

The structure of the nonsteroidal anti-inflammatory drug analogue 3-phenoxybenzoic acid bound to aldo–keto reductase 1C3 (AKR1C3) is presented as a lead for the discovery and development of novel agents for inhibiting AKR1C3, an anticancer target.
Keywords: nonsteroidal anti-inflammatory drugs, NSAIDs, aldo–keto reductase 1C3, AKR1C3, drug discovery
Abstract
Aldo–keto reductase 1C3 (AKR1C3) is a human enzyme that catalyzes the NADPH-dependent reduction of steroids and prostaglandins. AKR1C3 overexpression is associated with the proliferation of hormone-dependent cancers, most notably breast and prostate cancers. Nonsteroidal anti-inflammatory drugs (NSAIDs) and their analogues are well characterized inhibitors of AKR1C3. Here, the X-ray crystal structure of 3-phenoxybenzoic acid in complex with AKR1C3 is presented. This structure provides useful information for the future development of new anticancer agents by structure-guided drug design.
1. Introduction
Aldo–keto reductase 1C3 (AKR1C3) is a human enzyme belonging to the aldo–keto reductase superfamily which catalyzes the NADPH-dependent reduction of carbonyl moieties on a number of steroids and prostaglandins (Suzuki-Yamamoto et al., 1999 ▶), two examples of which are illustrated in Fig. 1 ▶. In most cases, the AKR1C3-dependent reduction of a ligand generates proliferative metabolites which exhibit greater potency and receptor-binding affinity than their precursors (reviewed in Byrns et al., 2011 ▶, and outlined briefly here). For example, AKR1C3 catalyzes the reduction of prostaglandins PGH2 and PGD2 to PGF2α and 9α,11β-PGF2, respectively; these products display increased F-prostanoid receptor affinity and enhanced proliferative activity. In the absence of AKR1C3 activity, PGD2 spontaneously dehydrates and isomerizes, forming anti-proliferative metabolites such as 15-deoxy-Δ12,14-PGJ2. Additionally, AKR1C3 catalyzes the reduction of Δ4-androstene-3,17-dione to testosterone, increasing androgen receptor affinity, of oestrone to 17β-oestradiol, increasing oestrogen receptor affinity, and of progesterone to 20α-hydroxyprogesterone, decreasing progesterone receptor affinity; consequently, hormone signalling is increased in some cells. Not surprisingly, AKR1C3 overexpression has been exhibited as a possible causal factor in the proliferation of many hormone-dependent cancers, most notably breast, endometrial and prostate cancers, and the extent of AKR1C3 overexpression appears to be positively correlated with both disease aggressiveness and poor patient prognosis (Guise et al., 2010 ▶; Hofland et al., 2010 ▶; Jansson et al., 2006 ▶; Lin et al., 2004 ▶; Nakamura et al., 2005 ▶; Stanbrough et al., 2006 ▶). These studies suggest that the discovery of drugs that target AKR1C3 may hold promise in the treatment of cancer.
Figure 1.

Examples of the reactions catalyzed by AKR1C3 and a schematic of the 3-phenoxybenzoic acid molecule. Testosterone is formed from Δ4-androstenedione and 9α,11β-PGF2 from PGD2 by AKR1C3-catalyzed reduction of the 17-carbonyl and 9-carbonyl groups, respectively. The 3-phenoxybenzoic acid molecule is an inhibitor of AKR1C3. This figure was drawn using ChemBioDraw Ultra 12.0 (CambridgeSoft)
Currently, the PDB contains ten different X-ray crystal structures of ternary complexes of AKR1C3 with ligands such as its cofactor NADP+, PGD2, 4-androsterone-3,17-dione, solvent molecules and inhibitors (Komoto et al., 2004 ▶, 2006 ▶; Lovering et al., 2004 ▶; Qiu et al., 2004 ▶, 2007 ▶). The AKR1C3 polypeptide folds to form an α8β8-barrel structure with an active-site pocket containing an oxyanion site formed by residues Tyr55 and His117, which in conjunction with the adjacent NADP+ molecule forms the catalytic centre targeted by known inhibitors of the enzyme.
Nonsteroidal anti-inflammatory drugs (NSAIDs) are some of the most potent inhibitors of AKR1C3 known, with two representative ternary structures currently deposited in the PDB containing either indomethacin or the N-phenylanthranilic acid flufenamic acid (Lovering et al., 2004 ▶). Indomethacin displays a marked selectivity for AKR1C3 compared with other AKR1C isoforms, while flufenamic acid is a potent nonselective inhibitor of the AKR1C1, AKR1C2 and AKR1C3 isoforms (Byrns et al., 2008 ▶; Lovering et al., 2004 ▶). Here, we report the X-ray crystal structure of an N-phenylanthranilic acid analogue, 3-phenoxybenzoic acid, bound to AKR1C3 with NADP for use in future structure-guided development of anticancer agents targeting AKR1C3.
2. Materials and methods
2.1. Expression and purification of AKR1C3
The AKR1C3 DNA sequence was purchased from GenScript Inc. and subcloned in a pET21b vector (Merck) utilizing NdeI and XhoI cloning sites. C-terminally His-tagged protein was obtained by leaky expression (not requiring induction) in Escherichia coli strain BL21 (DE3) using Terrific Broth containing 100 mg l−1 ampicillin; overnight cultures of 5 ml volume were transferred into 500 ml TB medium and incubated at 310 K with shaking at 160 rev min−1 for between 16 and 18 h. Bacterial cultures were pelleted by centrifugation (5000g, 30 min, 277 K) and disrupted by sonication in buffer A [40 mM Tris–HCl pH 7.5, 20% glycerol, 0.8% octyl β-d-glucopyranoside, 1 mM NADP+ and one whole EDTA-free protease-inhibitor tablet (Roche) per 50 ml volume]. The lysate was initially purified by centrifugation at 16 000 rev min−1 for a period of 25 min; the supernatant was applied onto an immobilized nickel-affinity chromatography column pre-equilibrated with buffer B (20 mM Tris–HCl pH 7.5, 10% glycerol, 150 mM NaCl) and eluted using a linear imidazole gradient (0–0.5 M). The fractions obtained by elution were immediately diluted 1:4 with buffer C (20 mM Tris–HCl pH 7.5, 10% glycerol, 0.5 mM EDTA, 1 mM DTT). The purified His-tagged protein was applied onto a Blue Sepharose affinity column pre-equilibrated with buffer C. Elution of the protein required a linear NaCl gradient (0–2 M). Following overnight storage at 277 K, the protein was buffer-exchanged extensively into buffer D (10 mM potassium phosphate buffer pH 7.0, 1 mM EDTA, 1 mM DTT, 0.005% decyl maltoside, 1.2 mM NADP+) using a 30 kDa molecular-weight cutoff spin column (Vivascience) and then concentrated to 25 mg ml−1.
2.2. Crystallization and data collection
Crystals were grown by the hanging-drop vapour-diffusion method from a solution consisting of 12.5 mg ml−1 protein, 5 mM potassium phosphate buffer pH 7.0, 0.5 mM EDTA, 0.5 mM DTT, 0.0025% decyl maltoside, 0.6 mM NADP+, 0.006% octyl β-d-glucopyranoside, 100 mM sodium acetate, 10% PEG 3350. The drop volumes were initially 2 µl, comprising 1 µl protein solution and 1 µl crystallization solution (200 mM sodium acetate, 20% PEG 3350), and were allowed to concentrate over a 0.5 ml volume of reservoir solution. Within 5 d, crystals had grown to maximum dimensions of 0.1 × 0.1 × 0.4 mm. A soaking solution was formed by dissolving the inhibitor 3-phenoxybenzoic acid (Sigma–Aldrich) in DMSO at 50 mM and adding 0.2 µl to 1.8 µl reservoir solution. Crystals were soaked for 3 d and were then briefly dipped into a cryoprotectant solution (200 mM sodium acetate, 20% PEG 3350, 20% ethylene glycol) before being flash-cooled in liquid nitrogen. Data were collected on a Rigaku MicroMax-007 HR rotating-anode instrument equipped with a marresearch mar345dtb detector and Oxford Cryosystem Cobra cooling system. Data-collection statistics are summarized in Table 1 ▶.
Table 1. Data-collection and refinement parameters.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Wavelength | 1.5418 [Cu Kα] |
| Space group | P212121 |
| Unit-cell parameters (Å) | a = 58.47, b = 64.70, c = 96.96 |
| Resolution (Å) | 53.82–1.68 (1.77–1.68) |
| Unique reflections | 42862 (6049) |
| Multiplicity | 21.8 (18.2) |
| Completeness (%) | 99.6 (97.6) |
| 〈I/σ(I)〉 | 39.4 (4.7) |
| Rmeas† | 0.062 (0.756) |
| Rp.i.m.‡ | 0.013 (0.174) |
| Wilson B (Å2) | 21.2 |
| Refinement | |
| Resolution (Å) | 53.82–1.68 |
| Rwork§ | 0.184 |
| Rfree§ | 0.214 |
| No. of working reflections | 40602 |
| No. of free reflections (free) | 2153 |
| Average B factors (Å2) | |
| Protein | 21.31 |
| Ligands | 15.5 |
| Waters | 27.2 |
| R.m.s.d. bonds (Å) | 0.028 |
| R.m.s.d. angles (°) | 2.230 |
| Ramachandran most favoured (%) | 96.8 |
| MolProbity rank | 97th percentile |
R
meas = 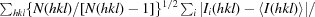
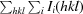 , where I
i(hkl) and 〈I(hkl)〉 are the observed and average intensities, respectively, and N is the multiplicity of reflection hkl.
, where I
i(hkl) and 〈I(hkl)〉 are the observed and average intensities, respectively, and N is the multiplicity of reflection hkl.
R
p.i.m. = 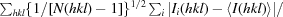 .
.
R
work = 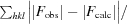
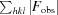 . R
work is calculated using 95% of the data selected randomly and used in refinement. R
free is calculated from the remaining 5% of the data not used in refinement.
. R
work is calculated using 95% of the data selected randomly and used in refinement. R
free is calculated from the remaining 5% of the data not used in refinement.
2.3. Structure determination, refinement and model quality
Data processing and scaling were performed using XDS (Kabsch, 2010 ▶) and SCALA (Evans, 2006 ▶). The structure of the AKR1C3–3-phenoxybenzoic acid complex was solved by molecular replacement using Phaser (McCoy et al., 2007 ▶) and the acetate/PEG AKR1C3 structure (PDB entry 2fgb; Qiu et al., 2007 ▶) with all nonprotein atoms removed. An NADP+ molecule was obtained from PDB entry 2fgb and was placed within well defined electron density visualized using Coot (Emsley et al., 2010 ▶). Atomic coordinates for 3-phenoxybenzoic acid were constructed and minimized using the PRODRG2 server (Schüttelkopf & van Aalten, 2004 ▶), and fitted into clearly defined and unambiguous electron density within the protein active site. The ternary model was refined by iterative steps of manual model building using Coot and maximum-likelihood refinement using REFMAC5 (Murshudov et al., 2011 ▶). Finally, the solvent structure was modelled and refined; difference density peaks greater than 1.8σ in F o − F c maps were identified as water molecules and included in the refinement. Two ethylene glycol molecules were identified adjacent to the inhibitor and were also included in the refinement. The resulting structure was ultimately refined using REFMAC TLS (translation–libration–screw) and maximum-likelihood refinement and the structure quality was evaluated using the MolProbity server (Chen et al., 2010 ▶). Some of the electron density was not interpretable and therefore residues 125–127 were left out of the final model. Final refinement statistics are given in Table 1 ▶.
3. Results and discussion
3.1. The three-dimensional structure and active site of AKR1C3
We have determined the X-ray crystal structure of AKR1C3 with the cofactor NADP+ and the drug-like inhibitor 3-phenoxybenzoic acid bound at a resolution of 1.68 Å in space group P212121; the model quality and stereochemistry were assessed by the MolProbity server at being in the 97th percentile (Table 1 ▶). The structure and data have been deposited in the PDB with code 3uwe. Following our standard protocol for low-affinity compounds, we soaked a high concentration of inhibitor into preformed AKR1C3 crystals for 3 d prior to data collection. This procedure displaces the octyl β-d-glucopyranoside and acetate molecules that would otherwise occupy the active site. The high affinity of 3-phenoxybenzoic acid for the protein might suggest the use of lower concentrations or shorter soak periods, but this was not investigated.
The asymmetric unit contains a single molecule of AKR1C3, one NADP+ molecule, one 3-phenoxybenzoic acid molecule, four ethylene glycol molecules and 160 water molecules. The AKR1C3 protein forms the archetypal and well characterized α8β8-barrel structure of this protein family, consisting of a cylindrical core of β-strands surrounded by α-helices (Fig. 2 ▶ a). The barrel structure is capped at the N-terminus by an antiparallel hairpin loop formed by two β-strands that are independent of the α8β8 barrel. In contrast, the C-terminal end of the barrel is organized into three flexible loops whose amino-acid side chains partially form the active site. Superposition of existing structures indicates that the flexibility of these loops in conjunction with the torsional capacity of individual amino-acid side chains allows changes in the active-site conformation that accommodate a large range of different ligands (including inhibitors) with differential affinity.
Figure 2.

The overall structure and active site of AKR1C3. (a) A ribbon diagram depicting the structure of AKR1C3 in complex with NADP+ and 3-phenoxybenzoic acid. The NADP+ molecule is shown as a multicoloured stick model with C atoms coloured white. The 3-phenoxybenzoic acid molecule is shown as a ball-and-stick model with C atoms coloured yellow. (b) Active-site structure showing electron density from a 2F o − F c OMIT map contoured at the 1.0σ level in Coot. The 3-phenoxybenzoic acid molecule is shown as a ball-and-stick model with the C atoms coloured yellow. The two ethylene glycol molecules found immediately adjacent to the inhibitor molecule are shown as green and red stick models and labelled EDO; water molecules are depicted as small red spheres. Hydrogen bonds are shown as dashed lines. These figures were generated using Incentive PyMOL v.1.3 (Schrödinger LLC).
3.2. AKR1C3 inhibition by 3-phenoxybenzoic acid
The AKR1C3 active site is large, with multiple ligand-binding cavities that have been annotated as the oxyanion site, steroid channel and subpockets SP1, SP2 and SP3 (Byrns et al., 2011 ▶). All of the previously characterized inhibitors share the common characteristic of an occupied SP1 site, which is lined by residues Phe306, Phe311, Tyr319, Ser118 and Asn167, and the current inhibitor is no exception. The 3-phenoxybenzoic acid molecule was fitted into clearly defined and unambiguous electron density within the active site, occupying both the oxyanion and SP1 sites (Fig. 2 ▶ b). The inhibitor carboxylic acid binds to the oxyanion site, in which the carboxylate group very closely overlays the acetate molecule found in other AKR1C3 structures (e.g. PDB entries 1s1p and 2fgb; Lovering et al., 2004 ▶; Qiu et al., 2007 ▶) and forms hydrogen bonds to the enzyme catalytic residues His117 and Tyr55, as well as to a conserved water network found in nearly all AKR1C3 crystal structures and located in and near the SP3 subpocket. The 3-phenoxy ring extends into the SP1 subpocket (Fig. 3 ▶) and makes van der Waals contacts with the aromatic residues Phe306, Phe311 and Tyr319 that line the pocket. In binding the two sites and forming appropriate nonbonded and hydrogen-bond contacts, the 3-phenoxybenzoic acid molecule displays a torsion angle of 28° about the C3—O3 bond and a 53° rotation about the O3—C7′ bond. In contrast, when compared with PDB entry 1s1p the protein active site undergoes only minor changes on binding, with the Cγ atoms of residues Phe311 and Phe317 displaced 0.5 Å and the side chain rotated 25° about the Cβ—Cγ bond, while the Trp227 Cβ atom is displaced 0.71 Å and the side chain rotated 15° about the Cβ—Cγ bond.
Figure 3.

Binding of 3-phenoxybenzoic acid and flufenamic acid in the AKR1C3 active site. The ligand molecules are shown as ball-and-stick models with the 3-phenoxybenzoic acid structure coloured black and the flufenamic acid structure coloured white. This figure was generated using Incentive PyMOL v.1.3 (Schrödinger LLC).
3-Phenoxybenzoic acid is related to a well characterized class of AKR1C3 inhibitors collectively known as N-phenylanthranilic acids, an example of which is flufenamic acid, the main difference being the use of an oxygen rather than an amino inter-ring linker. Also, the phenoxy/phenyl substituent is located at carbon 3 of the benzoic acid ring of 3-phenoxybenzoic acid (Fig. 1 ▶) and at carbon 2 in the case of the N-phenylanthranilic acids. Comparing the current structure with that containing flufenamic acid (PDB entry 1s2c; Lovering et al., 2004 ▶), we find many similarities. The benzoic acid rings bind very similarly in the oxyanion site and both the phenoxy and phenyl substituents bind in the SP1 pocket. Owing to their different substitution positions on the benzoic acid (at positions 3 and 2, respectively) the rings do not superimpose, but cross each other at an angle of approximately 40° (Fig. 3 ▶). The flufenamic acid molecule also contains a CF3 ring substituent that also allows it to extend further into the SP1 pocket than 3-phenoxybenzoic acid. The binding similarities are reflected in the similar IC50 values towards AKR1C3 of 0.68 and 1.7 µM for 3-phenoxybenzoic acid and flufenamic acid, respectively (Byrns & Penning, 2009 ▶; Gobec et al., 2005 ▶).
An additional class of compounds closely related to 3-phenoxybenzoic acid are the analogues of 3-(phenylamino)benzoic acid described by Adeniji et al. (2011 ▶). Like 3-phenoxybenzoic acid, the molecules characterized contain a phenyl subtituent in the 3-position (or meta position) but with an amide linker in common with flufenamic acid. The benzoic acid meta substitution alone, but particularly in conjuction with para substituents on the phenyl ring, produces submicromolar inhibitors of AKR1C3 with up to 360-fold selectivity (based on IC50 values) measured for the 1C3 isoform over 1C2 for a p-COCH3 substituent. While we have no comparable selectivity data for our meta-substituted molecule 3-phenoxybenzoic acid, we would predict a similar selectivity profile to the simple 3-(phenylamino)benzoic acid molecule (ratio of IC50 values for AKR1C2/AKR1C3 of 14). Additionally, our current crystal structure appears to be predictive for the binding mode of the 3-(phenylamino)benzoic acid class of compounds; allowing for minor inter-ring torsional adjustments, the para-phenyl substituents would bind deeply into the SP1 pocket without steric clash. The AKR1C2 SP1 pocket is smaller and more restrictive and would require much greater rearrangement of the inhibitor and/or protein active site.
Supplementary Material
PDB reference: AKR1C3 complexed with 3-phenoxybenzoic acid, 3uwe
Acknowledgments
The authors acknowledge funding from the New Zealand Lottery Health Fund (CJS), the Auckland Medical Research Foundation (CJS and JUF) and the Maurice Wilkins Centre for Molecular Biodiscovery (JUF and CJS), and the Faculty of Science, University of Auckland for providing a Summer Studentship for VJJ. The authors thank Dr Andrew Turnbull for invaluable discussions relating to AKR1C3 crystallography.
References
- Adeniji, A. O., Twenter, B. M., Byrns, M. C., Jin, Y., Winkler, J. D. & Penning, T. M. (2011). Bioorg. Med. Chem. Lett. 21, 1464–1468. [DOI] [PMC free article] [PubMed]
- Byrns, M. C., Jin, Y. & Penning, T. M. (2011). J. Steroid Biochem. Mol. Biol. 125, 95–104. [DOI] [PMC free article] [PubMed]
- Byrns, M. C. & Penning, T. M. (2009). Chem. Biol. Interact. 178, 221–227. [DOI] [PMC free article] [PubMed]
- Byrns, M. C., Steckelbroeck, S. & Penning, T. M. (2008). Biochem. Pharmacol. 75, 484–493. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gobec, S., Brozic, P. & Rizner, T. L. (2005). Bioorg. Med. Chem. Lett. 15, 5170–5175. [DOI] [PubMed]
- Guise, C. P., Abbattista, M. R., Singleton, R. S., Holford, S. D., Connolly, J., Dachs, G. U., Fox, S. B., Pollock, R., Harvey, J., Guilford, P., Doñate, F., Wilson, W. R. & Patterson, A. V. (2010). Cancer Res. 70, 1573–1584. [DOI] [PubMed]
- Hofland, J., van Weerden, W. M., Dits, N. F., Steenbergen, J., van Leenders, G. J., Jenster, G., Schröder, F. H. & de Jong, F. H. (2010). Cancer Res. 70, 1256–1264. [DOI] [PubMed]
- Jansson, A. K., Gunnarsson, C., Cohen, M., Sivik, T. & Stål, O. (2006). Cancer Res. 66, 11471–11477. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Komoto, J., Tamada, T., Watanabe, K. & Takusagawa, F. (2004). Biochemistry, 43, 2188–2198. [DOI] [PubMed]
- Komoto, J., Yamada, T., Watanabe, K., Woodward, D. F. & Takusagawa, F. (2006). Biochemistry, 45, 1987–1996. [DOI] [PubMed]
- Lin, H.-K., Steckelbroeck, S., Fung, K.-M., Jones, A. N. & Penning, T. M. (2004). Steroids, 69, 795–801. [DOI] [PubMed]
- Lovering, A. L., Ride, J. P., Bunce, C. M., Desmond, J. C., Cummings, S. M. & White, S. A. (2004). Cancer Res. 64, 1802–1810. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nakamura, Y., Suzuki, T., Nakabayashi, M., Endoh, M., Sakamoto, K., Mikami, Y., Moriya, T., Ito, A., Takahashi, S., Yamada, S., Arai, Y. & Sasano, H. (2005). Endocr. Relat. Cancer, 12, 101–107. [DOI] [PubMed]
- Qiu, W., Zhou, M., Labrie, F. & Lin, S.-X. (2004). Mol. Endocrinol. 18, 1798–1807. [DOI] [PubMed]
- Qiu, W., Zhou, M., Mazumdar, M., Azzi, A., Ghanmi, D., Luu-The, V., Labrie, F. & Lin, S.-X. (2007). J. Biol. Chem. 282, 8368–8379. [DOI] [PubMed]
- Schüttelkopf, A. W. & van Aalten, D. M. F. (2004). Acta Cryst. D60, 1355–1363. [DOI] [PubMed]
- Stanbrough, M., Bubley, G. J., Ross, K., Golub, T. R., Rubin, M. A., Penning, T. M., Febbo, P. G. & Balk, S. P. (2006). Cancer Res. 66, 2815–2825. [DOI] [PubMed]
- Suzuki-Yamamoto, T., Nishizawa, M., Fukui, M., Okuda-Ashitaka, E., Nakajima, T., Ito, S. & Watanabe, K. (1999). FEBS Lett. 462, 335–340. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: AKR1C3 complexed with 3-phenoxybenzoic acid, 3uwe