

The crystal structures of the C-terminal domain of S. cerevisiae Avo1 and of its human orthologue Sin1 have been determined. The structures show that the C-termini of the two proteins have the PH-domain fold with a putative binding site for phosphoinositides.
Keywords: TOR signalling, TOR complex 2, Avo1, PH domains, Sin1
Abstract
In eukaryotes, multiprotein complexes termed TOR complex 1 (TORC1) and TOR complex 2 (TORC2) function as major regulators of cell growth, metabolism and ageing. The C-terminal domain of the Saccharomyces cerevisiae TORC2 component Avo1 is required for plasma-membrane localization of TORC2 and is essential for yeast viability. X-ray crystal structures of the C-terminal domain of Avo1 and of its human orthologue Sin1 have been determined. The structures show that the C-termini of Avo1 and Sin1 both have the pleckstrin homology (PH) domain fold. Comparison with known PH-domain structures suggests a putative binding site for phosphoinositides.
1. Introduction
Target of rapamycin (TOR) is a highly conserved serine/threonine protein kinase and a central controller of the growth, metabolism and ageing of eukaryotic cells in response to nutrients, growth factors and cellular energy status (Wullschleger et al., 2006 ▶; Zoncu et al., 2011 ▶). TOR assembles into two functionally and structurally distinct protein complexes termed TOR complex 1 (TORC1) and TOR complex 2 (TORC2) (Loewith et al., 2002 ▶). These two protein complexes function as central nodes in a complex network of signal transduction pathways that are involved in normal physiological as well as pathogenic events. TORC1 mediates the rapamycin-sensitive signalling branch, which positively regulates anabolic processes such as translation and ribosome biogenesis and negatively regulates catabolic processes such as autophagy. TORC2 signalling is rapamycin-insensitive and regulates the spatial aspects of cell growth by controlling the organization of the actin cytoskeleton and cell polarity. TORC2 is also involved in the regulation of ceramide metabolism in Saccharomyces cerevisiae (Aronova et al., 2008 ▶). The two TOR complexes possess both overlapping and distinct components. In S. cerevisiae, TORC1 consists of the proteins Kog1, Lst8, Tco89 and either Tor1 or Tor2, whereas TORC2 is composed of the proteins Avo1, Avo2, Avo3, Bit61, Lst8 and Tor2. Avo1, Avo3 and Lst8 are essential conserved proteins that are required for kinase activity of TORC2. In contrast, Avo2 and Bit61 are not essential and no clear homologues have been identified in higher eukaryotes.
The precise mechanism by which the two TOR complexes achieve their different functions remains poorly understood, but it is likely that the subcellular localizations of the two TOR complexes are important for the detection of input signals and the determination of downstream signalling specificity. A previous live-cell imaging study showed that TORC1 and TORC2 have different localizations in S. cerevisiae: TORC1 is localized exclusively to the vacuolar membrane, whereas TORC2 is localized to the plasma membrane in a peculiar punctate pattern (Berchtold & Walther, 2009 ▶). The plasma-membrane localization of TORC2 is essential for yeast viability and is mediated by lipid binding of the C-terminal domain of Avo1, whose amino-acid sequence has limited similarity to that of known pleckstrin homology (PH) domains (Berchtold & Walther, 2009 ▶). The C-terminal domain of Sin1, the human orthologue of Avo1, also binds to lipids and may target mammalian TORC2 to the plasma membrane (Schroder et al., 2007 ▶).
The association of proteins with the surfaces of cellular membranes, or with specific phospholipid components of these membranes, is mediated by a growing number of modular membrane-targeting domains, including PH, PKC C2, C1, PX, FYVE and GRAM domains, which recognize specific lipid molecules in the membranes (Cho & Stahelin, 2005 ▶; Lemmon, 2008 ▶). The PH domain is a small domain of around 120 amino acids that was originally identified in the platelet protein pleckstrin and is found in many other proteins with membrane-associated functions. The PH-domain structures solved to date have essentially the same fold (Lemmon & Ferguson, 2000 ▶). The core of the PH domain is a seven-stranded antiparallel β-sandwich derived from two nearly orthogonal β-sheets. The β-sandwich is closed at one end by the C-terminal amphipathic α-helix. At the other end of the β-sandwich lie three variable loops (VL1–3) which constitute the positively charged face of the PH domain and mediate binding to phosphoinositides in many (but not all) PH domains. The inositol headgroups of phosphoinositides are differentially phosphorylated and the affinities and specificities of PH domains for phosphoinositides are diverse because of diversity in the sequence and length of the variable loops. Some PH domains bind specific phosphoinositides with sufficiently high affinity to mediate their membrane localization, whereas others possess weak affinities and need to form a multimer or require cooperation with other factors in order to associate with membranes.
High-resolution structures of TOR complexes and their subunits are required to advance molecular understanding of the mechanism underlying TOR signalling and also to develop new drugs to treat human diseases, including obesity, diabetes and cancer. In this study, as part of a structural characterization of TOR signalling components, we have determined the X-ray crystal structures of the C-terminal domains of S. cerevisiae Avo1 and human Sin1. The structures show that the C-termini of these proteins have a common PH-domain-like structure with a putative binding site for phosphoinositides.
2. Materials and methods
2.1. Protein expression and purification
To construct a plasmid for expression of the C-terminal domain (residues 1056–1176) of Avo1p (hereafter referred to as Avo1C) with an N-terminal His and S tag in Escherichia coli cells, the cDNA of Avo1C was PCR-amplified from yeast genomic DNA (Promega) and cloned into the BamHI/XhoI sites of pET30a-TEV (Matsuura & Stewart, 2004 ▶). The construct was verified by DNA sequencing. His/S-Avo1C was expressed in E. coli strain BL21-CodonPlus (DE3) RIL (Stratagene) at 298 K in LB medium. After harvesting, the cells were suspended in buffer A (30 mM Tris–HCl pH 7.5, 0.5 M NaCl, 10 mM imidazole, 1 mM PMSF, 2 mM β-mercaptoethanol) and lysed by sonication on ice. All subsequent purification steps were performed at 277 K. The clarified lysate was loaded onto Ni–NTA resin (Novagen), washed with buffer A containing 25 mM imidazole and eluted with buffer A containing 250 mM imidazole. His-TEV protease (5 µg ml−1) was added to the eluate to cleave off the His/S tag. The eluate was dialyzed against buffer B (30 mM Tris–HCl pH 8.0, 0.5 M NaCl, 2 mM β-mercaptoethanol) overnight, concentrated using an Amicon Ultra 3K concentrator and finally purified by size-exclusion chromatography on Superdex 75 (GE Healthcare) in buffer C (20 mM Tris–HCl pH 8.0, 0.5 M NaCl, 2 mM β-mercaptoethanol). The purified Avo1C was concentrated to 5 mg ml−1 using an Amicon Ultra 3K concentrator and used for crystallization.
To construct a plasmid for expression of the C-terminal domain (residues 372–493) of human Sin1 (hereafter referred to as hSin1C) with an N-terminal His and S tag in E. coli cells, the cDNA encoding hSin1C was amplified by RT-PCR from HeLa total RNA and cloned into the NcoI/SalI sites of pET30a-TEV (Matsuura & Stewart, 2004 ▶). The construct was verified by DNA sequencing. His/S-hSin1C was expressed and purified as described for His/S-Avo1C.
2.2. Crystallization
Crystals of Avo1C belonging to an orthorhombic crystal form (space group P212121; form 1) were grown at 293 K from 5 mg ml−1 Avo1C by sitting-drop vapour diffusion against a reservoir solution consisting of 0.1 M Tris–HCl pH 8.7, 32% MPD. Rod-shaped crystals grew to maximum dimensions of 0.1 × 0.1 × 0.2 mm in two weeks. The crystals were mounted in nylon loops and flash-cooled directly (without additional cryoprotectants) in liquid nitrogen.
Crystals of Avo1C belonging to a trigonal crystal form (space group P3121; form 2) were grown at 293 K from 5 mg ml−1 Avo1C by sitting-drop vapour diffusion against a reservoir solution consisting of 0.1 M Tris–HCl pH 7.0, 6% PEG 8000. Rod-shaped crystals grew to maximum dimensions of 0.05 × 0.05 × 0.2 mm in five months. The crystals were serially transferred to 0.1 M Tris–HCl pH 7.0, 0.3 M NaCl, 6% PEG 8000, 20% MPD in three steps and were flash-cooled in liquid nitrogen.
Crystals of hSin1C were grown at 293 K from 18 mg ml−1 hSin1C by sitting-drop vapour diffusion against a reservoir solution consisting of 0.1 M bis-tris pH 6.5, 1.26 M ammonium sulfate. Oval-shaped crystals grew to maximum dimensions of 0.05 × 0.05 × 0.1 mm in one month. The crystals were serially transferred to 0.1 M bis-tris pH 6.5, 0.4 M NaCl, 1.3 M ammonium sulfate, 24% glycerol in three steps and were flash-cooled in liquid nitrogen.
2.3. Data collection and structure determination
A 1.90 Å resolution data set was collected from an Avo1C crystal (form 1) at 100 K on Photon Factory beamline BL-5A and was processed using MOSFLM and CCP4 programs (Winn et al., 2011 ▶). The crystal had P212121 symmetry (unit-cell parameters a = 40.78, b = 75.70, c = 92.23 Å) with two molecules of Avo1C in the asymmetric unit. The structure was solved by molecular replacement with MOLREP (Vagin & Teplyakov, 2010 ▶) using an NMR structure of the PH domain of ARAP2 (PDB entry 2cod; RIKEN Structural Genomics/Proteomics Initiative, unpublished work) as a search model. Iterative cycles of model building using Coot (Emsley & Cowtan, 2004 ▶) and refinement using REFMAC5 (Murshudov et al., 2011 ▶) yielded a final model with an R free of 0.2703 (R cryst = 0.2140) that contained Avo1C residues 1066–1100, 1108–1174 (chain A), 1068–1098, 1109–1174 (chain B) and 116 water molecules. A TLSMD analysis (Painter & Merritt, 2006 ▶) was used to define TLS groups for the final cycles of refinement.
A 2.80 Å resolution data set was collected from an Avo1C crystal (form 2) at 100 K on Photon Factory beamline BL-5A and was processed using MOSFLM and CCP4 programs. The diffraction was severely anisotropic and the diffraction along the c* axis was superior to that in the other directions. Therefore, the data set was subjected to anisotropy correction using Phaser (McCoy et al., 2007 ▶) before molecular replacement and refinement. The crystal had P3121 symmetry (unit-cell parameters a = b = 60.92, c = 84.10 Å) with one molecule of Avo1C in the asymmetric unit. The structure was solved by molecular replacement with MOLREP using a globular PH domain (chain A residues 1080–1174 and chain B residues 1068–1076) of the Avo1C dimer from the form 1 crystal structure as a search model. Iterative cycles of model building using Coot and jelly-body refinement (suitable for low-resolution refinement) using REFMAC5 yielded a final model with an R free of 0.2888 (R cryst = 0.2436) that consisted of Avo1C residues 1065–1100, 1108–1138 and 1142–1174 and two water molecules.
A 2.00 Å resolution data set was collected from an hSin1C crystal at 100 K on SPring-8 beamline BL41XU and was processed using MOSFLM and CCP4 programs. The crystal had P43212 symmetry (unit-cell parameters a = b = 79.29, c = 123.60 Å) with two molecules of hSin1C in the asymmetric unit. The structure was solved by molecular replacement with MOLREP using a globular PH domain (chain A residues 1080–1174 and chain B residues 1068–1076) of the Avo1C dimer from the form 1 crystal structure as a search model. Iterative cycles of model building using Coot and refinement using REFMAC5 yielded a final model with an R free of 0.2239 (R cryst = 0.1934) that contained hSin1C residues 372–414 and 421–490 [with an additional three residues (Gly-Ala-Met) at the N-terminus owing to cloning] and 125 water molecules. Data-collection and refinement statistics are given in Table 1 ▶. Molecular-graphics figures were generated with PyMOL (DeLano, 2002 ▶), MOLSCRIPT (Kraulis, 1991 ▶) and RASTER3D (Merritt & Bacon, 1997 ▶).
Table 1. Crystallographic statistics.
Values in parentheses are for the highest resolution shell.
| Crystal | Avo1C, form 1 | Avo1C, form 2 | hSin1C |
|---|---|---|---|
| Data collection | |||
| X-ray source | PF BL-5A | PF BL-5A | SPring-8 BL41XU |
| Wavelength (Å) | 1.0 | 1.0 | 1.0 |
| Space group | P212121 | P3121 | P43212 |
| Unit-cell parameters (Å, °) | a = 40.78, b = 75.70, c = 92.23, α = β = γ = 90 | a = b = 60.92, c = 84.10, α = β = 90, γ = 120 | a = b = 79.29, c = 123.60, α = β = γ = 90 |
| Resolution (Å) | 37.85–1.90 (2.00–1.90) | 32.88–2.80 (2.95–2.80) | 41.53–2.00 (2.11–2.00) |
| Rmerge† | 0.051 (0.611) | 0.051 (0.595) | 0.105 (0.911) |
| Mean I/σ(I) | 17.8 (2.7) | 27.5 (3.7) | 13.5 (2.4) |
| Completeness (%) | 97.1 (92.3) | 96.9 (84.0) | 100.0 (100.0) |
| Multiplicity | 6.0 (5.5) | 9.8 (8.2) | 8.6 (8.4) |
| No. of reflections | 134028 (16709) | 44614 (4549) | 236919 (32924) |
| No. of unique reflections | 22483 (3043) | 4563 (554) | 27409 (3907) |
| Refinement | |||
| Resolution (Å) | 37.85–1.90 | 32.88–2.80 | 41.53–2.00 |
| Rcryst/Rfree‡ | 0.2140/0.2703 | 0.2436/0.2888 | 0.1934/0.2239 |
| No. of atoms | |||
| Protein | 1644 | 806 | 1805 |
| Water | 116 | 2 | 125 |
| No. of amino acids | 199 | 100 | 229 |
| Mean B factor (Å2) | 45.5 | 57.7 | 34.6 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.020 | 0.014 | 0.028 |
| Bond angles (°) | 1.937 | 1.905 | 2.461 |
| Ramachandran plot (%) | |||
| Favoured region | 97.2 | 97.8 | 95.0 |
| Allowed region | 2.2 | 2.2 | 5.0 |
| Disallowed region | 0.6 | 0.0 | 0.0 |
| PDB code | 3ulb | 3ulc | 3voq |
R
merge = 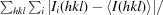
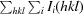 , where Ii(hkl) is the intensity measured for a given reflection and 〈I(hkl)〉 is the average intensity for multiple measurements of this reflection.
, where Ii(hkl) is the intensity measured for a given reflection and 〈I(hkl)〉 is the average intensity for multiple measurements of this reflection.
R
cryst = 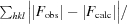
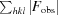 , where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively, for 95% of the reflection data used in the refinement. The free R factor was calculated using an equivalent equation to that used for R
cryst with 5% of the reflections that were omitted from the refinement.
, where |F
obs| and |F
calc| are the observed and calculated structure-factor amplitudes, respectively, for 95% of the reflection data used in the refinement. The free R factor was calculated using an equivalent equation to that used for R
cryst with 5% of the reflections that were omitted from the refinement.
2.4. Analytical size-exclusion chromatography
To analyze the oligomeric states of Avo1C and hSin1C in solution, we gel-filtered Avo1C and hSin1C in buffer D (20 mM HEPES pH 7.0, 0.5 M NaCl, 2 mM β-mercaptoethanol) with a Superdex 75 10/300 GL column (GE Healthcare) calibrated with globular proteins of known molecular weight. Each injection used 0.1 ml 0.11 mM protein and the elution from the column was monitored using ultraviolet absorbance at 280 nm.
3. Results and discussion
3.1. Crystal structure of the C-terminal domain of S. cerevisiae Avo1: a β-strand-swapped PH-domain homodimer
We obtained crystals of Avo1C (S. cerevisiae Avo1p residues 1056–1176) in two crystal forms. An orthorhombic P212121 crystal form (form 1) was obtained at pH 8.7 using MPD as a precipitant and a trigonal P3121 crystal form (form 2) was obtained at pH 7.0 using PEG 8000 as a precipitant. The form 1 crystals diffracted to 1.90 Å resolution and the structure was determined by molecular replacement (Fig. 1 ▶ a). The asymmetric unit of the form 1 crystal contained two molecules of Avo1C (chains A and B), which have essentially the same conformation (Fig. 1 ▶ d) and superpose with a Cα root-mean-square deviation of only 0.34 Å. The form 1 Avo1C structure exhibits a characteristic PH-domain fold with the notable exception that one of the seven β-strands that constitute the β-sandwich of the PH domain is donated by another molecule of Avo1C in the asymmetric unit to complete the PH-domain fold (Fig. 1 ▶ a). The core of one of the PH domains in the asymmetric unit is a β-sandwich formed by seven β-strands (the N-terminal third of β1/2 of chain B, the C-terminal third of β1/2 of chain A and β3–7 of chain A; see Figs. 1 ▶ b and 2 ▶ for the numbering of the secondary-structural elements). Similarly, the β-sandwich in the other PH domain in the asymmetric unit is formed by the N-terminal third of β1/2 of chain A, the C-terminal third of β1/2 of chain B and β3–7 of chain B. The long N-terminal β-strand (β1/2) of chain A and that of chain B form an antiparallel β-sheet along their entire length. Consequently, Avo1C forms a stable homodimer in which the two globular PH domains are connected by the long antiparallel N-terminal β-strands. The dimerization interface has a total buried surface area of 4100 Å2, which is much more extensive than the interaction interfaces of other proteins known to form stable interactions, which typically bury between 1200 and 2000 Å2 (Janin & Chothia, 1990 ▶). The central portion of the N-terminal β-strands is exposed to solvent but has a well defined electron density (Fig. 1 ▶ a) and serves as a spacer to separate the two globular PH domains by ∼30 Å. To our knowledge, the Avo1C structure reported here is the first structure of a domain-swapped PH-domain dimer.
Figure 1.

The X-ray crystal structures of Avo1C and hSin1C. (a) Avo1C crystal form 1. The two chains of Avo1C (chain A, cyan; chain B, green) in the asymmetric unit form a homodimer through swapping of the N-terminal β-strand. (b) A ribbon representation of Avo1C chain A in crystal form 1 with numbering of secondary-structure elements. (c) Avo1C crystal form 2. The asymmetric unit contains a single chain of Avo1C (chain A, magenta), which forms a β-strand-swapped dimer with another molecule (chain A′, grey) related by twofold crystallographic symmetry. In (a) and (c), two orthogonal views of the Avo1C homodimer rotated 90° around a horizontal axis are shown. 2mF o − DF c electron-density maps of the linker region contoured at 1.2σ are shown in the bottom panels. (d) A superposition of Avo1C crystal form 1 chain A (cyan) and chain B (green) and crystal form 2 chain A (magenta). (e) The two chains of hSin1C in the asymmetric unit. (f) A ribbon representation of hSin1C chain A with numbering of secondary-structure elements. (g) A superposition of chain A (orange) and chain B (yellow) of hSin1C. (h) A superposition of Avo1C (form 1; chain A, cyan; chain B, green) and hSin1C (chain A, orange).
Figure 2.

Amino-acid sequence alignment of the C-terminal PH domains of Avo1 orthologues. Hs, Homo sapiens; Xl, Xenopus laevis; Sp, Schizosaccharomyces pombe; Sc, Saccharomyces cerevisiae. Absolutely conserved residues are shaded in pink and homologous residues are shaded in yellow. The basic residues and hydrophilic residues which constitute a putative binding site for phosphoinositides are marked by blue and green triangles, respectively. Secondary-structure assignments of the crystal structures of hSin1C and Avo1C (form 1) are shown above and below the sequence alignment, respectively. Helices (α0, α1, α-helices; η1, 310-helix) are represented by cylinders and β-strands are shown as arrows.
The domain-swapping of Avo1C was also observed in another crystal form (form 2), which diffracted to 2.80 Å resolution. Although the form 2 crystal had only one molecule of Avo1C in the asymmetric unit, this molecule formed a β-strand-swapped dimer with another molecule related by twofold crystallographic symmetry (Fig. 1 ▶ c). Form 2 shares the same PH-domain fold with form 1 (Fig. 1 ▶ d). The only major conformational change between forms 1 and 2 was found in the long N-terminal linker strand between the two molecules of the dimer, which is melted in the central portion in form 2, giving rise to large differences between the two crystal forms in the relative orientation and distance between the two globular PH domains in the dimer (Figs. 1 ▶ a, 1 ▶ c and 1 ▶ d). It should be noted that even though the central portion of the linker deviates from the β-strand conformation in form 2, it had well defined electron density and therefore the chain tracing was unambiguous (Fig. 1 ▶ c). Thus, the central portion of the N-terminal strand β1/2 seems to function as a rather flexible linker between the two globular PH domains of the Avo1C dimer, enabling packing of the dimer in two different crystal forms.
3.2. The crystal structure of the C-terminal domain of human Sin1 has a monomeric PH-domain fold
We next determined the crystal structure of hSin1C (human Sin1 residues 372–493), the C-terminal domain of the human orthologue of Avo1, at 2.0 Å resolution. In contrast to the dimeric Avo1C crystal structures, domain-swapping was not observed in the hSin1C crystal structure (Fig. 1 ▶ e). The two molecules of hSin1C in the asymmetric unit are monomers which have the classical PH-domain fold composed of seven β-strands and a C-terminal α-helix, with an additional α-helix (α0) at the N-terminus (Figs. 1 ▶ e and 1 ▶ f). The two molecules of hSin1C in the asymmetric unit have essentially the same structure (Fig. 1 ▶ g), with the only major difference being found in the orientation of the N-terminal helix α0. Helix α0 protrudes out of the core PH domain and thus the difference in the α0 orientation is probably caused by different crystal-packing interactions. The core PH-domain structure of hSin1C superimposes well with that of Avo1C (Fig. 1 ▶ h), with a Cα root-mean-square deviation of 1.0 Å. The level of structural similarity between Avo1C and hSin1C is remarkable considering that their amino-acid sequence identity is only 23% (Fig. 2 ▶). Thus, apart from the fact that domain-swapping was observed in Avo1C but not in hSin1C, the crystal structures of the two proteins have the PH-domain structure in common.
3.3. The isolated C-terminal PH domain of S. cerevisiae Avo1 and human Sin1 is monomeric in solution
The observation that Avo1C, but not hSin1C, crystallizes as a β-strand-swapped dimer raises the question of whether the domain-swapping is species-specific or is an artifact of crystallization. We used analytical size-exclusion chromatography (SEC) to examine the oligomeric states of Avo1C and hSin1C in solution. As shown in Fig. 3 ▶, Avo1C and hSin1C migrated with apparent molecular masses of 13.9 and 14.7 kDa, respectively, when applied onto a Superdex 75 column and both proteins eluted as a single peak close to RNase A (13.7 kDa). The true molecular masses of Avo1C and hSin1C are 14.6 and 14.1 kDa, respectively, and therefore the SEC analysis suggests that both Avo1C and hSin1C are most likely to be monomeric in solution, at least under the assay conditions. Thus, while Avo1C crystallizes as a domain-swapped dimer, we do not observe dimerization in solution and therefore conclude that the dimeric arrangement of Avo1C is probably an artifact of the crystallization process. Based on the structural and biochemical data, we predict that the solution structure of Avo1C is probably monomeric, as observed in the crystal structure of hSin1C. It is conceivable that the high concentration (32%) of MPD which was used to crystallize Avo1C in crystal form 1 was sufficiently high to partially denature Avo1C, yielding crystals of the domain-swapped dimer. The form 2 crystals of Avo1C took a long time (five months) to develop and this may be a consequence of a requirement for instability of the protein in order to adopt this crystal packing.
Figure 3.

Size-exclusion chromatography (SEC) of Avo1C and hSin1C. (a) Overlay of the elution profiles. (b) SEC analysis. The apparent molecular mass was determined from a calibration curve established using globular proteins of known molecular mass. Avo1C and hSin1C eluted with apparent molecular masses of 13.9 and 14.7 kDa, respectively, consistent with both proteins existing as monomers in solution.
It is not uncommon that a protein behaves as a monomer in solution but crystallizes as a domain-swapped dimer, and similar observations of domain-swapping as an artifact of crystallization have been reported for the N-terminal CAP-Gly domain of the dynactin large subunit p150glued (Honnappa et al., 2006 ▶) and the Src homology 2 (SH2) domain of interleukin-2 tyrosine kinase (Joseph et al., 2012 ▶).
However, we also note that the structural and biochemical analyses presented here were made using the isolated PH domains of Avo1 and Sin1, and the oligomeric state of these domains in TORC2 still remains to be elucidated in vivo. Previous gel-filtration experiments showed that TORC2 is oligomeric in solution and is likely to exist as a TORC2–TORC2 dimer (Wullschleger et al., 2005 ▶). A live-cell imaging study suggested that TORC2 is oligomeric on the surface of the plasma membrane in yeast (Berchtold & Walther, 2009 ▶). We cannot exclude the possibility that interactions with the other subunits of TORC2 may result in the formation of a domain-swapped dimer of the C-terminal PH domain of Avo1, as observed in the crystal structures of Avo1C, stabilizing the plasma-membrane binding of TORC2 owing to dimerization. An increase in the avidity of membrane binding caused by the oligomerization of PH domains has been observed for dynamins (Klein et al., 1998 ▶) and could be a common strategy employed by proteins with membrane-associated functions to regulate membrane targeting.
3.4. Comparison with related PH-domain structures suggests a binding site for phosphoinositides
PH domains most often bind phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2], a plasma-membrane-specific lipid in yeast (Lemmon, 2008 ▶), and a previous liposome-flotation assay showed that Avo1C binds to PtdIns(4,5)P2 and, to a much lesser extent, to PtdIns(3,4)P2 (Berchtold & Walther, 2009 ▶). Analysis of the electrostatic surface potential shows that Avo1C possesses a conspicuous basic pocket on its surface (Fig. 4 ▶ a) which is formed in much the same way as the variable loops (VL1–3) form a basic pocket that constitutes the binding site for phosphoinositides in many other PH domains of membrane-associated proteins, including protein kinase B (PKB; Thomas et al., 2002 ▶; Fig. 4 ▶ e), phospholipase C (Ferguson et al., 1995 ▶), Bruton’s tyrosine kinase (BTK; Baraldi et al., 1999 ▶) and general receptor for phosphoinositides-1 (GRP1; Ferguson et al., 2000 ▶). The spatial arrangement of the residues composing the basic pocket of Avo1C is similar to the phosphoinositide-binding site of the PH domain of PKB (Figs. 5 ▶ a and 5 ▶ c). The basic pocket of Avo1C is an area of clustered basic residues (Arg1073, His1083, Arg1085, Lys1110, Lys1112 and Lys1145) and a polar residue (Tyr1147). All of these residues belong to the same chain except for Arg1073, which belongs to strand β1/2 of the other chain. The Avo1C residues Arg1073, Arg1085 and Tyr1147 are equivalent to Lys14, Arg25 and Arg86 of PKB, respectively (Figs. 5 ▶ a and 5 ▶ c). These three basic residues of PKB are key residues that form ionic interactions with the D3 and D4 phosphate groups, and mutagenesis of these residues affects the affinity of PKB for PtdIns(3,4,5)P3 and PtdIns(3,4)P2 (Thomas et al., 2002 ▶). Therefore, the corresponding residues of Avo1 could be important for the localization of Avo1 and TORC2 to the plasma membrane via phosphoinositide binding. Thus, the electrostatic surface potential and the similarity to other PH domains suggest that the basic pocket of Avo1C is likely to be a binding site for phosphoinositides.
Figure 4.

Comparison of the electrostatic surfaces of the PH domains of Avo1C (crystal form 1), hSin1C and PKB (PDB entry 1h10; Thomas et al., 2002 ▶). (a) Electrostatic surface of the Avo1C homodimer (crystal form 1) with the molecule in the same orientation as in the uppermost panel of Fig. 1 ▶(a). (b) A ribbon diagram of hSin1C shown in the same orientation as chain A of Avo1C in (a). (c) Electrostatic surface of hSin1C with the molecule in the same orientation as in (b). (d) A ribbon diagram of the PH domain of PKB in the same orientation as in (b). Ins(1,3,4,5)P4 bound to the pocket formed by VL1–3 is shown as a stick model. (e) Electrostatic surface of the PH domain of PKB with the molecule in the same orientation as in (d). In (a), (c) and (e) protein surfaces are coloured by the electrostatic potential of the solvent-accessible surface. Positively charged regions are coloured blue, negatively charged regions red and neutral regions white. The colour ramp is from −3kT to +3kT, where k is the Boltzmann constant and T is the absolute temperature. The surface potential was calculated with APBS (Baker et al., 2001 ▶) assuming a solvent of 150 mM NaCl. PQR files used for calculation were converted from PDB files using PDB2PQR (Dolinsky et al., 2004 ▶). In (a) and (c) the region marked by a dashed circle is the basic pocket that may be a binding site for phosphoinositides.
Figure 5.

Comparison of the residues of the basic pockets of (a) Avo1C crystal form 1 (chain A, cyan; chain B, green), (b) hSin1C (chain A, orange) and (c) the PH domain of PKB (PDB entry 1h10; wheat colour) complexed with Ins(1,3,4,5)P4. Hydrogen bonds are shown as black dashed lines.
Similarly, hSin1C has a basic pocket, as highlighted by a red dashed circle in Fig. 4 ▶(c), raising the possibility that this pocket of hSin1C is also a binding site for phosphoinositides. However, close examination of the basic pocket reveals that the residues comprising the basic surface of this pocket are not highly conserved between Avo1 and Sin1 (the only conserved residues are Lys428 in Sin1 (Lys1110 in Avo1) and Lys464 in Sin1 (Lys1145 in Avo1) (Fig. 2 ▶) and the spatial arrangement of the basic residues in hSin1C is quite different from that in Avo1C and PKB (Fig. 5 ▶). At this level of structural similarity, it is impossible to predict how Sin1 binds phosphoinositides. Thus, although Avo1C and hSin1C share the PH-domain fold and previous biochemical studies have shown that both proteins bind phosphoinositides (Schroder et al., 2007; Berchtold & Walther, 2009), the precise mechanism of the recognition of phosphoinositides may be highly variable.
Supplementary Material
PDB reference: Avo1 PH domain, crystal form 1, 3ulb
PDB reference: Avo1 PH domain, crystal form 2, 3ulc
PDB reference: Sin1 PH domain, 3voq
Acknowledgments
We thank our colleagues in Nagoya, especially Hidemi Hirano, Masako Koyama, Natsumi Saito, Junya Kobayashi and Tatsuo Hikage, for assistance and discussion. We are also indebted to the staff of Photon Factory and SPring-8 for assistance during data collection.
References
- Aronova, S., Wedaman, K., Aronov, P. A., Fontes, K., Ramos, K., Hammock, B. D. & Powers, T. (2008). Cell Metab. 7, 148–158. [DOI] [PMC free article] [PubMed]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed]
- Baraldi, E., Djinovic Carugo, K., Hyvönen, M., Surdo, P. L., Riley, A. M., Potter, B. V., O’Brien, R., Ladbury, J. E. & Saraste, M. (1999). Structure, 7, 449–460. [DOI] [PubMed]
- Berchtold, D. & Walther, T. C. (2009). Mol. Biol. Cell, 20, 1565–1575. [DOI] [PMC free article] [PubMed]
- Cho, W. & Stahelin, R. V. (2005). Annu. Rev. Biophys. Biomol. Struct. 34, 119–151. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Dolinsky, T. J., Nielsen, J. E., McCammon, J. A. & Baker, N. A. (2004). Nucleic Acids Res. 32, W665–W667. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Ferguson, K. M., Kavran, J. M., Sankaran, V. G., Fournier, E., Isakoff, S. J., Skolnik, E. Y. & Lemmon, M. A. (2000). Mol. Cell, 6, 373–384. [DOI] [PubMed]
- Ferguson, K. M., Lemmon, M. A., Schlessinger, J. & Sigler, P. B. (1995). Cell, 83, 1037–1046. [DOI] [PubMed]
- Honnappa, S., Okhrimenko, O., Jaussi, R., Jawhari, H., Jelesarov, I., Winkler, F. K. & Steinmetz, M. O. (2006). Mol. Cell, 23, 663–671. [DOI] [PubMed]
- Janin, J. & Chothia, C. (1990). J. Biol. Chem. 265, 16027–16030. [PubMed]
- Joseph, R. E., Ginder, N. D., Hoy, J. A., Nix, J. C., Fulton, D. B., Honzatko, R. B. & Andreotti, A. H. (2012). Acta Cryst. F68, 145–153. [DOI] [PMC free article] [PubMed]
- Klein, D. E., Lee, A., Frank, D. W., Marks, M. S. & Lemmon, M. A. (1998). J. Biol. Chem. 273, 27725–27733. [DOI] [PubMed]
- Kraulis, P. J. (1991). J. Appl. Cryst. 24, 946–950.
- Lemmon, M. A. (2008). Nature Rev. Mol. Cell Biol. 9, 99–111. [DOI] [PubMed]
- Lemmon, M. A. & Ferguson, K. M. (2000). Biochem. J. 350, 1–18. [PMC free article] [PubMed]
- Loewith, R., Jacinto, E., Wullschleger, S., Lorberg, A., Crespo, J. L., Bonenfant, D., Oppliger, W., Jenoe, P. & Hall, M. N. (2002). Mol. Cell, 10, 457–468. [DOI] [PubMed]
- Matsuura, Y. & Stewart, M. (2004). Nature (London), 432, 872–877. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Merritt, E. A. & Bacon, D. J. (1997). Methods Enzymol. 277, 505–524. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Painter, J. & Merritt, E. A. (2006). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Schroder, W. A., Buck, M., Cloonan, N., Hancock, J. F., Suhrbier, A., Sculley, T. & Bushell, G. (2007). Cell. Signal. 19, 1279–1289. [DOI] [PubMed]
- Thomas, C. C., Deak, M., Alessi, D. R. & van Aalten, D. M. (2002). Curr. Biol. 12, 1256–1262. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wullschleger, S., Loewith, R. & Hall, M. N. (2006). Cell, 124, 471–484. [DOI] [PubMed]
- Wullschleger, S., Loewith, R., Oppliger, W. & Hall, M. N. (2005). J. Biol. Chem. 280, 30697–30704. [DOI] [PubMed]
- Zoncu, R., Efeyan, A. & Sabatini, D. M. (2011). Nature Rev. Mol. Cell Biol. 12, 21–35. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: Avo1 PH domain, crystal form 1, 3ulb
PDB reference: Avo1 PH domain, crystal form 2, 3ulc
PDB reference: Sin1 PH domain, 3voq