

Abstract
Previous studies have shown that modulation of the receptor-mediated cannabinoid system during neuroinflammation can produce potent neuroprotective and anti-inflammatory effects. However, in this context, little is known about how selective activation of the cannabinoid type-2 receptor (CB2R) affects the activated state of the brain endothelium and blood–brain barrier (BBB) function. Using human brain tissues and primary human brain microvascular endothelial cells (BMVECs), we demonstrate that the CB2R is highly upregulated during inflammatory insult. We then examined whether the CB2R agonists could attenuate inflammatory responses at the BBB using a mouse model of LPS-induced encephalitis and highly selective CB2R agonists. Visualization by intravital microscopy revealed that administration of JWH133 [(6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran] or a novel resorcinol-based compound, O-1966 (1-[4-(1,1-dimethyl-heptyl)-2,6-dimethoxy-phenyl]-3-methyl-cyclohexanol), greatly attenuated leukocyte adhesion in surface pial vessels and in deep ascending cortical postcapillary venules. BBB permeability assessments with small and large fluorescent tracers showed that CB2R agonists were effective at preventing barrier leakiness after LPS administration. To determine whether the effects by CB2R agonists on barrier protection are not only due to the CB2R modulation of immune cell function, we tested the agonists in vitro with barrier-forming primary BMVECs. Remarkably, the addition of CB2R agonist increased transendothelial electrical resistance and increased the amount of tight junction protein present in membrane fractions. Furthermore, CB2R agonists decreased the induction of intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 surface expression in BMVECs exposed to various proinflammatory mediators. Together, these results suggest that pharmacological CB2R ligands offer a new strategy for BBB protection during neuroinflammation.
Introduction
The blood–brain barrier (BBB) shields the brain parenchyma from immune cells and toxins in the blood, thus allowing the adequate environment needed for normal neuronal and glial cell function. During neuroinflammation, the BBB can become impaired by the triggered inflammatory response from all the cells of the neurovascular unit (Neuwelt et al., 2008), and particularly from immune–endothelial cell engagement (Persidsky et al., 2006a). Therapeutic strategies that prevent the immune–endothelial interactions and maintain tightness of the barrier are critical in neuroprotection (Greenwood et al., 2002). One promising therapeutic avenue could be activation of the endogenous cannabinoid system, which has been shown previously to induce both anti-inflammatory and neuroprotective effects.
There are two well-characterized cannabinoid receptors with distinct physiological properties. The psychoactive effects of cannabinoids are associated with the cannabinoid type-1 receptor (CB1R), while the CB2R mainly mediates anti-inflammatory actions (Miller and Stella, 2008). In the brain, the anti-inflammatory and neuroprotective effects of cannabinoids have been confirmed in animal models of multiple sclerosis (MS), Alzheimer's disease (AD), stroke, and amyotrophic lateral sclerosis (ALS) (for review, see Cabral and Griffin-Thomas, 2009; Zhang et al., 2009b). These effects have been observed with pharmacological ligands that act on either the CB1R, CB2R, or both. For example, in a viral model of MS, CB1R agonists have been shown to reduce perivascular CD4+ T lymphocyte infiltration, inhibit microglial responses, and suppress the upregulation of adhesion molecules in the brain endothelium (Mestre et al., 2009). However, because of their psychoactive effects, the uses of CB1R agonists have a limited therapeutic potential, leaving CB2R agonists as the practical option.
Although the modulatory effects of the CB2R on immune cells have been studied, not much is known about its effects on brain endothelial cells. Therefore, the improved pathological outcomes observed in vivo with cannabinoids may be a sum effect of the attenuation on leukocyte and endothelial cell activation. CB2R has been found in brain endothelium (Golech et al., 2004; Lu et al., 2008) and endothelial cells from other organs (Rajesh et al., 2007). Attenuation of endothelial activation by CB2R has been observed in nonbrain endothelial cells; as such, synthetic CB2R agonists were shown to prevent TNFα-induced activation of human coronary artery endothelial cells in vitro (Rajesh et al., 2007).
In this study, we address the effects of CB2R agonists on the brain endothelium and BBB function. First, we found enhanced CB2R receptor expression in cerebral vessels of human brain tissue affected by neuroinflammation [HIV-1 encephalitis (HIV-E)]. We also tested CB2R agonists, in an LPS-induced encephalitis model to evaluate the effects on leukocyte–endothelial cell adhesion in both surface and ascending cortical vessels. We showed that a significant level of leukocyte adhesion was prevented in the encephalitic brain following administration of CB2R agonist. CB2R activation reduced expression of adhesion molecules needed for leukocyte engagement in TNFα- or LPS-activated primary human brain microvascular endothelial cells (BMVECs) and in brain endothelium of animals after LPS administration. Also, for the first time, our results show that the BBB protective effects of CB2R agonists are due to the upregulated expression of tight junction (TJ) proteins.
Materials and Methods
Reagents and experimental animals.
The selective CB2 receptor agonist, (6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran (JWH133), was purchased from Tocris Bioscience. The selective CB2 receptor agonist, O-1966 (1-[4-(1,1-Dimethyl-heptyl)-2,6-dimethoxy-phenyl]-3-methyl-cyclohexanol), was acquired from Organix and synthesized as described previously (Wiley et al., 2002). The selective CB2 receptor antagonist, 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide (SR144528) was purchased from Cayman Chemical. JWH133 was solubilized in Tocrisolve-100 (Tocris Bioscience), an aqueous soluble emulsion composed of a 1:4 ratio of soya oil to water that is emulsified with the block copolymer Pluronic F68. Crystalline O-1966 was dissolved in a pure ethanol/emulphor/saline mixed solution at 1:1:18. SR144528 was solubilized in a 1:1 solution of ethanol/1× PBS. Escherichia coli LPS from E. coli 0127:B8 was purchased from Sigma-Aldrich.
All intravital microscopy and whole-brain permeability analysis was performed on either 8-week-old male C57BL/6 mice or the CB2 receptor knock-out mice (strain B6.129P2-Cnr2tm1Dgen/J) purchased from the Jackson Laboratory. PCR genotyping of the knock-out mice was performed with DNA extracted (from the tails) using the following specific PCR primers: moIMR0086 (GGG GAT CGA TCC GTC CTG TAA GTC T; mutant forward), oIMR7552 (GAC TAG AGC TTT GTA GGT AGG CGG G; common backward), and oIMR7565 (GGA GTT CAA CCC CAT GAA GGA GTA C; wild-type forward). The expected results were one band of ∼550 bp for mutant, one band of 385 bp for wild-type, and two bands of 385 bp and ∼550 bp for heterozygote. Animals were housed and allowed to acclimate for 1–2 weeks in the Temple University Central Animal Facility. The animals were provided standard environmental enrichment conditions and were fed with a commercial pellet diet and water ad libitum. All in vivo experiments were conducted in accordance with the guidelines approved by the Institutional Animal Care and Use Committee at Temple University.
Cell culture.
BMVECs were supplied by Michael Bernas and Dr. Marlys Witte (University of Arizona, Tucson, AZ). Isolation and the BMVEC culture procedure have been detailed previously (Bernas et al., 2010). Briefly, BMVECs were isolated from vessels of normal tissue derived from brain resections performed for the treatment of intractable epilepsy. The BMVEC cultures were used until passage 5 and were expanded in DMEM/F-12 media supplemented with 10% heat-inactivated fetal bovine serum (FBS), endothelial cell growth supplement (ECGS; BD Biosciences), heparin (1 mg/ml; Sigma), amphotericin B (2.5 μg/ml; Invitrogen), penicillin (100 U/ml; Invitrogen), and streptomycin (100 μg/ml; Invitrogen). Before experimentation, BMVEC monolayers were placed in medium containing the above supplements but lacking ECGS and heparin. Under these conditions, the BMVEC cultures were routinely evaluated for the presence of endothelial markers and barrier formation (Bernas et al., 2010).
Primary human monocytes were supplied by the Human Immunology Core at the University of Pennsylvania (Philadelphia, PA). The cells were isolated by countercurrent centrifugal elutriation and maintained in DMEM containing heat inactivated 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), and l-glutamine (2 mm) and were used within 24 h of isolation.
Intravital microscopy.
Intravital microscopy for in vivo leukocyte adhesion and vascular permeability was performed on adult male mice weighing between 20 and 25 g. Following institutional regulations and guidelines for mouse survival surgery, intravital microscopy was performed on animals that underwent craniotomy and cranial window implantation as described previously (Ramirez et al., 2010b). At the start of the procedure, the animals were placed under anesthesia [ketamine (100 mg/ml, i.p.) and xylazine (20 mg/kg, i.p.) mixture (1:1) at a dose of 1 ml/kg] and depth of anesthesia was monitored by both toe and pinna pinch tests. Maintenance of normal body temperature was ensured with the use of a rodent thermal blanket. Once fully anesthetized, the head was positioned in a stereotaxic frame (Stoelting) with the nose bar set at zero. The craniotomy was performed lateral to the sagittal suture, and centered between lambda and bregma. A 1 cm square area of skin on the dorsal surface of the skull over the right cortical hemisphere was excised and the periosteum exposed. The craniotomy was performed by generating a 4 mm diameter circular osteotomy with a rechargeable cordless micro drill (Stoelting) over the right parietal cortex extending from attachment of the temporal muscle to the midpoint of the sagittal suture in the coronal direction and aligned to the middle of the sagittal suture. Of note, for intravital microscopy using wide-field epifluorescence, the dura was carefully removed; however, when using multiphoton microscopy, the dura was left untouched. A 5 mm coverslip was then introduced over the exposed brain, and an airtight seal was generated using Nexaband Quick Gel (which shows no histotoxicity). The tissue adhesive quickly created a waterproof seal between the edge of the skin and the glass coverslip. A recovery period of at least 4 d was allowed between implantation of the cranial window and intravital microscope observation.
On the day of microscopy observation, animals were anesthetized and immobilized as described above. Observation of surface cerebral vessels through the cranial window was performed with a Stereo Discovery V20 epifluorescence microscope (Carl Zeiss Microimaging) equipped with a AxioCam MR digital camera (Carl Zeiss Microimaging) that was connected to a PC for image acquisition and analysis using Axiovision imaging software (Carl Zeiss Microimaging). For deep-tissue imaging, we used a Leica TCS SP5 II MP multiphoton microscope configured with a tunable femtosecond pulsed Mai Tai Ti:Sapphire laser (Spectra Physics), a resonant scanner (allowing acquisition of 29 frames per second), and NDD detectors. Dental cement was used around the cranial window to create a well in which distilled water was placed to permit the use of the 20× dipping objective (NA, 0.95).
For evaluation of adhesion, leukocytes were stained in vivo with an intravenous bolus injection of 50 μl of a 0.01% solution of rhodamine 6G (Sigma-Aldrich). Identification of the brain vasculature and evaluation of vessel permeability was performed using intravenously injected 70 kDa fluorescein-conjugated dextrans (at 10 mg/ml in a 100 μl saline solution). For wide-field epifluorescence, leukocytes were visualized by fluorescent light (601 nm excitation). Selective filtering allowed visualization of the fluorescent cells on a dark background. A 30 s video (time-series image set between 16 and 20 frames per second) was captured using the digital high-speed recorder Axiovision module (Carl Zeiss Microimaging). Adherent leukocytes were defined as the total number of the leukocytes firmly attached to the endothelium that did not change their location during the entire 30 s observation period. Adherent leukocytes were scored as the number of cells per square millimeter of the vascular surface area, calculated from the diameter and standardized length (100 μm) of the vessel segment under observation. For multiphoton imaging, randomly selected cortical postcapillary venules (20–30 μm in diameter and down to 600 μm below the cortical surface) were selected for imaging. Z stacks of 400 μm thickness at 1 μm steps were collected. Rendered three-dimensional constructs using the Imaris imaging software (Bitplane) were analyzed to obtain the number of adherent leukocytes per 100 μm of vessel segment.
Immunohistochemistry and immunofluorescence.
Evaluation of CB2R expression was performed on postmortem paraffin-embedded brain tissue sections from four HIV-1-infected patients with severe encephalitis (Ramirez et al., 2010a) and from four seronegative age-matched controls provided by the National NeuroAIDS Consortium (Washington, DC). Using standard immunohistochemistry methods, immunolabeling was performed on serial sections cut at 5 μm in thickness. The following antibodies and dilutions were used: polyclonal antibodies against the human CB2R (Thermo Scientific) was diluted at 1:100 with overnight incubation and polyclonal antibodies to the ionized calcium-binding adaptor molecule-1 (Iba-1; Wako Chemical) was used at 1:200 with overnight incubation. Detection was performed with biotinylated anti-mouse secondary antibodies (Dako) using the Strep-ABC complex (Dako) and the DAB peroxidase substrate from Vector Laboratories. Counterstaining of the nuclei was performed using hematoxylin (Dako).
Immunofluorescence for intercellular adhesion molecule 1 (ICAM-1) was performed on mouse frozen sections cut to 5 μm in thickness. Polyclonal antibodies to ICAM-1 (Santa Cruz Biotechnology) were used at 1:100 with overnight incubation. To observe the vessels, dual labeling was performed with mouse monoclonal antibodies to the glucose transporter-1 (Abcam) used at 1:50 with overnight incubation. Secondary antibodies anti-rabbit Alexa-488 and anti-mouse Alexa-594 (Invitrogen) were used at 1:400 for 1 h. The slides were mounted with Prolong antifade reagent containing DAPI (Invitrogen).
CB2R and ZO-1 protein expression was evaluated by immunofluorescence on monolayers of BMVECs plated on type I collagen-coated coverslips (BD Biosciences). The cells were fixed with 3% (v/v) formaldehyde (Polysciences) and permeabilized with 0.1% Triton X-100 in PBS. Primary antibodies and dilutions used included polyclonal antibodies to ZO-1 (1:25; Invitrogen) and CB2R (1:100; Thermo Scientific). The cells and primary antibodies were incubated overnight at 4°C. Secondary antibodies anti-rabbit Alexa-488 and anti-mouse Alexa-594 (Invitrogen) were used at 1:400 for 1 h. Cells were then washed and mounted onto slides with Prolong antifade containing DAPI (Invitrogen).
Immunohistochemistry and immunofluorescence were visualized with a Nikon Eclipse 80i wide-field fluorescence microscope fitted with a color DS-Fi1 camera for imaging of chromagen stained slides and a Coolsnap EZ CCD camera (Photometrics) for fluorescence imaging. Imaging software NIS Elements BR (Nikon Instruments) was used for image acquisition and processing.
Western blot.
Low-passage BMVECs or isolated brain microvessels were used for the Western blots. BMVEC cellular fractions were collected using the ProteoExtract kit (EMD Chemicals) as outlined by the manufacturer's protocol. Brain microvessels were isolated as described previously (Dogrukol-Ak et al., 2009), and lysates were prepared using the CelLytic-M reagent (Sigma). The protein concentrations in fractions were estimated using the BCA method (Thermo Scientific). Cellular fractions were loaded at 50 μg per lane and resolved by SDS-PAGE on 4–20% precast gradient gels (Thermo Scientific). Proteins were transferred to nitrocellulose membranes and incubated overnight with polyclonal antibodies against CB2R (1:1000; Cayman Chemicals), occludin (1:500; Invitrogen), claudin-5 (1:300; Invitrogen), and sodium potassium ATPase (1:1000; Abcam). Secondary antibodies conjugated to HRP (Thermo Scientific) were then added for 1 h and detected using Supersignal West Pico chemiluminescence substrate (Thermo Scientific). Chemiluminescence signal detection was performed with the gel documentation system G:Box Chemi HR16 (Syngene). Densitometry analysis was performed with the GeneTools software package (Syngene).
Flow cytometry.
Surface expression of adhesion molecules was measured in BMVECs by flow cytometry. Briefly, following various treatments, 2 × 106 cells were placed in staining solution (2% BSA in PBS with 0.5% NaN3) containing fluorophore-conjugated antibodies to CD54 (anti-ICAM-1-APC; BD Biosciences) and CD106 (anti-VCAM-1-PE; BD Biosciences) for 30 min on ice. Cells were then washed and fixed in 2% methanol-free formaldehyde (Thermo Scientific) in 1× PBS. Acquisition and analysis of the labeled cells were then performed using a FACSCalibur flow cytometer (BD Biosciences). Acquisition parameters and gating were controlled by CellQuest software (BD Biosciences). Data analysis was performed with FlowJo software (Tree Star). The data represent the fluorescence intensity of gated populations (as determined by isotype-matched controls) of at least 10,000 events recorded in a single experiment that was repeated at least three times.
Measurement of transendothelial electrical resistance.
To determine the integrity of brain endothelial monolayers, transendothelial electrical resistance (TEER) measurements were performed using the 1600R electric cell-substrate impedance sensing (ECIS) system (Applied Biophysics) as described previously (Ramirez et al., 2009). The ECIS system provides real-time monitoring of changes in TEER. In brief, BMVECs at 1 × 105 per well were plated on collagen type I-coated 8W10E+ electrode arrays (Applied Biophysics). The cells were then allowed to form monolayers reaching stable TEER values. After 7 d (with medium change every 3 d), the monolayers were exposed to the specific treatment (indicated in the figure). The readings were acquired continuously at 4 kHz at 30 min intervals. Confluent BMVEC monolayers demonstrated baseline TEER readings between 1500 and 2400 Ω. The data are shown as the percentage change from the baseline TEER along with the SEM from at least three independent experiments containing three condition replicates.
Adhesion assays.
Quantitative adhesion assays were performed as described previously (Ramirez et al., 2008). BMVECs were seeded on collagen type I-coated 96-well black-wall, clear-bottom plates (Corning Costar) at a density of 2.5 × 104 cells per well. Confluent monolayers were then exposed to the indicated experimental treatments. Freshly isolated human monocytes at 5 × 106 cells/ml were labeled with 5 μm of the fluorescent tracer calcein AM (Invitrogen). All treatments were removed from the endothelial cells before adding monocytes at 2.5 × 105 cells per well. The monocytes and endothelial cells were incubated together for 15 min and then rinsed three times with 1× PBS to eliminate nonadherent monocytes. The fluorescence of adherent monocytes was measured using a Spectramax M5 fluorescence plate reader (Molecular Devices). The number of adherent monocytes was determined from external standards of known numbers of labeled monocytes. The results for adhesion are represented as the mean ± SEM-fold adhesion (number of adherent monocytes for each experimental condition divided by the basal adhesion of the untreated control).
BBB permeability in vivo.
Changes in BBB permeability were assessed using the fluorescent tracer sodium fluorescein (Na-F); the procedure performed was a modification of methods described previously (Hawkins and Egleton, 2006). Briefly, animals were injected intravenously with 50 μl of 2% Na-F in PBS. The tracer was allowed to circulate for 30 min. The mice were anesthetized and then transcardially perfused with PBS until colorless perfusion was visualized. The animals were then decapitated and the brains quickly isolated. After removal of the meninges, cerebellum, and brain stem, the tissue was weighed and homogenized in a 10× volume of 50% trichloroacetic acid. The homogenate was centrifuged for 10 min at 13000 × g, and the supernatant was neutralized with 5 m NaOH (1:0.8). Measurement of Na-F fluorescence was determined at excitation/emission wavelengths of 440/525 nm using a SpectraMax M5 microplate reader (Molecular Devices). Fluorescent dye content was calculated using external standards with a range of 10–200 ng/ml. The data are expressed as the fold (compared to control) amount of tracer found per mg of tissue.
Statistical analysis.
The values shown in all figures and those mentioned in the text represent the average ± SEM of experiments that were conducted multiple times (as indicated). Statistical significance (p < 0.05) was determined by performing unpaired two-tailed Student's t test or ANOVA using Prism version 5 software (GraphPad Software).
Results
CB2R is expressed on brain endothelium
Previous studies have demonstrated that expression of the CB2R is significantly increased in human brain tissues affected by MS, ALS, and AD (Benito et al., 2005, 2007, 2008; Yiangou et al., 2006). In the case of MS, CB2R was found on microglia, perivascular macrophages, and T cells (Benito et al., 2007). Given the important role the BBB plays during inflammation, we sought to characterize whether the expression level of CB2R changes in brain endothelium that is under inflammatory insult. To test this possibility, we investigated the presence of CB2R in human brains from patients with neuroinflammation, namely, HIV-1 CNS infection (n = 4), and seronegative controls (n = 3). Immunoreactivity in control brain tissues with no evidence of microglia activation showed minimal staining of CB2R on the cerebral microvasculature (Fig. 1A). However, enhanced expression of CB2R was detected on brain endothelial cells present on brain tissues sections from cases of HIV-E (Fig. 1A). Iba-1, a well-known specific marker for microglia, demonstrated prominent inflammation in the parenchyma of encephalitic cases. Of note, CB2R expression was also increased on microglial cells in HIV-E as reported previously (Persidsky et al., 2011). Because inflammatory responses in the brain constitutes a complexity of signaling molecules originating from various cell types, we sought to determine in culture whether a specific inflammatory stimulus would be enough to induce the expression of CB2R in endothelial cells. Using confluent cultures of primary human BMVECs, the basal expression of CB2R was evaluated by immunofluorescence. We performed double immunostaining for CB2R and ZO-1, a highly expressed TJ protein in barrier-forming BMVECs. Figure 1B shows moderate immunostaining for the CB2R in the cultured BMVECs (derived from different donors) along with strong intercellular expression of ZO-1. CB2R staining appeared diffused in the cytosolic and membranous compartments and did not stain positive in interconnecting areas (Fig. 1B). Next, the CB2R protein in membrane fractions was evaluated by Western blot from BMVECs exposed to various proinflammatory stimuli. Confirming the immunostaining analysis, BMVECs express CB2R under resting untreated culture conditions (Fig. 1C). Similar to brain tissues affected by HIV-E (Fig. 1A), CB2R expression on BMVECs was induced by proinflammatory cytokines (IL-1β, 50 ng/ml; TNFα, 25 ng/ml for 24 h) or LPS (50 ng/ml for 24 h). Although all proinflammatory stimuli tested raised the expression of CB2R expression, ranging between 1.71- and 3.36-fold over untreated BMVECs, TNFα and LPS consistently produced the highest induction of CB2R. These data suggest that CB2R is expressed in brain endothelium and is upregulated by inflammatory insults.
Figure 1.
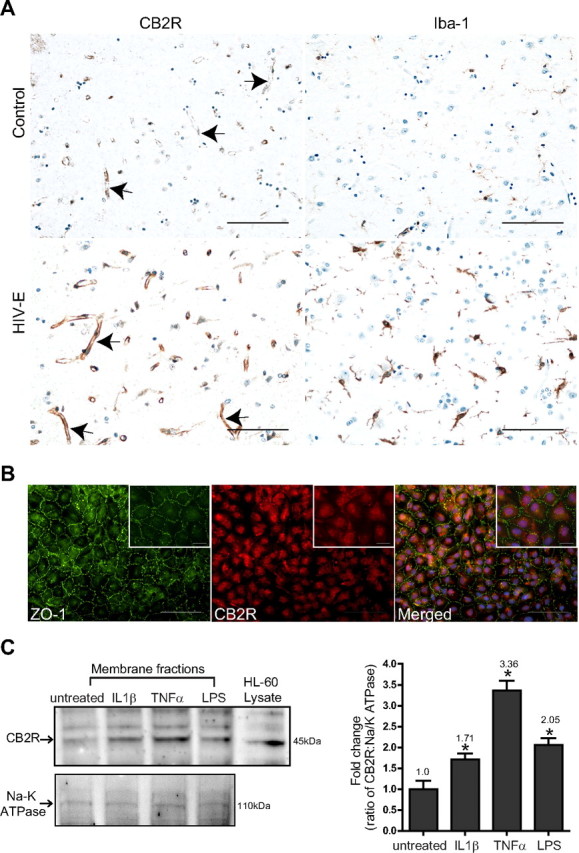
Upregulation of CB2R expression in brain endothelium. A, Brain tissue from frontal cortical regions was obtained from four cases of HIV-E and from seronegative age-matched controls (n = 3). H&E and immunohistochemistry were performed for CB2R and Iba-1 (arrows indicate CB2R-immunopositive vessels). Neuroinflammation was confirmed by induced expression of Iba-1 in reactive microglia (a feature of HIV-E). The images were taken under 20× objective power. B, Representative images showing immunofluorescence labeling of tight junction protein ZO-1 and CB2R on cultured primary human BMVECs. Note the continuous pattern of ZO-1 immunostaining depicting the location of endothelial tight junctions. The images shown were taken under 20× and 60× (inset) objective power. C, Analysis of CB2R expression in membrane fractions from BMVECs exposed to proinflammatory stimuli. BMVECs were exposed to the following inflammatory mediators for 24 h: IL-1β at 50 ng/ml, TNFα at 25 ng/ml, and LPS at 50 ng/ml). Densitometry values for CB2R expression are also shown and were calculated from the ratio of CB2R expression to that of Na/K ATPase (loading control). The HL-60 lysate is shown as a reference (positive control) to indicate the presence of CB2R. The values are represented as fold change relative to the untreated control. Scale bars: A, 100 μm; B, 100 μm; insets, 25 μm. Asterisk denotes statistical significance (p < 0.05).
CB2R activation decreases leukocyte adhesion in surface (pial) cortical vessels
CB2R stimulation has been reported to diminish inflammation in several animal models (Benito et al., 2008; Mestre et al., 2009; Zhang et al., 2009a,b), while the mechanism associated with such effects remains largely obscure. Increased expression of CB2R on brain endothelium could serve as a target for anti-inflammatory effects of CB2R agonists via diminishing endothelial cell engagement by leukocytes, leading to their migration and associated tissue injury. To explore this idea, we investigated whether CB2R stimulation would diminish leukocyte adhesion in vivo, using an animal model of systemic inflammatory responses. We used intravital microscopy via cranial window to allow the direct observation of cerebral vascular changes and leukocyte–endothelial cell interactions in real time (Ramirez et al., 2010b). A week after cranial window implantation, LPS (from E. coli 0127:B8) was injected to induce systemic inflammation and cytokine production. Figure 2A shows the schematic of the study design with the time points when visualizations were performed. Selective CB2R agonist (JWH133) was injected simultaneously with LPS (6 mg/kg), and leukocyte adhesion on surface pial vessels was monitored at 0, 4, and 24 h by intravital fluorescence microscopy (Fig. 2B) and quantitated by image analysis as described in Materials and Methods. The first row (Fig. 2B) shows the baseline leukocyte adhesion before LPS insult, which was minimal. At 4 h after LPS injection, leukocyte adhesion was markedly increased (Fig. 2B, middle, first column), showing a 30-fold induction in adhesion over baseline (Fig. 2C). Of note, leukocyte marginalization occurred only in the veins and not in arterial vessels, which have a more straight and consistent vessel caliber than veins. Administration of the CB2R agonist JWH133 in combination with LPS produced a significant reduction in leukocyte–endothelial cell interaction (Fig. 2B, middle, second column). JWH133 attenuated leukocyte adhesion by 56% (p < 0.001; Fig. 2C) after LPS injection. At 24 h, adhesion still remained increased by ∼18-fold compared to baseline, and JWH133 diminished immune cell adhesion by 65% (p < 0.001; Fig. 2C). The effects of the JWH133 agonist on reducing leukocyte adhesion appeared to be specific to CB2R, since nearly complete reversal was observed when the potent CB2R antagonist SR144528 was also present (Figs. 2B, middle, third column, 3C). Of note, whether at 4 or 24 h, systemic inflammatory response produced no observable diapedesis of immune cells into the brain parenchyma by these methods.
Figure 2.
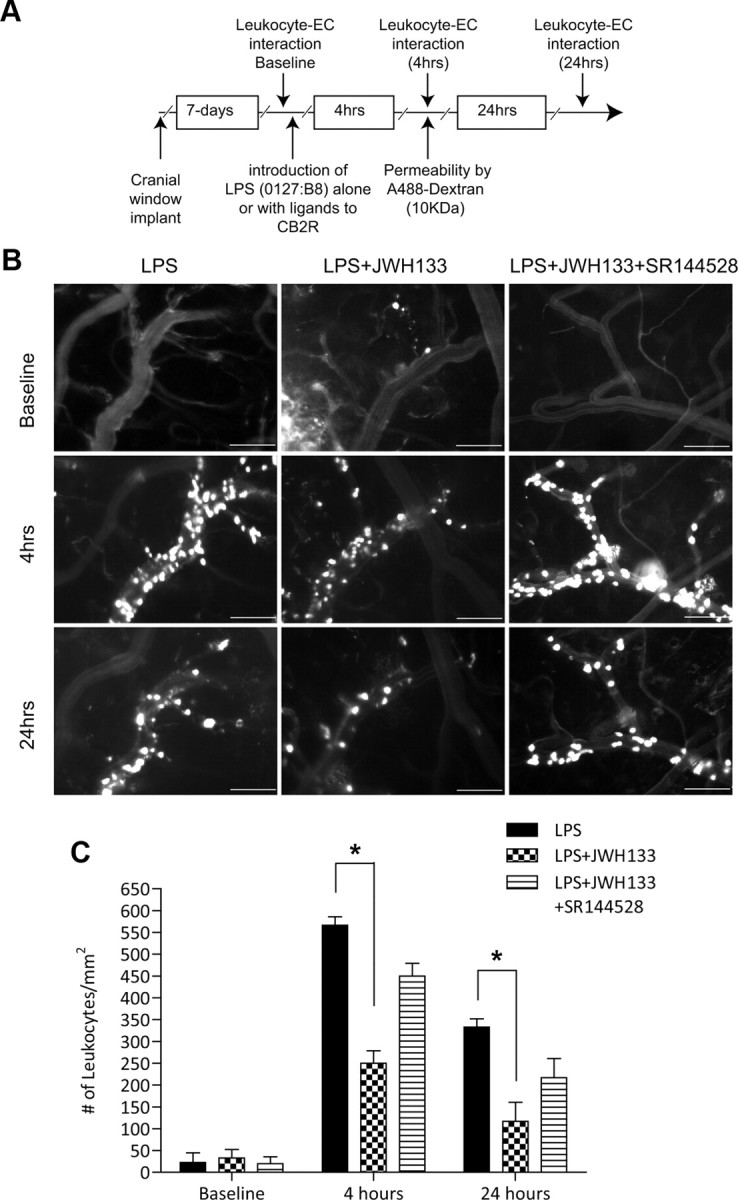
CB2R agonist reduces leukocyte adhesion in LPS-associated encephalitis. A, Experimental flow schematic of treatments and intravital microscopy endpoints. C57BL/6 mice were implanted with cranial windows as described in Materials and Methods. B, Seven days after the cranial window procedure, animals were placed in the following experimental conditions: LPS alone (6 mg/kg, i.p.), LPS with CB2 agonist JWH133 (10 mg/ml, i.p.), and LPS with JWH133 and the CB2R antagonist SR144528 (10 mg/ml, i.p.). Representative images from a video of leukocytes labeled with the fluorescent compound rhodamine 6G were visualized for adhesion in pial venules at 0, 4, and 24 h by intravital microscopy. The top row shows the baseline leukocyte adhesion. The middle and bottom rows represent images taken at 4 and 24 h, respectively. Scale bars: 100 μm. C, Quantitative measurements of leukocytes under firm adhesion (not rolling) during an observation period of 30 s. Adhering leukocytes from multiple vessels and from at least three fields of view per animal (three animals per group) were scored as the number of cells per square millimeter of the vascular surface area, calculated from the diameter and standardized length (100 μm) of the vessel segment under investigation. The results are shown as the mean adhesion ± SEM (n = 3). *p < 0.001 [difference between the groups compared (brackets)].
Figure 3.

A novel resorcinol CB2R agonist, O-1966, reduces LPS-induced leukocyte adhesion in cerebral vessels. A, Intravital microscopy analysis of endothelial-leukocyte adhesion events in animal groups injected with either LPS (6 mg/kg, i.p.), LPS with O-1966 (5 μm, i.p.), and LPS with O-1966 and SR144528 (10 μm, i.p.). Shown are representative images of fluorescently labeled leukocytes taken by intravital video microscopy. Scale bars: 100 μm. B, Measurements calculated from image and video analysis of immune cells attached to the brain endothelium during the indicated time points. The results are shown as the mean adhesion ± SEM (n = 3). *p < 0.001 [difference between the groups compared (brackets)].
Given the response observed with the commercially available CB2R agonist JWH133, we sought to test the same in vivo parameters with a novel resorcinol derivative CB2 agonist (O-1966) in a similar fashion (Wiley et al., 2002). Figure 3 shows that this highly selective CB2R agonist also markedly reduced adhesion of leukocytes to cerebral vessels at 4 and 24 h (by 50 and 60%, respectively; p < 0.001). Interestingly, during dose–response analysis (data not shown) of O-1966, we found that only half a dose of O-1966 (5 μm) was needed to achieve the maximal inhibition of leukocyte adhesion compared to that of JWH133 (10 μm). Again, the CB2R antagonist SR144528 injected simultaneously reversed the effects of O-1966.
To further assure that the effects of the CB2 agonist are indeed mediated by the CB2R, we used CB2R −/− animals. Figure 4A shows the genotype of the animals used. The knock-out was generated by targeted gene disruption (insertion) of the Cnr2 gene (Basu and Dittel, 2011). The presence of a 385 bp band corresponds to the wild-type allele, whereas the presence of a 550 bp band corresponds to the mutant allele. Animals that lack the expression of the CB2R show only the 550 bp mutant allele and not the wild-type allele (Fig. 4A). CB2R knock-out mice were implanted with cranial windows, and leukocyte adhesion was observed after LPS injection with either CB2R agonist, JWH133 or O-1966. At both time points, the CB2R knock-out animals treated with LPS only had a similar level of leukocyte adhesion as wild-type animals, resulting in ∼30-fold increase by 4 h and 18-fold by 24 h (Fig. 4B,C) compared to baseline. However, complete resolution of inflammation after the single exposure of LPS did take longer in the knock-out animals, extending to 72 h compared to 48 h for the wild-type (data not shown). As expected, the CB2R agonists had no effect on minimizing leukocyte–endothelial cell interaction in the CB2R knock-out animals (Fig. 4B,C). Therefore, the effects of either JWH133 or O-1966 appear to be entirely dependent on the presence of the CB2 receptor.
Figure 4.
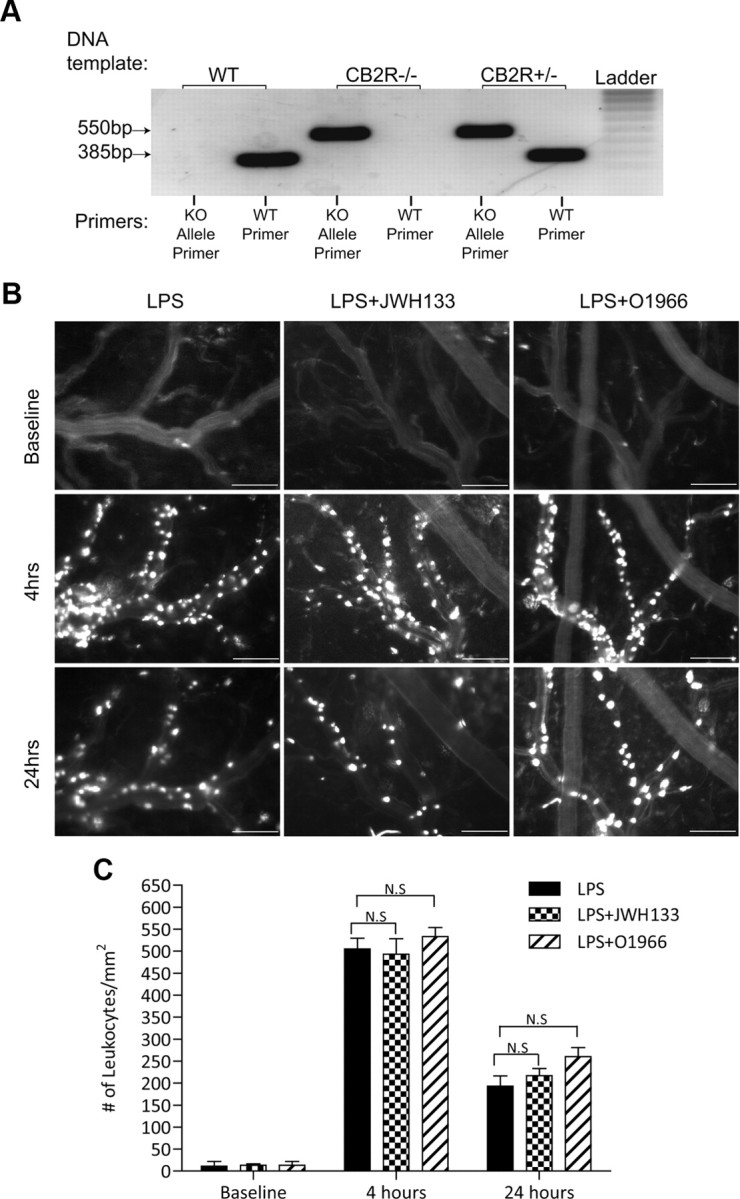
CB2R agonists are ineffective at reducing immune–endothelial interaction during neuroinflammation in CB2 receptor knock-out (KO) animals. A, Standard genotype PCR for the Cnr2-targeted mutation in wild-type (WT), heterozygous, and CB2R knock-out animals. A specific primer set for the normal allele shows a 385 bp fragment where as the primer set that amplifies the insertional mutation appears at 550 bp. CB2R knock-out animals show only the mutant allele and not the wild-type allele. B, Shows representative images from intravital microscopy analysis of adherent leukocytes in CB2R knock-out animals that were administered LPS (6 mg/kg, i.p.) alone, LPS with JWH133 (10 mg/ml, i.p.), or LPS with O-1966 (5 mg/ml, i.p.). Scale bars: 100 μm. C, Measurements calculated from image and video analysis of immune cells attached to the brain endothelium during the indicated time points. The results are shown as the mean adhesion ± SEM (n = 3). N.S., Not significant.
CB2R activation decreases leukocyte adhesion in deep cortical ascending postcapillary venules
Since the brain vasculature differs depending on neuroanatomical location, it is possible that the observation made on surface pial vessels with the CB2R agonist may not apply to vessels that are part of the neurovascular unit (Owens et al., 2008). Because conventional wide-field fluorescence microscopy is limited with respect to the depth to which an object can be imaged, analysis of leukocyte adhesion in brain parenchymal vessels was visualized with multiphoton microscopy. Animals were prepared for intravital microscopy as before but without the dura removal, and animals were treated as indicated (Fig. 5). Imaging was performed down to 800 μm below the surface of the cortex on postcapillary venules that ranged between 20 and 30 μm in diameter. Leukocytes and the vasculature were concomitantly imaged with rhodamine 6G and high-molecular-weight FITC-dextrans. Volume acquisition (z stack) and time (t series) of ascending venules were rendered in 3D and analyzed for the number of attached leukocytes. Using tracking software, the distinction between adhesion and rolling was discerned by evaluating the four-dimensional movement of leukocytes inside the vessels. Figure 5A (top) shows the lack of adhesion in untreated animals, with streaking leukocytes in the lumen. As expected, the LPS insult revealed significant immune–endothelial interaction (Fig. 5A, middle), with red-stained leukocytes attached to the endothelium. Introduction of O-1966 with LPS reduced the number of adherent cells, thus confirming the previous results in surface vessels (Figure 5A, bottom). Of note, similar results were achieved with JWH133 (data not shown). Figure 5B provides a quantitative assessment of the findings from multiphoton microscopy.
Figure 5.
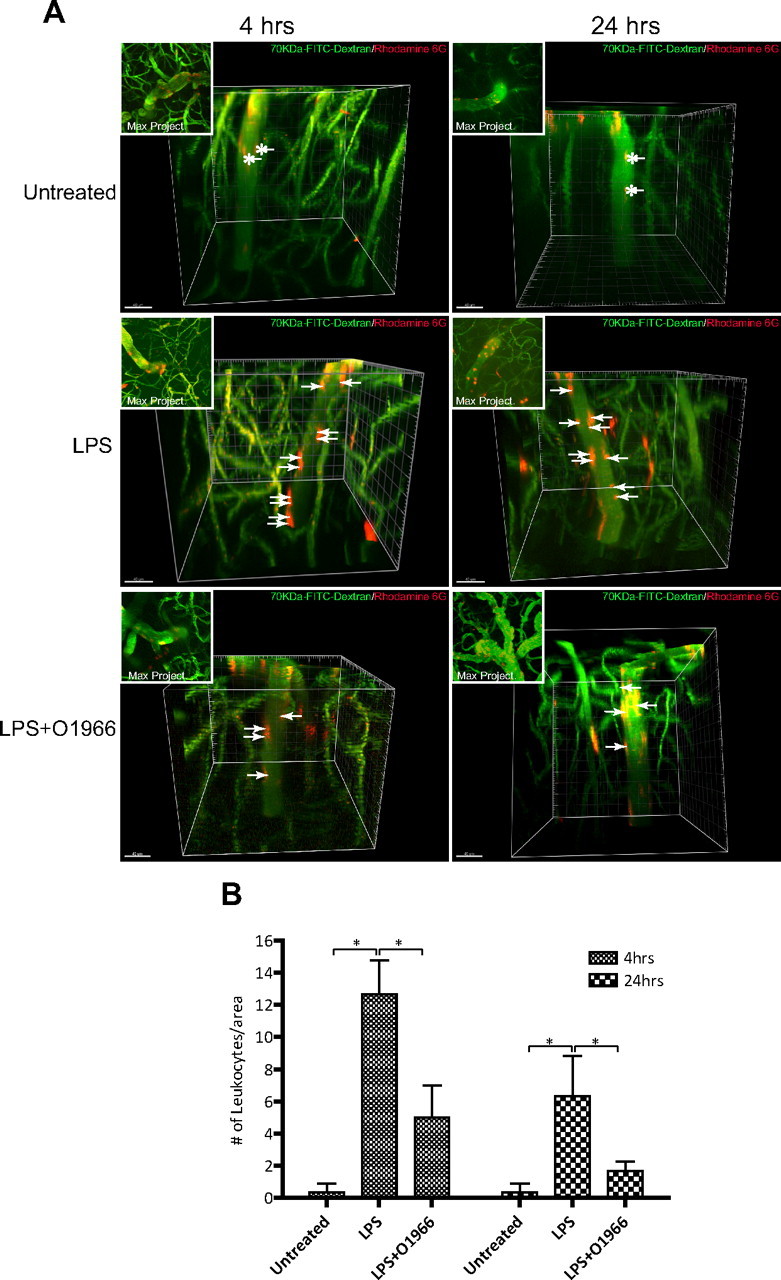
Resorcinol-based CB2R agonist inhibits leukocyte adhesion in ascending postcapillary venules. A, Representative images of 3D reconstructions of z stacks taken by multicolor two-photon intravital microscopy using animals with the indicted experimental conditions. The intraparenchymal microvasculature of the brain cortex (down to 800 μm below the cortex surface) was outlined by intravenous injection of 70 kDa FITC-conjugated dextrans (green). Leukocytes were fluorescently labeled by intravenous injection of rhodamine 6G (red). Leukocyte adhesion events were identified in ascending postcapillary venules ranging from 20 to 30 μm in diameter. In untreated control animals, only moving leukocytes were apparent (streaking cells; asterisks). Animals where LPS (6 mg/kg) was administered showed significant leukocyte adhesion (arrows). Insets, Maximum projection composites providing a top view of the z stack. Scale bars: 40 μm. B, The results of particle counting, providing a measure of the number of attached leukocytes per vessel surface area (assuming cylindrical dimensions and using 100 μm of vessel length). The results are shown as the mean adhesion ± SEM (n = 3). *p < 0.001 [difference between the groups compared (bracket)].
CB2R agonists prevent the upregulation of adhesion molecules in activated brain endothelial cells
Adhesion molecules play an essential role in the engagement of immune cells to the endothelium. Therefore, the interfering effects on adhesion seen with these pharmacological ligands of CB2R may be the result of a lower expression profile of adhesion molecules, such as ICAM-1 and vascular cell adhesion molecule-1 (VCAM-1). To test this possibility, immunolabeling for ICAM-1 and VCAM-1 was performed in brain tissue sections from control, LPS-treated, or LPS/O-1966-treated animals. Shown in Figure 6A are representative images from low- (20×) and high-magnification (100×) images of ICAM-1 immunolabeling of cortical brain regions. Although present, ICAM-1 basal expression is low in the brain endothelium (Fig. 6A,B). The presence of LPS clearly shows the upregulation in ICAM-1 at 4 h (the time point of maximal leukocyte adhesion in this model). When LPS was coadministered with O-1966, ICAM-1 immunoreactivity was considerably lower and close to the levels observed under basal conditions.
Figure 6.

Inhibition of ICAM-1 and VCAM-1 expression by CB2R agonist in activated brain endothelium. A, Immunofluorescence histology was performed on frozen brain tissue sections (15 μm thick) from animals treated as indicated. The sections were stained with DAPI and immunolabeled for ICAM-1. Representative images at 20× and 100× objective power are shown. Sections from animals with LPS-associated encephalitis displayed a high level of immunopositive staining for ICAM-1 in the vasculature (arrows) compared to the marginally positive staining in the control animals (middle and top, respectively). Sections from animals where simultaneous introduction of O-1966 and LPS was used showed levels of ICAM-1 similar to the untreated control (bottom). Scale bars: 20× magnification, 100 μm; 100× magnification, 25 μm. B, Brain microvessels were isolated, and Western blots for ICAM-1, CD31, and β-actin were performed. Corresponding densitometry results are shown as the fold change from the untreated control (mean ± SD). C, Adhesion assays performed with primary human BMVECs and primary human monocytes as described in Materials and Methods. BMVECs were exposed to TNFα (20 ng/ml) alone or with increasing concentrations of 1–50 μm of either CB2R agonist, JWH133 or O-1966. BMVECs were treated for 4 h, and the treatments were removed before the addition of monocytes. The data are represented as the fold difference (mean ± SEM) of adhesion, which is the number of adherent cells from the treated condition divided by the basal adhesion in untreated cells. All data collected were from at least three different BMVEC donors and performed in triplicate. D, Representative histograms of FACS analyses of adhesion molecules ICAM-1 and VCAM-1 on brain endothelial cells after TNFα (20 ng/ml) or LPS (50 ng/ml) exposure with the presence of CB2R agonist O-1966. TNFα and LPS induced similar surface expression of ICAM-1 and VCAM-1 after 4 h exposure. The upregulation of adhesion molecules induced by TNFα or LPS was suppressed by O-1966. The data are represented as the MFI of ICAM-1 or VCAM-1 as a percent of the maximum intensity. For all the graphs, the asterisk denotes statistical significance (p < 0.05).
Next, we performed experiments in primary human BMVECs to (1) determine whether the observations in vivo could be confirmed in human brain endothelial cells, (2) isolate the effects of the CB2R agonist on endothelial cells from leukocytes, and (3) determine whether the same effects on adhesion molecule expression are also observed. BMVEC monolayers were stimulated with TNFα (20 ng/ml) for 4 h with increasing concentrations of JWH133 or O-1966. Purified primary human monocytes were placed on the BMVECs to initiate adhesion only after all treatments were removed and the medium was changed. Unlike the in vivo experiments, the change in medium before the monocyte introduction allows for the evaluation of the effects of the CB2R agonist on BMVECs only. As shown in Figure 6C, the results from the adhesion assays indicate that human brain endothelial cells respond similarly to CB2R agonists in preventing immune cell adhesion. In addition, the effects with either CB2R agonist are dose dependent for the reduction in adhesion. Exposure to TNFα induced an average fourfold increase in the number of monocytes attaching to the BMVEC monolayers. O-1966 diminished monocyte adhesion from 2.8- to 1.5-fold, whereas JWH133 was ineffective at the lowest concentration, attenuating adhesion from 2.8- to 2.1-fold at the higher doses.
To confirm whether a similar decrease in adhesion molecules can also be observed in human BMVECs treated with CB2R agonist, FACS analysis was performed. BMVECs were treated as indicated (Fig. 6D), and surface expression of ICAM-1 and VCAM-1 was evaluated.
The cells were stained using antibodies to ICAM-1, VCAM-1, or isotype-matched nonimmune antibodies. Treatment of BMVECs with TNFα (Fig. 6D, top) resulted in 5- and 10-fold increases in ICAM-1/VCAM-1 mean fluorescence intensity (MFI) for TNFα and LPS, respectively. The upregulation of ICAM-1 and VCAM-1 by either inflammatory insult was nearly completely inhibited by the addition of O-1966. Thus, CB2R activation during inflammatory insult can regulate the expression profile of adhesion molecules leading to the inhibition in immune–endothelial interaction.
CB2R agonists prevent BBB dysfunction
Loss of BBB integrity results in the leakiness of blood components from the vessel lumen into the brain parenchyma. LPS exposure induces an acute opening of the BBB, which is also potentiated by the presence of inflammatory mediators and activated immune cells. To assess permeability, animals were injected intravenously with fluorescein-labeled 70 kDa dextran, and extravasation was visualized by intravital microscopy. Figure 7A shows that there was no leakage of FITC-dextran from the cerebral vessels at baseline. In contrast, 4 h after LPS injection, animals had considerable vascular permeability, which was greatly attenuated by the presence of O-1966 (Fig. 7A). To arrive at a more precise quantitative comparison of permeability, animals were injected with the small molecular tracer Na-F (376 Da), and the brain tissue content of Na-F was measured (Fig. 7B). Tracer content in the whole brain was elevated by threefold in the LPS condition compared to naive animals. Animals administered with either O-1966 or JWH133 showed reduced LPS-enhanced permeability by 50% when compared to the LPS animals. Taken together, the results indicate that CB2R agonist provides a barrier protective effect, thus limiting the degree of permeability that is induced during inflammation.
Figure 7.
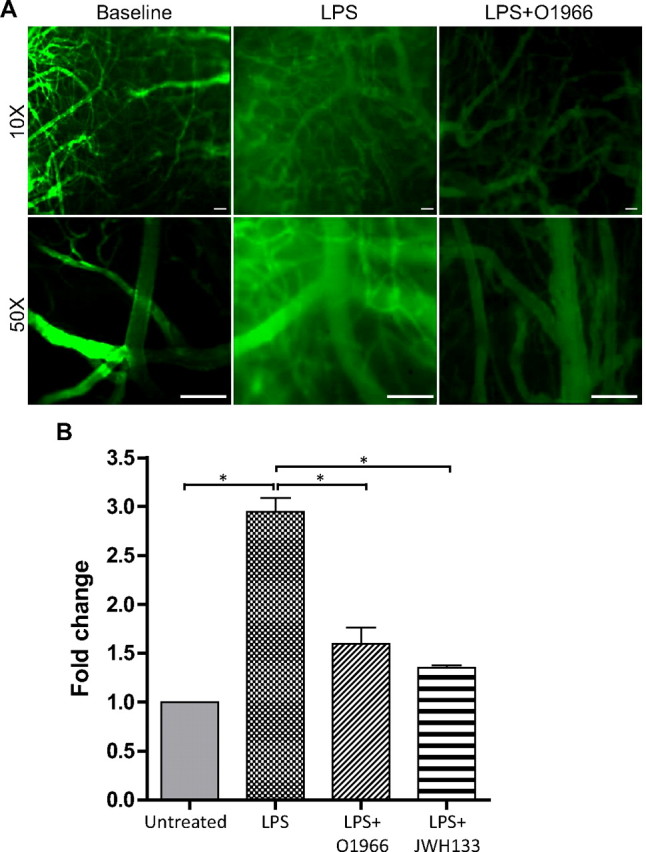
CB2R agonists maintain BBB integrity in vivo. LPS induction of BBB leakiness in vivo was visualized by FITC-dextran extravasation or measured by whole-brain Na-F content. A, Images from intravital microscopy show no dextran permeability out of the vessel at baseline, whereas LPS significantly induced dextran leakage into perivascular spaces. The dextran leakage was attenuated by the treatment with O-1966. Images were taken at low magnification (10×) and high magnification (50×). Scale bars: 100 μm. B, BBB permeability was evaluated by administration of the tracer Na-F in saline via an intraperitoneal route as described in Materials and Methods. Following perfusion, the content of Na-F in homogenized brain tissue was measured. LPS significantly increased Na-F leakage into the brain parenchyma compared to untreated mice, which was attenuated by treatment with O-1966 or JWH133. The results are expressed in fold change from calculating the relative fluorescence (RFU) in milligrams of brain tissue and dividing the treated by the untreated. The graph shows the results as the average ± SEM. *p < 0.001.
CB2R agonists improve BBB function by promoting TJ protein increase in membrane fractions
Next, we sought to determine whether a CB2R agonist could protect BBB integrity after disruption by proinflammatory stimulus (LPS). Assessment of BBB leakiness and integrity can be measured via TEER. BMVECs were grown on ECIS array electrodes, and TEER was monitored continuously over the course of 24 and 36 h (Fig. 8A–C). Treatment of BMVEC monolayers with LPS (50 ng/ml) induced a rapid drop in resistance (80% of control) over time, which was significantly attenuated by O-1966 (Fig. 8A; p < 0.05). These data indicate that CB2R stimulation can provide BBB protection under inflammatory conditions, supporting our in vivo observations (prevention of BBB disruption after LPS injection in mice).
Figure 8.
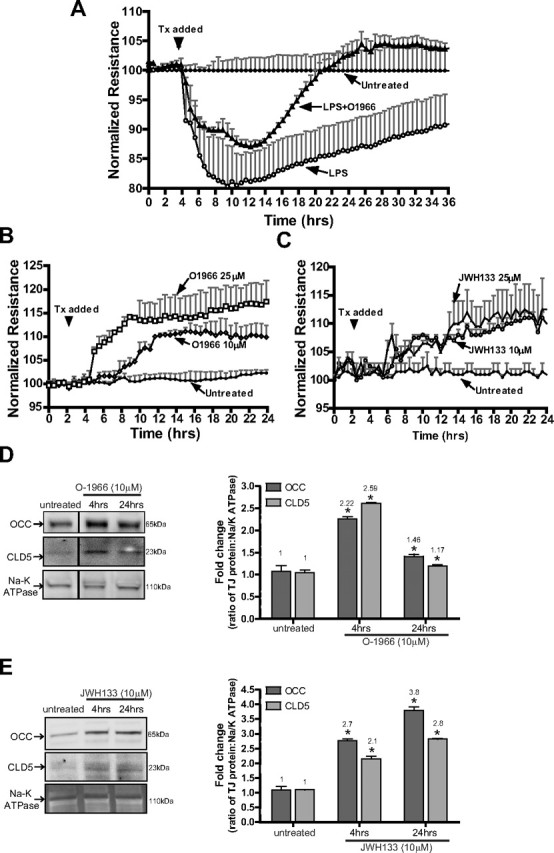
CB2R agonist protects and enhances barrier function in human BMVECs. A, TEER, an indicator of barrier integrity, was measured (by ECIS) in monolayers untreated or treated with either LPS (50 ng/ml) or LPS with O-1966 (10 μm). The resistance was measured at 4 kHz in 30 min intervals for the duration of the time shown. Treatments (Tx) were initiated (arrow) after stable resistance was reached. The data are presented as the percentage change from baseline, which is the resistance measured after treatment divided by the resistance acquired before treatment introduction. B, TEER measurements in BMVECs treated with low (10 μm) and high (25 μm) concentrations of O-1966. C, TEER measurements in BMVECs treated with low (10 μm) and high (25 μm) concentrations of JWH133. CB2R agonists induced a further tightening of the barrier, evident by a rise in the degree of resistance above the basal level (B, C). D, E, Western blots of BMVEC membrane fractions show the tight junction protein levels of occludin (OCC) and claudin-5 (CLD5) in untreated BMVECs and BMVECs treated with O-1966 (D) or JWH133 (E) for 4 h. Densitometry values, indicated to the right, were calculated using the Na/K ATPase loading control, and the data are represented as the fold change from the untreated cells. The asterisk denotes statistical significance (p < 0.05).
Our next set of experiments was designed to understand whether CB2R activation could increase barrier structural integrity under physiologic conditions. Application of the highly selective CB2R agonists (O-1966, JWH133) resulted in dose- and time-dependent augmentation of tightness of BMVEC monolayers (11–15%, p < 0.05; Fig. 8B,C). Although various signaling mechanisms can influence BBB tightness, the culminating effect is on TJ proteins located between brain endothelial cells assuring tightness of the BBB. Therefore, TJ protein amount in BMVEC membrane fractions was evaluated after treatment with CB2R agonists. Four hour stimulation of CB2R with O-1966 or JWH133 caused a 2.2- to 2.7-fold increase in the amount of occludin and claudin-5 (Fig. 8D,E) present in the membranous fraction of BMVEC lysates. Longer exposure to JWH133 resulted in further increases in occludin (3.8-fold) and claudin-5 (2.8-fold), while O-1966 increased occludin only by 50% and claudin-5 by 20% when compared to untreated controls. In summary, these results suggest CB2R protective effects on BBB function are mediated in part by the increased presence of TJ proteins in the membrane fraction of brain endothelium.
Discussion
Previous studies have shown that activation of CB2R by pharmacological ligands promotes anti-inflammatory outcomes. Although suppression of inflammatory responses resulting from CB2R activation has been observed in various cell types, little is known about how it affects the brain endothelium or the BBB. Here we provide compelling evidence that CB2R ligands, including a novel resorcinol-based agonist, not only decrease immune–endothelial interactions, but also render direct BBB protection during neuroinflammation.
Previous studies indicated that CB2R expression was restricted to peripheral tissues, particularly in immune cells (Cabral and Griffin-Thomas, 2009). However, it is now known that CB2R can also be found in cells of the CNS, but predominantly as a consequence of neuroinflammation (for review, see Benito et al., 2008). Thus far, this augmentation in CB2R expression has been reported to be largely localized to microglial cells and perivascular macrophages. In this study, our findings point to the induction of CB2R in yet another cell type, the brain endothelial cell. Under normal physiological conditions, the vessels of the brain express very low levels of CB2R (Schley et al., 2009). Basal expression of CB2R in endothelial cells has also been observed in vitro, including in BMVECs (Mestre et al., 2006), human umbilical vein endothelial cells, and pulmonary artery endothelial cells (Haraldsen et al., 1996; Zoratti et al., 2003). Our observations confirm this expression; however, analysis of brain tissue sections from cases of HIV-1 encephalitis revealed a strong upregulated immunopositive staining for CB2R on the vasculature. To our knowledge, this is the first demonstration of increased CB2R levels in brain endothelium during neuroinflammation. Another study also showed an increase in CB2R in vessels of the brain, but only in proliferating microvessels of glioblastomas (Schley et al., 2009). Given the complexity of the inflammatory response within the neurovascular unit, we sought to determine whether isolated human BMVECs would also upregulate CB2R by proinflammatory factors. Indeed, all tested inflammatory insults enhanced CB2R in the cultured cells with varying degrees of expression. Whether the inducibility of CB2R triggered by inflammation is part of the pathogenesis process or part of the mechanism to resolve the injury remains unclear. Evidence for the latter suggests that endothelial production of endocannabinoids after inflammatory insult may act to regulate endothelial activation (Golech et al., 2004). In any case, the presence of upregulated CB2R offers the opportunity to target the receptor with synthetic and highly selective CB2R agonists to dampen neuroinflammation.
To characterize the effects of CB2R on immune–endothelial interaction and BBB function, we used the CB2R agonists JWH133 and O-1966 (Wiley et al., 2002). O-1966 is a novel synthetic dimethoxy-resorcinol-dimethylheptyl analog that has approximately a 225-fold greater CB2R selectivity (Ki = 22.5 nm) over CB1R (Wiley et al., 2002). Using LPS-induced encephalitis and intravital microscopy, we showed that leukocyte adhesion in cerebral vessels was significantly reduced after JWH133 or O-1966 administration. Of note, only half the dose of O-1966 was required to attenuate immune cell adhesion compared to JWH133 (5 vs 10 mg/kg, respectively). Interestingly, a recent study using cannabidiol, a CB1R antagonist and a CB2R inverse agonist, in LPS-associated encephalitis was shown to decrease leukocyte adhesion and vasodilatation (Ruiz-Valdepeñas et al., 2011). Given that the brain endothelium also expresses the CB1R (Golech et al., 2004), it remains unknown which receptor provides the endothelial protective effects with cannabidiol. In fact, the effects reported here with JWH133 or O-1966 are specific to CB2R since both compounds were ineffective when coadministered in the presence of CB2R antagonist SR144528 or when used in CB2R knock-out mice. The changes in adhesion with CB2R agonists were noted both in pial vessels and deep cortical ascending post capillary venules, the sites of most leukocyte–endothelial cell interaction during neuroinflammation (Owens et al., 2008). Evaluation of CB2R agonists in surface and deep cerebral vessels was important because surface pial vessels do not have an integrated neurovascular unit, and thus anti-inflammatory responses differ between the two sites (Persidsky et al., 2006a).
Previous studies in ischemia/reperfusion injury and experimental autoimmune encephalomyelitis have shown the effectiveness of CB2R agonists to reduce leukocyte adhesion (Zhang et al., 2009b; Murikinati et al., 2010). However, these investigations did not address whether the effects of CB2R apply only to the immune cell, the endothelial cell, or both, given that both cell types express CB2R. Consistent with previous reports (Yamashita et al., 2000), administration of LPS induces massive cytokine production and elevated levels of adhesion molecule expression. Histological examination revealed a low basal level in ICAM-1 expression in cortical vessels and the expected induction in ICAM-1 expression after LPS (Fig. 6). The novel CB2R agonist, O-1966, prevented the induction of ICAM-1. Thus, the reduction in leukocyte adhesion seen in the intravital analysis was due in part to the lack of firm attachment between endothelial adhesion molecules and immune cell integrins. Interestingly, it is possible that intercellular adhesion is abrogated by CB2R signaling even before adhesion (i.e., rolling), since along with adhesion, rolling (data not shown) was also reduced in animals with both LPS and agonist. Therefore, further analyses need be performed to identify whether regulation of selectins may also be a target of CB2R activation. These results complement previous work in human coronary artery endothelial cells in which induction of ICAM-1 and VCAM-1 expression was also attenuated following LPS and JWH133 treatment (Rajesh et al., 2007). Therefore, signaling events triggered by CB2R agonists that prevent the increase in surface expression of adhesion molecules may apply universally to endothelial cells of brain and nonbrain origin.
Undoubtedly, activated immune cells are affected by CB2R agonists in many ways, including cytokine production and adhesion/migration ability (Pacher and Steffens, 2009). For instance, adoptive transfer of immune cells exposed to the CB2R agonist JWH133 prevented the attachment and transendothelial migration of leukocytes in inflamed retinas (Xu et al., 2007). In an attempt to uncouple the effects of CB2R agonists on both immune and endothelial cells, we continued our investigations in vitro with primary human BMVECs. Therefore, BMVECs were activated with cytokines in the presence of CB2R agonist, and then the agonist was removed and adhesion with human monocytes evaluated. Our experiments revealed a dose-dependent decrease in monocyte adhesion to cytokine activated endothelium when CB2R agonists were present. Flow cytometry confirmed that the decrease in monocyte adhesion was due to the CB2R-mediated diminution of ICAM-1 and VCAM-1 surface expression induced by LPS or TNFα (Fig. 6). The results presented clarify that these effects are not only one sided (i.e., immune cells), but that the brain endothelium also clearly responds to CB2R agonists and prevents the induction of adhesion molecules under inflammatory insult.
BBB permeability is increased in neuroinflammation and is one of the contributing factors associated with neurodegeneration. Distinct from the vascular beds in the periphery, the vasculature of the brain features a high concentration of intercellular tight junctions that form the BBB (Abbott, 2005). To study the role of CB2R in BBB function, we assessed BBB permeability in vivo. We examined whether a breach in the BBB, as a result of LPS-induced encephalitis, could be abrogated. We observed that CB2R agonists did maintain barrier integrity as visualized by the absence of fluorescence-labeled high-molecular-weight dextrans (70 kDa) entering the brain parenchyma (Fig. 7). Since BBB permeability evaluations with large molecular tracers may fail to detect permeability to smaller molecules, we also used a low-molecular-weight tracer. Although significantly reduced, global assessment of BBB permeability with sodium fluorescein (376 Da) showed that permeability still remained at half a fold higher than the basal level. Perhaps this is due to the low number of leukocytes that still remained adherent. Our previous work clearly demonstrates the effect that leukocyte adhesion/engagement has on barrier tightness (Ramirez et al., 2009). To correlate our in vivo results, we used BMVECs and transendothelial electrical resistance (Bernas et al., 2010). The measurements showed the expected sustained decrease in TEER after LPS; conversely, O-1966 was able to prevent the full drop in resistance and allowed the barrier to rebound back to normal levels more rapidly (Fig. 8). Surprisingly, the protection in barrier integrity was achieved beyond the anti-inflammatory effects of CB2R, since the TEER showed a further enhancement of barrier tightness with just the CB2R agonist present. These results are reminiscent of other barrier-enhancing agents such as steroids, cAMP, or growth factors (Liu et al., 2002; Helms et al., 2010; Kashiwamura et al., 2011). We also found that the barrier-enhancing effects could be explained in part by the augmentation in the TJ proteins, occludin and claudin-5, in membrane fractions of CB2R agonist treated BMVECs. Together, these analyses provide compelling evidence that CB2R agonists, independent of their anti-inflammatory effects, promote and enhance BBB integrity.
At present, it is unclear how CB2R signaling cascades may act on both the TJ complex and inflammatory pathways. It is possible that CB2R activation in BMVECs, as has been shown in other cell types, may affect MAPK signaling that promotes the production of anti-inflammatory mediators such as IL-10 (for review, see Basu and Dittel, 2011). Whether concurrently or sequentially, it is also possible that CB2R activation inhibits pathways that alter normal TJ protein expression or stability. One of the pathways described previously to be critical for tight junction protein stability is the Rho GTPase pathway (Persidsky et al., 2006b). Specifically, RhoA activity negatively impacts the stability of the TJ complex. This is a reasonable possibility in light of a recent report by Kurihara et al. (2006) describing that in T-cells the activity of RhoA decreases in response to CB2R stimulation. Alternatively, CB2R signaling may attenuate oxidative stress signals that are known (for example, via matrix metalloproteinases) to affect the status of tight junction proteins (Lischper et al., 2010; Batkai et al., 2011). Undoubtedly future investigations will help shed light on how CB2R signaling events in the BMVECs contribute to anti-inflammation and barrier protection.
In summary, CB2R receptors may provide a therapeutic target, not only for their effects on attenuating immune cell activation, but also in protecting the BBB during neuroinflammation. Specifically, our findings demonstrate that CB2R agonists can dampen the activation of brain endothelial cells, while preventing barrier disruption via induction of membrane-bound TJ protein presence. Our findings also have much broader pathophysiological implications, indicating that the cannabinoid system is an integral part of inflammatory responses in endothelial cells.
Footnotes
This work was supported in part by NIH Grants RO1MH065151 R01DA025566, and R37AA015913 (Y.P.) and a Temple University development grant (S.H.R.). J.H. was supported by a fellowship from the Hungarian-American Enterprise Scholarship Fund. We acknowledge the NIH National NeuroAIDS Consortium for brain tissue specimens used in this study. We thank Dr. Ellen Unterwald (Temple University School of Medicine) for assistance with the CB2R knock-out animals.
References
- Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5–23. doi: 10.1007/s10571-004-1374-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res. 2011;51:26–38. doi: 10.1007/s12026-011-8210-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batkai S, Mukhopadhyay P, Horvath B, Rajesh M, Gao RY, Mahadevan A, Amere M, Battista N, Lichtman AH, Gauson LA, Maccarrone M, Pertwee RG, Pacher P. Delta(8)-tetrahydrocannabivarin protects against hepatic ischemia/reperfusion injury by attenuating oxidative stress and inflammatory response involving CB(2) receptors. Br J Pharmacol. 2011 doi: 10.1111/j.1476-5381.2011.01410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito C, Kim WK, Chavarria I, Hillard CJ, Mackie K, Tolón RM, Williams K, Romero J. A glial endogenous cannabinoid system is upregulated in the brains of macaques with simian immunodeficiency virus-induced encephalitis. J Neurosci. 2005;25:2530–2536. doi: 10.1523/JNEUROSCI.3923-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito C, Romero JP, Tolón RM, Clemente D, Docagne F, Hillard CJ, Guaza C, Romero J. Cannabinoid CB1 and CB2 receptors and fatty acid amide hydrolase are specific markers of plaque cell subtypes in human multiple sclerosis. J Neurosci. 2007;27:2396–2402. doi: 10.1523/JNEUROSCI.4814-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito C, Tolón RM, Pazos MR, Núñez E, Castillo AI, Romero J. Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008;153:277–285. doi: 10.1038/sj.bjp.0707505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernas MJ, Cardoso FL, Daley SK, Weinand ME, Campos AR, Ferreira AJ, Hoying JB, Witte MH, Brites D, Persidsky Y, Ramirez SH, Brito MA. Establishment of primary cultures of human brain microvascular endothelial cells to provide an in vitro cellular model of the blood-brain barrier. Nat Protoc. 2010;5:1265–1272. doi: 10.1038/nprot.2010.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral GA, Griffin-Thomas L. Emerging role of the cannabinoid receptor CB2 in immune regulation: therapeutic prospects for neuroinflammation. Expert Rev Mol Med. 2009;11:e3. doi: 10.1017/S1462399409000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogrukol-Ak D, Kumar VB, Ryerse JS, Farr SA, Verma S, Nonaka N, Nakamachi T, Ohtaki H, Niehoff ML, Edwards JC, Shioda S, Morley JE, Banks WA. Isolation of peptide transport system-6 from brain endothelial cells: therapeutic effects with antisense inhibition in Alzheimer and stroke models. J Cereb Blood Flow Metab. 2009;29:411–422. doi: 10.1038/jcbfm.2008.131. [DOI] [PubMed] [Google Scholar]
- Golech SA, McCarron RM, Chen Y, Bembry J, Lenz F, Mechoulam R, Shohami E, Spatz M. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res Mol Brain Res. 2004;132:87–92. doi: 10.1016/j.molbrainres.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Greenwood J, Etienne-Manneville S, Adamson P, Couraud PO. Lymphocyte migration into the central nervous system: implication of ICAM-1 signalling at the blood-brain barrier. Vascul Pharmacol. 2002;38:315–322. doi: 10.1016/s1537-1891(02)00199-4. [DOI] [PubMed] [Google Scholar]
- Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P. Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells. J Immunol. 1996;156:2558–2565. [PubMed] [Google Scholar]
- Hawkins BT, Egleton RD. Fluorescence imaging of blood-brain barrier disruption. J Neurosci Methods. 2006;151:262–267. doi: 10.1016/j.jneumeth.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Helms HC, Waagepetersen HS, Nielsen CU, Brodin B. Paracellular tightness and claudin-5 expression is increased in the BCEC/astrocyte blood-brain barrier model by increasing media buffer capacity during growth. AAPS J. 2010;12:759–770. doi: 10.1208/s12248-010-9237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwamura Y, Sano Y, Abe M, Shimizu F, Haruki H, Maeda T, Kawai M, Kanda T. Hydrocortisone enhances the function of the blood-nerve barrier through the up-regulation of claudin-5. Neurochem Res. 2011;36:849–855. doi: 10.1007/s11064-011-0413-6. [DOI] [PubMed] [Google Scholar]
- Kurihara R, Tohyama Y, Matsusaka S, Naruse H, Kinoshita E, Tsujioka T, Katsumata Y, Yamamura H. Effects of peripheral cannabinoid receptor ligands on motility and polarization in neutrophil-like HL60 cells and human neutrophils. J Biol Chem. 2006;281:12908–12918. doi: 10.1074/jbc.M510871200. [DOI] [PubMed] [Google Scholar]
- Lischper M, Beuck S, Thanabalasundaram G, Pieper C, Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res. 2010;1326:114–127. doi: 10.1016/j.brainres.2010.02.054. [DOI] [PubMed] [Google Scholar]
- Liu F, Schaphorst KL, Verin AD, Jacobs K, Birukova A, Day RM, Bogatcheva N, Bottaro DP, Garcia JG. Hepatocyte growth factor enhances endothelial cell barrier function and cortical cytoskeletal rearrangement: potential role of glycogen synthase kinase-3beta. FASEB J. 2002;16:950–962. doi: 10.1096/fj.01-0870com. [DOI] [PubMed] [Google Scholar]
- Lu TS, Avraham HK, Seng S, Tachado SD, Koziel H, Makriyannis A, Avraham S. Cannabinoids inhibit HIV-1 Gp120-mediated insults in brain microvascular endothelial cells. J Immunol. 2008;181:6406–6416. doi: 10.4049/jimmunol.181.9.6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre L, Correa F, Docagne F, Clemente D, Guaza C. The synthetic cannabinoid WIN 55,212–2 increases COX-2 expression and PGE2 release in murine brain-derived endothelial cells following Theiler's virus infection. Biochem Pharmacol. 2006;72:869–880. doi: 10.1016/j.bcp.2006.06.037. [DOI] [PubMed] [Google Scholar]
- Mestre L, Docagne F, Correa F, Loría F, Hernangómez M, Borrell J, Guaza C. A cannabinoid agonist interferes with the progression of a chronic model of multiple sclerosis by downregulating adhesion molecules. Mol Cell Neurosci. 2009;40:258–266. doi: 10.1016/j.mcn.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Miller AM, Stella N. CB2 receptor-mediated migration of immune cells: it can go either way. Br J Pharmacol. 2008;153:299–308. doi: 10.1038/sj.bjp.0707523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murikinati S, Jüttler E, Keinert T, Ridder DA, Muhammad S, Waibler Z, Ledent C, Zimmer A, Kalinke U, Schwaninger M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010;24:788–798. doi: 10.1096/fj.09-141275. [DOI] [PubMed] [Google Scholar]
- Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, Engelhardt B, Grammas P, Nedergaard M, Nutt J, Pardridge W, Rosenberg GA, Smith Q, Drewes LR. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7:84–96. doi: 10.1016/S1474-4422(07)70326-5. [DOI] [PubMed] [Google Scholar]
- Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol. 2008;67:1113–1121. doi: 10.1097/NEN.0b013e31818f9ca8. [DOI] [PubMed] [Google Scholar]
- Pacher P, Steffens S. The emerging role of the endocannabinoid system in cardiovascular disease. Semin Immunopathol. 2009;31:63–77. doi: 10.1007/s00281-009-0145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006a;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across blood-brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006b;107:4720–4730. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Ho W, Ramirez SH, Potula R, Abood ME, Unterwald E, Tuma R. HIV-1 infection and alcohol abuse: neurocognitive impairment, mechanisms of neurodegeneration and therapeutic interventions. Brain Behav Immun. 2011;25(Suppl 1):S61–S70. doi: 10.1016/j.bbi.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Bátkai S, Haskó G, Liaudet L, Huffman JW, Csiszar A, Ungvari Z, Mackie K, Chatterjee S, Pacher P. CB2-receptor stimulation attenuates TNF-alpha-induced human endothelial cell activation, transendothelial migration of monocytes, and monocyte-endothelial adhesion. Am J Physiol Heart Circ Physiol. 2007;293:H2210–H2218. doi: 10.1152/ajpheart.00688.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Heilman D, Morsey B, Potula R, Haorah J, Persidsky Y. Activation of peroxisome proliferator-activated receptor gamma (PPARgamma) suppresses Rho GTPases in human brain microvascular endothelial cells and inhibits adhesion and transendothelial migration of HIV-1 infected monocytes. J Immunol. 2008;180:1854–1865. doi: 10.4049/jimmunol.180.3.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Potula R, Fan S, Eidem T, Papugani A, Reichenbach N, Dykstra H, Weksler BB, Romero IA, Couraud PO, Persidsky Y. Methamphetamine disrupts blood-brain barrier function by induction of oxidative stress in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:1933–1945. doi: 10.1038/jcbfm.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Fan S, Dykstra H, Reichenbach N, Del Valle L, Potula R, Phipps RP, Maggirwar SB, Persidsky Y. Dyad of CD40/CD40 ligand fosters neuroinflammation at the blood-brain barrier and is regulated via JNK signaling: implications for HIV-1 encephalitis. J Neurosci. 2010a;30:9454–9464. doi: 10.1523/JNEUROSCI.5796-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Fan S, Zhang M, Papugani A, Reichenbach N, Dykstra H, Mercer AJ, Tuma RF, Persidsky Y. Inhibition of glycogen synthase kinase 3beta (GSK3beta) decreases inflammatory responses in brain endothelial cells. Am J Pathol. 2010b;176:881–892. doi: 10.2353/ajpath.2010.090671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Valdepeñas L, Martínez-Orgado JA, Benito C, Millán A, Tolón RM, Romero J. Cannabidiol reduces lipopolysaccharide-induced vascular changes and inflammation in the mouse brain: an intravital microscopy study. J Neuroinflammation. 2011;8:5. doi: 10.1186/1742-2094-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schley M, Ständer S, Kerner J, Vajkoczy P, Schüpfer G, Dusch M, Schmelz M, Konrad C. Predominant CB2 receptor expression in endothelial cells of glioblastoma in humans. Brain Res Bull. 2009;79:333–337. doi: 10.1016/j.brainresbull.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Beletskaya ID, Ng EW, Dai Z, Crocker PJ, Mahadevan A, Razdan RK, Martin BR. Resorcinol derivatives: a novel template for the development of cannabinoid CB(1)/CB(2) and CB(2)-selective agonists. J Pharmacol Exp Ther. 2002;301:679–689. doi: 10.1124/jpet.301.2.679. [DOI] [PubMed] [Google Scholar]
- Xu H, Cheng CL, Chen M, Manivannan A, Cabay L, Pertwee RG, Coutts A, Forrester JV. Anti-inflammatory property of the cannabinoid receptor-2-selective agonist JWH-133 in a rodent model of autoimmune uveoretinitis. J Leukoc Biol. 2007;82:532–541. doi: 10.1189/jlb.0307159. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Kawashima S, Ohashi Y, Ozaki M, Ueyama T, Ishida T, Inoue N, Hirata K, Akita H, Yokoyama M. Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation. 2000;101:931–937. doi: 10.1161/01.cir.101.8.931. [DOI] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Adler MW, Abood ME, Ganea D, Jallo J, Tuma RF. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc Res. 2009a;78:86–94. doi: 10.1016/j.mvr.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Martin BR, Adler MW, Razdan RJ, Kong W, Ganea D, Tuma RF. Modulation of cannabinoid receptor activation as a neuroprotective strategy for EAE and stroke. J Neuroimmune Pharmacol. 2009b;4:249–259. doi: 10.1007/s11481-009-9148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti C, Kipmen-Korgun D, Osibow K, Malli R, Graier WF. Anandamide initiates Ca(2+) signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br J Pharmacol. 2003;140:1351–1362. doi: 10.1038/sj.bjp.0705529. [DOI] [PMC free article] [PubMed] [Google Scholar]