

AAC(6′)-Im is an N-acetyltransferase enzyme responsible for aminoglycoside resistance in E. faecium and E. coli isolates. Crystals of the kanamycin complex of this enzyme have been prepared and preliminary X-ray diffraction experiments have been undertaken.
Keywords: N-acetyltransferases, AAC(6′)-Im, aminoglycoside resistance
Abstract
Bacterial resistance to the aminoglycoside antibiotics is primarily the result of enzymatic deactivation of the drugs. The aminoglycoside N-acetyltransferases (AACs) are a large family of bacterial enzymes that are responsible for coenzyme-A-facilitated acetylation of aminoglycosides. The gene encoding one of these enzymes, AAC(6′)-Im, has been cloned and the protein (comprising 178 amino-acid residues) was expressed in Escherichia coli, purified and crystallized as the kanamycin complex. Synchrotron diffraction data to approximately 2.0 Å resolution were collected from a crystal of this complex on beamline BL12-2 at SSRL (Stanford, California, USA). The crystals belonged to the hexagonal space group P65, with approximate unit-cell parameters a = 107.75, c = 37.33 Å, and contained one molecule in the asymmetric unit. Structure determination is under way using molecular replacement.
1. Introduction
Ever since antimicrobial compounds were first used in a clinical setting to treat bacterial infections, there has been the concomitant emergence of bacterial strains that are resistant to these compounds. There are now bacterial isolates that are resistant to almost every antibacterial compound on the market, and such bacteria are increasingly causing life-threatening infections. One such family of antibacterials to which there is now universal widespread resistance are the aminoglycosides, which were originally isolated from soil bacteria (Greenwood, 1995 ▶). Streptomycin, the archetypal member of the aminoglycosides, was discovered in 1943 and was the first compound found to be effective against Mycobacterium tuberculosis (Greenwood, 1995 ▶). The aminoglycoside family, which includes a number of clinically relevant drugs such as gentamicin, tobramycin, kanamycin and amikacin, are targeted to the 30S ribosome, where drug binding leads to mistranslation of the mRNA (Carter et al., 2000 ▶; Vakulenko & Mobashery, 2003 ▶).
Acquired resistance to the aminoglycosides primarily occurs through enzymatic modification of the drug and involves three different classes of enzyme, each with a number of variants. There are now 80 or more such bacterial enzymes; they are predominantly carried on mobile genetic elements, which facilitates their spread throughout both Gram-positive and Gram-negative bacterial populations. These enzymes fall into three classes: the coenzyme-A-dependent acetyltransferases (AACs) and the ATP-dependent nucleotidyltransferases (ANTs) and phosphotransferases (APHS) (Davies & Wright, 1997 ▶; Smith & Baker, 2002 ▶; Vakulenko & Mobashery, 2003 ▶; Kim & Mobashery, 2005 ▶; Ramirez & Tolmasky, 2010 ▶). They can be classified into subtypes according to the position on the drug at which the modification occurs and also according to their resistance profile, which specifies a subset of aminoglycosides that they are able to modify. Broad-spectrum resistance to the aminoglycosides in Gram-positive bacteria is dominated by the bifunctional enzyme AAC(6′)-Ie-APH(2′′)-Ia (Ferretti et al., 1986 ▶) which comprises acetyltransferase and phosphotransferase functional domains and is the product of a single fused gene with a single start and stop codon. Despite this enzyme being the most important with respect to acquired resistance to the aminoglycides, its structure has remained elusive to date. Difficulties in obtaining diffraction-quality crystals of the entire AAC(6′)-Ie-APH(2′′)-Ia may result from significant differences in the optimal crystallization conditions for the acetyltransferase and phosphotransferase functionalities of this bifunctional enzyme or from conformational mobility in the interdomain linker which could render the enzyme highly flexible. On the other hand, attempts to separate the AAC(6′)-Ie domain of the bifunctional enzyme for structural studies led to significant impairment of the catalytic activity of the truncated acetyltransferase compared with the full-length enzyme (Boehr et al., 2004 ▶). The structures of three AAC(6′) enzymes have been reported to date: AAC(6′)-Ib (Vetting et al., 2008 ▶), AAC(6′)-Ii (Burk et al., 2003 ▶) and AAC(6′)-Iy (Vetting et al., 2004 ▶). All three enzymes have similar structures and are members of the large GCN5-related N-acetyltransferase (GNAT) superfamily. Alignment of the sequences of the known AAC(6′) structures with AAC(6′)-Ie indicates rather low identity (ranging from 7 to 24%). However, the monofunctional AAC(6′)-Im enzyme shows 58% sequence identity and 80% sequence similarity to the AAC(6′)-Ie domain of the bifunctional enzyme (Fig. 1 ▶). AAC(6′)-Im has been identified in clinical Escherichia coli and Enterococcus faecium isolates (Chow et al., 2001 ▶). Owing to the high amino-acid sequence similarity between AAC(6′)-Im and AAC(6′)-Ie, the AAC(6′)-Im enzyme may represent a good structural model for the acetyltransferase component of the bifunctional enzyme. In this regard, we have initiated structural analysis of this enzyme and have cloned, expressed, purified and crystallized the AAC(6′)-Im enzyme (178 amino-acid residues; MW 21 600) from E. faecium.
Figure 1.

Sequence alignment between AAC(6′)-Im (AAC-Im) and AAC(6′)-Ie (AAC-Ie). Residues which are identical in the two sequences are designated by asterisks and residues which are similar are designated by colons.
2. Materials and methods
2.1. Cloning, expression and purification of AAC(6′)-Im
The gene for the AAC(6′)-Im enzyme (GenBank accession No. AF337947) was custom-synthesized (GenScript) for optimal expression in E. coli and cloned into the unique NdeI and HindIII sites of pET22b(+) expression vector (Novagen) to generate the pET22:AAC(6′)-Im plasmid. For protein expression and purification, this vector DNA was transformed into E. coli BL21 (DE3) strain and clones were selected on LB agar supplemented with 100 µg ml−1 ampicillin. E. coli BL21 (DE3) cells harboring the pET22:AAC(6′)-Im plasmid were grown overnight in LB medium supplemented with 100 µg ml−1 ampicillin. The bacterial suspension was diluted 100-fold in 150 ml medium and grown in a shaker incubator at 310 K until the optical density reached 0.6 at 600 nm. Protein expression was induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside and the culture was incubated for an additional 20 h at 295 K. Cells were collected by centrifugation (5000g, 10 min) and disrupted by sonication in buffer A (25 mM HEPES pH 7.5, 1 mM EDTA, 0.2 mM DTT). The cell lysate was centrifuged (20 000g, 30 min) and the nucleic acids were precipitated from the supernatant with 1.5% streptomycin sulfate at 277 K. After centrifugation (20 000g, 30 min), the soluble protein fraction was dialyzed in 25 mM HEPES pH 7.5. The AAC(6′)-Im enzyme was purified on an Affi-Gel 15/kanamycin A affinity column and was eluted with a linear gradient of NaCl. The fractions were analyzed by acetyltransferase assay (Kim et al., 2007 ▶) and by SDS–PAGE (Fig. 2 ▶). All fractions containing the enzyme were pooled, concentrated to 3 mg ml−1 and dialyzed against 25 mM HEPES pH 7.5. The enzyme was further concentrated to 12 mg ml−1 (0.5 mM) in the presence of 10 mM kanamycin A.
Figure 2.

12% SDS–PAGE of AAC(6′)-Im under reducing conditions. Lane 1, molecular-weight marker (labeled in kDa); lanes 2 and 3, soluble proteins from cell lysate; lanes 4, 5 and 6, unbound proteins (flowthrough fractions) during purification by affinity chromatography; lane 7, purified AAC(6′)-Im.
2.2. Crystallization
Initial coarse screens for apo and the kanamycin complex of AAC(6′)-Im were performed with commercially available sparse-matrix screens (Crystal Screen, Crystal Screen 2, PEG/Ion and PEG/Ion 2; Hampton Research) using the sitting-drop method and resulted in a number of different conditions that gave rise to crystals. Crystals were grown at 277 K in Intelli-Plates (Art Robbins Instruments) using a reservoir volume of 75 µl and drops consisting of 1 µl protein complex solution and 1 µl reservoir solution. Initial screening experiments gave several different crystal forms ranging in shape from blocks to needles. Crystals from four conditions were flash-cooled in liquid nitrogen and stored in a sample cassette designed for use with the Stanford Automated Mounting (SAM) system (Cohen et al., 2002 ▶) for subsequent diffraction screening experiments.
2.3. Data collection and preliminary X-ray analysis
Initial screening of the crystals for diffraction quality was carried out on SSRL beamline BL11-1. Complete X-ray diffraction data were collected from a single AAC(6′)-Im–kanamycin crystal on SSRL beamline BL12-2 using a Pilatus 6M pixel array detector. A total of 650 images were collected with an oscillation angle of 0.2° and an exposure time of 0.2 s per image. The data were processed and scaled with the XDS/XSCALE programs (Kabsch, 2010 ▶). A summary of the data-collection statistics is given in Table 1 ▶. Preliminary molecular-replacement calculations were attempted using the programs MOLREP (Vagin & Teplyakov, 2010 ▶) and Phaser (McCoy et al., 2007 ▶), utilizing the structure of AAC(6′)-Ib (Vetting et al., 2008 ▶) as a search model.
Table 1. Statistics of native data collection.
Values in parentheses are for the highest resolution shell.
| Wavelength (Å) | 1.0332 |
| Space group | P65 |
| Unit-cell parameters (Å) | a = 107.75, c = 37.33 |
| Resolution range (Å) | 35.3–2.0 (2.05–2.00) |
| Mosaicity (°) | 0.5 |
| Observed reflections | 121419 |
| Unique reflections | 17083 |
| Average multiplicity | 7.1 |
| Rmerge† (%) | 6.7 (60.0) |
| 〈I/σ(I)〉 | 18.1 (3.9) |
| Completeness (%) | 99.7 (97.1) |
R
merge = 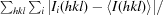
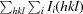 , where I
i(hkl) is the observed intensity of a given reflection and 〈I(hkl)〉 is the mean intensity for all observations of that reflection.
, where I
i(hkl) is the observed intensity of a given reflection and 〈I(hkl)〉 is the mean intensity for all observations of that reflection.
3. Results and discussion
The AAC(6′)-Im enzyme was prepared either as the apo form or as the kanamycin complex, which proved to be very beneficial in that only the kanamycin–AAC(6′)-Im complex yielded diffraction-quality crystals. Four conditions gave crystals of the complex which diffracted to 2.0–2.7 Å resolution (Fig. 3 ▶): form I, 0.2 M potassium sodium tartrate, 0.1 M sodium citrate pH 5.6, 2.0 M ammonium sulfate; form II, 0.2 M MgCl2, 0.1 M Tris–HCl pH 8.5, 30% PEG 4000; form III, 0.1 M MES pH 6.5, 12% PEG 20 000; form IV, 0.05 M HEPES pH 7.0, 20% PEG 3350, 1% tryptone. The form I crystals were block-like and required separation with a needle. Crystals of forms II, III and IV were all long thin needles which were readily separable and easily mounted. Preliminary screening of the crystals showed that crystal forms I, II and III had different space groups and unit-cell parameters and that form IV was identical to form III. Form I crystals belonged to space group P2 or P21, with unit-cell parameters a = 54.5, b = 68.5, c = 72.1 Å, β = 106.8°, and diffracted to approximately 2.7 Å resolution. Calculation of the Matthews coefficient (V M; Matthews, 1968 ▶) using an estimated molecular mass of 21.7 kDa gave a value of 3.0 Å3 Da−1 (59% solvent content) assuming two molecules per asymmetric unit. Form II crystals belonged to space group C2, with unit-cell parameters a = 161.3, b = 35.0, c = 67.3 Å, β = 102.6°, and diffracted to around 2.1 Å resolution with two molecules in the asymmetric unit (V M = 2.1 Å3 Da−1; 42% solvent content). Form III and IV crystals belonged to either a trigonal or hexagonal space group with either two molecules or one molecule in the asymmetric unit. The unit-cell parameters were approximately a = 107.8, c = 37.3 Å and the crystals diffracted to better than 2.0 Å resolution.
Figure 3.

The four crystal forms of AAC(6′)-Im: (a) form I crystals, (b) form II crystals, (c) form III crystals, (d) form IV crystals. The four panels are on the same scale; a scale bar is shown in (a).
A single flash-cooled form III crystal in a cryoprotectant composed of the crystallization buffer with 20% glycerol was used to collect a complete X-ray data set. The crystal diffracted to approximately 1.9 Å resolution, but the high-resolution limit of the final data set was truncated at 2.0 Å because the value of R merge in the 2.0–1.9 Å resolution shell was deemed to be too high at around 80%. A representative image is shown in Fig. 4 ▶ and the results are summarized in Table 1 ▶. Following data processing with XDS and XSCALE, the program POINTLESS (Evans, 2006 ▶) was used to determine the Laue group, resulting in two possible space groups: P61 and P65. Calculation of the Matthews coefficient (V M) gave a value of 2.9 Å3 Da−1 (57% solvent content) assuming one molecule per asymmetric unit.
Figure 4.

Diffraction image of the kanamycin complex of AAC(6′)-Im, crystal form III. The resolution circle is at approximately 2.0 Å resolution.
Although the level of amino-acid sequence identity between the members of the AAC(6′) subfamily is low, it was thought that molecular replacement using AAC(6′)-Ib as a search model might give reasonable starting phases for the AAC(6′)-Im structure. The AAC(6′)-Ib model was modified with the program CHAINSAW from the CCP4 suite (Winn et al., 2011 ▶), truncating the nonconserved residues at the Cβ atom. Structural comparison of the three known AAC(6′) structures [AAC(6′)-Ib, PDB entry 1v0c (Vetting et al., 2008 ▶); AAC(6′)-Ii, PDB entry 1n71 (Burk et al., 2003 ▶); AAC(6′)-Iy, PDB entry 1s3z (Vetting et al., 2004 ▶)] showed that although the main core of the molecule was structurally conserved, some of the surface loops had different lengths and adopted different conformations in the three models. Several different models were produced from this CHAINSAW model with loop truncations. Preliminary molecular-replacement calculations were performed using the programs MOLREP (Vagin & Teplyakov, 2010 ▶) and Phaser (McCoy et al., 2007 ▶), testing the two possible hexagonal space groups suggested by POINTLESS; the best solution was obtained in space group P65 by both programs. The R factor and score from MOLREP were 0.51 and 0.26, respectively, and the log-likelihood gain and Z score following the translation function from Phaser were 80 and 10.9, respectively. Refinement of the solution is currently being undertaken.
Acknowledgments
This work was supported by Grant No. AI057393 from the National Institutes of Health (SBV) and by Grant No. 5 P41 RR001209 from the National Center for Research Resources (SSRL). Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy (BES, BER) and by the National Institutes of Health (NCRR, BTP, NIGMS). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH.
References
- Boehr, D. D., Daigle, D. M. & Wright, G. D. (2004). Biochemistry, 43, 9846–9855. [DOI] [PubMed]
- Burk, D. L., Ghuman, N., Wybenga-Groot, L. E. & Berghuis, A. M. (2003). Protein Sci. 12, 426–437. [DOI] [PMC free article] [PubMed]
- Carter, A. P., Clemons, W. M., Brodersen, D. E., Morgan-Warren, R. J., Wimberly, B. T. & Ramakrishnan, V. (2000). Nature (London), 407, 340–348. [DOI] [PubMed]
- Chow, J. W., Kak, V., You, I., Kao, S. J., Petrin, J., Clewell, D. B., Lerner, S. A., Miller, G. H. & Shaw, K. J. (2001). Antimicrob. Agents Chemother. 45, 2691–2694. [DOI] [PMC free article] [PubMed]
- Cohen, A. E., Ellis, P. J., Miller, M. D., Deacon, A. M. & Phizackerley, R. P. (2002). J. Appl. Cryst. 35, 720–726. [DOI] [PMC free article] [PubMed]
- Davies, J. & Wright, G. D. (1997). Trends Microbiol. 5, 234–240. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ferretti, J. J., Gilmore, K. S. & Courvalin, P. (1986). J. Bacteriol. 167, 631–638. [DOI] [PMC free article] [PubMed]
- Greenwood, D. (1995). Antimicrobial Chemotherapy, edited by D. Greenwood, pp. 32–48. Oxford University Press.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kim, C. & Mobashery, S. (2005). Bioorg. Chem. 33, 149–158. [DOI] [PubMed]
- Kim, C., Villegas-Estrada, A., Hesek, D. & Mobashery, S. (2007). Biochemistry, 46, 5270–5282. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Ramirez, M. S. & Tolmasky, M. E. (2010). Drug Resist. Updat. 13, 151–171. [DOI] [PMC free article] [PubMed]
- Smith, C. A. & Baker, E. N. (2002). Curr. Drug Targets Infect. Dis. 2, 143–160. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Vakulenko, S. B. & Mobashery, S. (2003). Clin. Microbiol. Rev. 16, 430–450. [DOI] [PMC free article] [PubMed]
- Vetting, M. W., Magnet, S., Nieves, E., Roderick, S. L. & Blanchard, J. S. (2004). Chem. Biol. 11, 565–573. [DOI] [PubMed]
- Vetting, M. W., Park, C. H., Hegde, S. S., Jacoby, G. A., Hooper, D. C. & Blanchard, J. S. (2008). Biochemistry, 47, 9825–9835. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.