is unfolded,
is the
free energy of unfolding in water, and m is the constant of
proportionality. At [U]50%, the concentration
of urea at which 50% of the protein is unfolded,
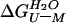 =
m[U]50%. Thus
=
m[U]50%. Thus
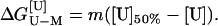 |
2 |
The denaturation curves were fitted to an equation derived from
Eq. 2, which yields the values of
[U]50% and m and their standard
errors. All mutants exhibited values of m that were the same
within error as the value for wild type, indicating that the mutations
do not significantly change the structure of the native or denatured
states. Therefore, the value of
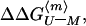 , the change on
mutation in the free energy of unfolding of the monomer at a mean value
of the midpoint of unfolding, can be calculated from the equation:
, the change on
mutation in the free energy of unfolding of the monomer at a mean value
of the midpoint of unfolding, can be calculated from the equation:
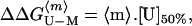 |
3 |
where m is the average value of m, obtained from measurements on all of the mutant proteins and repetitive runs on wild type, of 1.60 ± 0.01 kcal⋅mol M−1 (16). The use of a mean value of m allows calculation of the change in the free energy of unfolding on mutation with a low standard error.
Calculation of the Change in Free Energy of Unfolding of Dimeric p13suc1 on Mutation.
The dissociation constants, Kd, of the mutant protein can be combined with that of the wild type to obtain ΔΔGD-M, the effect of mutation on the equilibrium between monomer and dimer, as follows:
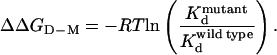 |
4 |
This parameter can be combined with the change in free energy of unfolding of the monomer on mutation, ΔΔGU-M, to give the change in free energy of unfolding of the dimer on mutation, ΔΔGU-D:
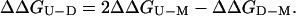 |
5 |
Note that the Kds were measured at elevated temperatures (to have a relatively fast equilibration time), but the free energy of unfolding of the monomers was measured at 25°C. However, it can reasonably be assumed that the change in free energy of unfolding on mutation (of a conservative nature) is independent of temperature, because the heat capacity of the native state of the protein is unlikely to change on mutation. Further, the values of ΔΔGU-M obtained for a number of suc1 mutants from calorimetry measurements at elevated temperatures are the same within error as those obtained from standard urea denaturation experiments at 25°C (unpublished results). This conclusion was also reached on detailed analysis of the protein chymotrypsin inhibitor 2 (19).
Measurement of Aggregation Kinetics.
The kinetics of aggregation was measured as follows: the absorbance at 600 nm was monitored as a function of time in a 1-cm quartz cuvette thermostated at 50°C. Protein concentrations were 1 mM, with the exception of PA92. This mutant aggregated so quickly that the concentration had to be lowered to 700 μM to allow the measurement to be made.
Results and Discussion
suc1 Domain Swaps via the Denatured State.
A general feature of domain swapping is that monomeric and oligomeric forms are separated by a high-energy barrier. In the case of suc1, equilibration starting from 2 mM of dimer in 50 mM Tris⋅HCl, pH 7.5, takes 3 months at 25°C. Such energy barriers make it difficult to measure the equilibria involved in domain swapping and thus preclude an energetic analysis. We propose that the energy barrier in suc1 arises because interconversion between monomer and dimer occurs via the denatured state (Fig. 1B). The evidence for this is as follows.
Figure 1.

Schematic representation of the structures of monomer and dimer forms of suc1 and model of domain swapping in suc1. (A) Monomer and dimer forms of suc1 coexist in solution in the absence of higher-order oligomers and aggregate over a wide range of conditions. The monomer form comprises a four-stranded β-sheet capped at one end by three short α-helices (26). In the dimer form, however, β-strand 4 (shown in red) originates from the other monomer in the dimer pair (27). There is no significant change in the fold except in the region that mediates the process, namely the hinge loop between β-strands 3 and 4. This loop (also colored red) adopts an extended conformation allowing β-strand 4 to exchange in the dimer, while it folds back on itself to form a β-hairpin in the monomer. The frequent occurrence of prolines in the hinge loops of domain-swapping proteins has been reported before (14). The β-hinge motif HxPEPH (where x is any residue) is conserved in the cks family. (B) Interconversion between monomer and dimer occurs via the denatured state. The residual interactions between β2 and the exchanging strand β4 that are retained in the denatured state are highlighted. (C) SDS/PAGE of wild-type suc1 at 0.6 mM protein concentration, showing two bands corresponding approximately to the molecular weights of monomer (13.3 kDa) and dimer (26.6 kDa). The protein sample was incubated in 2.5% SDS/6% β-mercaptoethanol at 95°C for 5 min before loading, and the gels were run by using the Phastgel System (AmershamPharmacia). Both bands were confirmed to be suc1 by N-terminal sequencing (Department of Biochemistry, University of Cambridge).
(i) The part of the structure exchanged in suc1 is a single β-strand, β4, centrally located in the 4-stranded β-sheet between strands 2 and 3 (Fig. 1A). A protein engineering analysis of the folding pathway of monomeric suc1 showed that β4 is a part not only of the hydrophobic core of the protein but also of the folding nucleus, and so it unfolds late, only after the transition state for unfolding (16). Moreover, an intermediate is transiently populated in the refolding reaction of suc1, and the interactions involving β4 are retained substantially in this species also.
(ii) We can also show that the dimer folds directly from the unfolded state by an independent pathway and not via initial formation of the monomer: protein at a concentration of 500 μM was unfolded in 30 mM HCl (pH 1.5) and then refolded by mixing with an equal volume of renaturing buffer to give a final pH of 7.5, 50 mM Tris buffer/1 mM EDTA (25°C). The proportions of monomer and dimer formed, determined immediately as described in Materials and Methods, were 80% monomer and 20% dimer. In contrast, a sample of monomer at the same temperature, pH, and protein concentration takes several months to reach equilibrium with the dimer form.
(iii) Another line of evidence comes from studies of the denatured state of suc1. Monomeric wild-type suc1 at a high concentration gives two bands on a 2% SDS gel, corresponding in their molecular weights to monomer and dimer, and both were confirmed subsequently to be suc1 by N-terminal sequencing (Fig. 1C). Because we know by fluorescence and NMR that suc1 is unfolded in 2% SDS (data not shown), we propose that the dimeric band corresponds to two unfolded molecules associating via residual interactions between β2 and the exchanging strand β4 (Fig. 1B). This conclusion is supported by molecular dynamics simulations of the unfolding pathway of monomeric suc1, showing that native-like interactions between β2 and β4 are retained even in the denatured state (20).
Therefore, we can conclude that strand exchange must occur from a state significantly more unfolded than the major kinetic intermediate. In other words, the interconversion of monomer and dimer occurs through the denatured state (Fig. 1B). Thus, suc1 is a protein with two distinct folding pathways leading to two different native states.
As a consequence of interconversion via the denatured state, the kinetic barrier can be lowered by shifting conditions to those in which the rate of unfolding is increased, by altering conditions such as temperature, pH, or denaturant concentration. For monomeric suc1, which has a melting temperature of 68°C, equilibration at 50°C is complete within 5 min at 2 mM protein concentration and a pH of 7.5. A similar rate of equilibration is observed in 3.4 M urea at 25°C (pH 7.5), the midpoint of unfolding of suc1 being 4.4 M. Likewise, a pH of 5 (25°C) speeds up the equilibration, whereas the unfolding midpoint of suc1 is at pH 3.8. Under all these conditions, the unfolding rates are increased, leading to fast equilibration. Likewise, domain swapping in other proteins has been shown to occur by a short exposure to mildly denaturing conditions (2, 21, 22). An appropriate choice of conditions thus allows the study of the equilibrium between monomer and dimer and the determination of the dissociation constant (Kd = 1.84 ± 0.03 mM at 50°C, pH 7.5).
Two Proline Residues Control Partitioning Between Monomer and Dimer.
Domain swapping in suc1 must be explained solely by conformational changes in the hinge loop, which is defined as those residues for which the change in (φ,ψ) angles is greater than 20° (2). Thus, the hinge loop comprises residues 88–92 in suc1, but to verify the boundaries, we included residues 87 and 93 in our analysis. All of the residues in the hinge were mutated to alanine, and the dissociation constants, Kd, of the mutant proteins were measured at 50°C (where equilibration is sufficiently fast to be measured easily). The value obtained for each mutant was combined with the Kd of the wild type to determine ΔΔGD-M, the effect of mutation on the equilibrium between monomer and dimer, by using Eq. 4 (Table 1). The effect of the mutation on the stability of monomer and dimer forms, ΔΔGU-M and ΔΔGU-D, respectively, can then be dissected by using Eq. 5 (Table 1) (Fig. 2).
Table 1.
Thermodynamic data for domain swapping of suc1 wild type and mutants
| Kd, μM | ΔΔGD-M/2* | ΔΔGU-M | ΔΔGU-D/2* | |
|---|---|---|---|---|
| Wild type | 1,850 ± 30 | |||
| VA87 | 1,970 ± 80 | −0.02 ± 0.03 | −0.12 ± 0.02 | −0.10 ± 0.04 |
| HA88 | 5,140 ± 240 | −0.33 ± 0.03 | −0.14 ± 0.02 | 0.19 ± 0.04 |
| VA89 | 4,370 ± 380 | −0.28 ± 0.05 | 0.24 ± 0.03 | 0.52 ± 0.06 |
| PA90† | 889 × 10† | −1.98 | −1.19 ± 0.04 | 0.79 |
| EA91 | 640 ± 50 | 0.34 ± 0.05 | 0.22 ± 0.03 | −0.12 ± 0.06 |
| PA92 | 180 ± 10 | 0.74 ± 0.04 | −0.07 ± 0.04 | −0.81 ± 0.06 |
| HA93 | 1,780 ± 90 | 0.01 ± 0.03 | 0.91 ± 0.03 | 0.89 ± 0.04 |
| EP91‡ | 0.1 | 2.98 | ||
| Δ8789‡ | 0.001 | 4.41 |
Kd is the dissociation constant for the monomer–dimer equilibrium, obtained as described in Methods. ΔΔGD-M is the change in stability of the monomer relative to the dimer on mutation, calculated by using Eq. 4. ΔΔGU-M is the change in the free energy of unfolding of the monomer on mutation, obtained from fluorescence-monitored urea denaturation experiments. ΔΔGU-D is the change in the free energy of unfolding of the dimer on mutation, calculated by combining ΔΔGD-M and ΔΔGU-M, by using Eq. 5.
Values quoted are per subunit.
The Kd for this mutant is very high, and therefore, because measurements could not be made at concentrations in the range of the Kd, it is an estimate only.
The Kds for these mutants are very low, and therefore, because measurements could not be made at concentrations in the range of the Kd, these are estimates only.
The dissociation constants of the mutants VA87 and HA93 are close to that of wild type, as expected, because these side chains occupy similar environments in monomer and dimer. In contrast, large changes in the dissociation constant are observed on mutation of the hinge residues, with PA90 and PA92 displaying the most dramatic effects. The mutation PA90 stabilizes the monomer by >1 kcal⋅mol−1 and destabilizes the dimer by 0.75 kcal⋅mol−1 per subunit. In contrast, PA92 has a negligible effect on the monomer stability and stabilizes the dimer by 0.8 kcal⋅mol−1 per subunit. Further, double mutant cycle analysis shows that the two prolines interact unfavorably in the monomer with a coupling energy of 0.4 kcal⋅mol−1 (23), whereas they interact favorably in the dimer with a coupling energy of −0.3 kcal⋅mol−1. These data demonstrate that proline 90 introduces strain in the hinge of the monomer, whereas it stabilizes the hinge in the dimer. In contrast, proline 92 has no net effect on the monomer hinge conformation, but it introduces strain in the hinge of the dimer. The double mutant cycle analysis shows there is a strain between the two prolines in the monomer, which is released in the dimer. Because the two proline side chains do not contact each other, the observed effect must be caused by backbone strain.
suc1 Aggregates by Domain Swapping.
Mutation of the other hinge residues results in less pronounced but still significant changes in the dissociation constant compared with wild type. Mutants HA88 and VA89 have higher dissociation constants, whereas that of EA91 is lower than wild type. Remarkably, we observe a qualitative correlation between the dissociation constants and the aggregation propensities: wild-type suc1 aggregates visibly when equilibrated at 50°C for more than a few minutes. Mutants HA88, VA89, and PA90, which domain swap less than wild type, have a reduced tendency to aggregate; mutants EA91 and PA92, which domain swap more than wild type, have an increased aggregation propensity (Fig. 3A). Thus, residues E91 and P92 specifically direct dimer formation in suc1 while preventing aggregation. Because domain swapping and aggregation are affected similarly by mutation, we propose they share a common mechanism; strand exchange can occur between two monomers to form a “closed-ended” dimer pair (3), or it can result in “open-ended” high-order oligomers (Fig. 3B).
Figure 3.

Relationship between domain swapping and aggregation in suc1. (A) Kinetics of aggregation of wild-type suc1 (crosses) and hinge mutants HA88 (closed triangles), VA89 (diamonds), PA90 (open triangles), EA91 (circles), and PA92 (squares). Mutation to alanine of the residues in the first half of the hinge (H88, V89, P90) slows down aggregation of suc1, whereas it is accelerated by mutation of the residues in the second half of the hinge (E91, P92). (B) Schematic showing how strand exchange can occur between two monomers to form a “closed-ended” dimer pair or between adjacent monomers in an “open-ended” high-order oligomer.
Redesigning suc1 Assembly by Manipulating the Strain.
Our results rationalize the frequent occurrence of prolines in the hinge loop of domain-swapping proteins (14). We show how the restrictions that prolines impose on local loop conformations favor the structure that minimizes loop strain while respecting the overall topology (Fig. 4). Therefore, the main consideration in the rational design of domain-swapping molecules is the energy balance between the two conformations of the hinge sequence: the introduction of strain in the hinge loop of the monomer favors oligomerization. We have applied this principle in two ways. The first is by shortening the hinge to make it more difficult to fold back on itself, as suggested in theory and shown by experiment previously (12, 24, 25). We made two variants in which three consecutive residues in the hinge were deleted. Deletion of residues V87, H88, and V89 (named Δ8789) resulted in 99% dimer formation at 20 μM protein concentration (Figs. 4 and 5), corresponding to an approximate Kd of 1 nM (Table 1). Deletion of residues P90, E91, and P92 (Δ9092) did not completely prevent monomer from being formed (data not shown). This result highlights the critical role of P90, because hinge shortening per se is not sufficient to drive the equilibrium toward dimer. The second design approach is the introduction of a third proline in the hinge, at position 91, to make the hinge more rigid. EP91 was ≈95% dimeric at 20 μM protein concentration (Figs. 4 and 5), corresponding to a Kd of 0.1 μM (Table 1).
Figure 4.

Scheme showing how proline residues exploit backbone strain. The hinge residues are represented as color-coded bricks. Prolines are shown in dark blue, and their preceding peptide bond is in bold to indicate restrictions imposed on it by the ring of the proline. Positions of backbone strain are shown by yellow zig-zag lines. There is an equilibrium in the wild-type protein between the monomer conformation in which P90 is strained and the dimer conformation in which P92 is strained. The equilibrium can be shifted by manipulating the strain in the following ways: (i) Mutation of P90 to alanine removes the strain from the monomer hinge conformation, thereby shifting the equilibrium almost completely toward the monomer. (ii) Deletion of three residues (Δ8789) results in a very short hinge that is severely strained by two prolines. It cannot easily fold back on itself in the monomer, and therefore the equilibrium is shifted completely toward dimer. (iii) Likewise, introducing a third proline into the hinge in the mutant EP91 results in increased strain in the monomer hinge conformation.
Figure 5.

Redesigning suc1 assembly. Elution profile of suc1 mutants shows that the mutant protein PA90 is >99% monomeric (2 mM protein concentration), whereas the mutant proteins EP91 is ≈95% dimeric (20 μM protein concentration), and Δ8789 is ≈99% dimeric (20 μM protein concentration).
Summary
Pro90 is energetically unfavorable in the monomer hinge loop conformation, whereas Pro92 is energetically unfavorable in the dimer hinge loop conformation. These two prolines therefore control the equilibrium between the two loop conformations, which in turn determines the equilibrium between monomeric and domain-swapped states of suc1, because both states have equivalent interactions outside the loop. The importance of prolines for domain swapping is indicated by their frequent occurrence in hinge loops. Our results rationalize this observation and suggest that the same mechanism may apply in other domain-swapped proteins. The strain imposed by the prolines is of greater magnitude in the monomer than the dimer, and we therefore propose that hinge loops act as “molecular springs” that release tension by adopting an alternative conformation, while respecting the overall structure of the protein.
We have used our results to rationally manipulate the strain in the hinge loop of suc1 and redesign its assembly state. Thus, domain swapping is a suitable strategy for evolving oligomers from monomers in a stepwise manner. We have shown that single point mutations are sufficient to tip the balance toward oligomers, and there is no need for preexistent complementary binding interfaces, although the subsequent accumulation of mutations could result in new interfaces that further stabilize the oligomeric form or enhance allosteric signal transduction.
Acknowledgments
This work was supported by the Medical Research Council (MRC) of the United Kingdom. L.S.I. was supported by a Beit Memorial Fellowship in Medical Research (U.K.) and a Career Development Award from the MRC of the U.K. F.R. and J.W.H.S. were supported by Marie Curie Training and Mobility of Research Fellowships from the European Community. S. M. V. Freund is gratefully acknowledged for acquiring the NMR spectra and C. M. Johnson for calorimetry measurements. We thank A. R. Fersht, J. Clarke, and M. Bycroft for reading the manuscript, and L. Serrano for helpful discussions and for suggestion of the mutation EP91.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
SaintJean, A. P. & Creighton, D. J. (1999) FASEB J. 13, A1560 (abstr.).
References
- 1.Bennett M J, Choe S, Eisenberg D. Protein Sci. 1994;3:1444–1463. doi: 10.1002/pro.5560030911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bennett M J, Choe S, Eisenberg D. Proc Natl Acad Sci USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlunegger M P, Bennett M J, Eisenberg D. Adv Protein Chem. 1997;50:61–122. doi: 10.1016/s0065-3233(08)60319-8. [DOI] [PubMed] [Google Scholar]
- 4.Crestfield A M, Stein W H, Moore S. Arch Biochem Biophys Suppl. 1962;1:217–222. [PubMed] [Google Scholar]
- 5.Fink A L. Folding Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 6.Siddhanta U, Presta A, Fan B C, Wolan D, Rousseau D L, Stuehr D J. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 7.Vitagliano L, Adinolfi S, Sica F, Merlino A, Zagari A, Mazzarella L. J Mol Biol. 1999;293:569–577. doi: 10.1006/jmbi.1999.3158. [DOI] [PubMed] [Google Scholar]
- 8.Nurizzo D, Cutruzzola F, Arese M, Bourgeois D, Brunori M, Cambillau C, Tegoni M. Biochemistry. 1998;37:13987–13996. doi: 10.1021/bi981348y. [DOI] [PubMed] [Google Scholar]
- 9.Mannervik B, Cameron A D, Fernandez E, Gustafsson A, Hansson L O, Jemth P, Jiang F Y, Jones T A, Larsson A K, Nilsson L O, et al. Chem Biol Interact. 1998;112:15–21. doi: 10.1016/s0009-2797(97)00147-6. [DOI] [PubMed] [Google Scholar]
- 10.Cameron A D, Olin B, Ridderstrom M, Mannervik B, Jones T A. EMBO J. 1997;16:3386–3395. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Didonato A, Cafaro V, Romeo I, Dalessio G. Protein Sci. 1995;4:1470–1477. doi: 10.1002/pro.5560040804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray A J, Head J G, Barker J J, Brady R L. Nat Struct Biol. 1998;5:778–782. doi: 10.1038/1816. [DOI] [PubMed] [Google Scholar]
- 13.Hayes M V, Sessions R B, Brady R L, Clarke A R. J Mol Biol. 1999;285:1857–1867. doi: 10.1006/jmbi.1998.2415. [DOI] [PubMed] [Google Scholar]
- 14.Bergdoll M, Remy M-H, Cagnon C, Masson J-M, Dumas P. Structure (London) 1997;5:391–401. doi: 10.1016/s0969-2126(97)00196-2. [DOI] [PubMed] [Google Scholar]
- 15.Rousseau F, Schymkowitz J W H, Sanchez del Pino M, Itzhaki L S. J Mol Biol. 1998;284:503–519. doi: 10.1006/jmbi.1998.2173. [DOI] [PubMed] [Google Scholar]
- 16.Schymkowitz J W H, Rousseau F, Irvine L R, Itzhaki L S. Struct Folding Des. 2000;8:89–100. doi: 10.1016/s0969-2126(00)00084-8. [DOI] [PubMed] [Google Scholar]
- 17.Gill C S, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 18.Clarke J, Fersht A R. Biochemistry. 1993;32:43322–4329. doi: 10.1021/bi00067a022. [DOI] [PubMed] [Google Scholar]
- 19.Oliveberg M, Tan Y J, Silow M, Fersht A R. J Mol Biol. 1998;277:933–943. doi: 10.1006/jmbi.1997.1612. [DOI] [PubMed] [Google Scholar]
- 20.Alonso D O V, Alm E, Daggett V. Struct Folding Des. 2000;8:101–110. doi: 10.1016/s0969-2126(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 21.Essig N Z, Wood J F, Howard A J, Raag R, Whitlow M. J Mol Biol. 1993;234:897–901. doi: 10.1006/jmbi.1993.1638. [DOI] [PubMed] [Google Scholar]
- 22.Kortt A A, Malby R L, Caldwell J B, Gruen L C, Ivancic N, Lawrence M C, Howlett G J, Webster R G, Hudson P J, Colman P M. Eur J Biochem. 1994;221:151–157. doi: 10.1111/j.1432-1033.1994.tb18724.x. [DOI] [PubMed] [Google Scholar]
- 23.Schymkowitz J W H, Rousseau F, Itzhaki L S. J Mol Biol. 2000;301:201–206. doi: 10.1006/jmbi.2000.3958. [DOI] [PubMed] [Google Scholar]
- 24.Green S M, Gittis A G, Meeker A K, Lattman E E. Nat Struct Biol. 1995;2:746–751. doi: 10.1038/nsb0995-746. [DOI] [PubMed] [Google Scholar]
- 25.Raag R, Whitlow M. FASEB J. 1995;9:73–80. doi: 10.1096/fasebj.9.1.7821762. [DOI] [PubMed] [Google Scholar]
- 26.Endicott J A, Noble M E, Garman E F, Brown N, Ramussen R, Nurse P, Johnson L N. EMBO J. 1995;14:1004–1014. doi: 10.1002/j.1460-2075.1995.tb07081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourne Y, Arvai A S, Bernstein S L, Watson M H, Reed S I, Endicott J A, Noble M E, Johnson L N, Tainer J A. Proc Natl Acad Sci USA. 1995;92:10232–10236. doi: 10.1073/pnas.92.22.10232. [DOI] [PMC free article] [PubMed] [Google Scholar]