

Abstract
Aurora kinases are serine/threonine protein kinases that are involved in cancer development and are important targets for cancer therapy. By high throughput screening of a chemical library we found that benzo[e]pyridoindole derivatives inhibited Aurora kinase. The most potent compound (compound 1) was found to be an ATP competitive inhibitor, which inhibited in vitro Aurora kinases at the nanomolar range. It prevented, ex vivo, the phosphorylation of Histone H3, induced mitosis exit without chromosome segregation, known phenomena observed upon Aurora B inactivation. This compound was also shown to affect the localization of Aurora B, since in the presence of the inhibitor the enzyme was delocalized on the whole chromosomes and remained associated with the chromatin of newly formed nuclei.
In addition, compound 1 inhibited the growth of different cell lines derived from different carcinoma. Its IC50 for H358 NSCLC (Non Small Cancer Lung Cells), the most sensitive cell line, was 145 nM. Furthermore compound 1 was found to be efficient towards multicellular tumour spheroid growth. It exhibited minimal toxicity in mice while it had some potency towards aggressive NSCLC tumours. Benzo[e]pyridoindoles represent thus a potential new lead for the development of Aurora kinase inhibitors.
Keywords: Animals; Cell Line, Tumor; Chromatin; metabolism; Chromosome Segregation; HeLa Cells; Histones; metabolism; Humans; Indoles; chemistry; pharmacology; Inhibitory Concentration 50; Mice; Phosphorylation; Protein Kinase Inhibitors; chemistry; pharmacology; Protein-Serine-Threonine Kinases; antagonists & inhibitors; metabolism; Pyridones; chemistry; pharmacology; Small Molecule Libraries
Keywords: Mitosis, Aurora kinase, small-molecule inhibitors, chromosomal passenger complex, pyridoindoles, mitotic slippage.
Introduction
Aurora kinases are a family of serine/threonine protein kinases that play a key role in mitosis progression (1, 2). Aurora A is found to be associated with both centrosomes and microtubules and it is required for centrosome duplication, entry into mitosis, formation of bipolar spindle and mitotic checkpoint (3–5). Aurora B exhibits typical passenger protein behaviour during mitosis. Initially, the kinase associates with centromeres, and, as mitosis proceeds, it relocates to the central spindle and the midbody. Aurora B is essential for chromosome condensation, kinetochore functions, spindle checkpoint activation and cytokinesis completion (2, 6–8).
Aurora A and B are over-expressed in many cancers, including primary colon and breast cancer (1, 9). Furthermore, the human Aurora A gene is localised to the 20q13 amplicon, which is associated with a poor prognosis in breast cancer (9). Xenografts of mouse NIH-3T3 cells overexpressing Aurora A give rise to tumours in nude mice, suggesting that Aurora A behaves as an oncogene (10). Under similar conditions, over-expression of Aurora B may induce metastasis (11). In the light of these observations, Aurora kinases have emerged as druggable targets for cancer therapy and thus, identification of Aurora kinase small molecule inhibitors is of particular interest (12–14).
Several Aurora A and B inhibitors, including ZM477439, Hesperadin, VX-680 (MK-0457), MLN8054, PHA-739358 have been described (15–20). Most of them have been included in clinical trials. VX-680, considered as the Aurora reference inhibitor, suppresses tumour growth in vivo and encouraging results were reported for three patients with refractory Chronic Myeloid Leukemia (21, 22) perhaps partly through the inhibition of the T315I mutant BCR-ABL (23).
The in vitro high throughput screening of the proprietary Institut Curie-CNRS small molecule library of 6560 compounds allowed the identification of benzo[e]pyridoindoles as new inhibitors of Aurora kinases. Ex vivo assays in HeLa cells showed that the identified best hit (compound 1, C1) inhibited the activity of Aurora B and mimicked the phenotype obtained after siRNA suppression of Aurora B expression. Experiments with several other tumour cell lines demonstrated that the C1 significantly affected tumour cell growth in either two- or three-dimensional cultures. C1 when used in the treatment of mice bearing H358 tumours with C1 suppressed tumour cell growth. Benzo[e]pyridoindoles are thus proposed as leads for the development of Aurora kinase inhibitors.
Materials and Methods
Recombinant proteins
Recombinant Histone H3 and kinase domain of Aurora A were expressed in E. coli and purified to homogeneity. VX-680 was purchased from Kava technology Inc, while paclitaxel and nocodazole were from Sigma.
Protein kinase assay
The protein kinase assay was performed in 20 mM Tris-HCl, 20 mM KCl, 20 mM MgCl2, 0.4 mM ATP, 0.4 mM DTT, pH 7.5. Recombinant histone H3 was used as substrate. The reaction started by the addition of the recombinant enzyme. After 1 hour of incubation at 37 °C the remaining ATP was monitored by addition of kinase-Glo™ (Promega, France) under the conditions suggested by the supplier. Ten minutes later the fluorescence was recorded with a Fluostar Optima (BMG Labtechnologies). Staurosporine (0.5 mM) was used as positive control.
High throughput screening
The assay was performed as described above in black 96-well plates and started with the addition of the Aurora A kinase domain. The Z-factor of the assay was estimated to be 0.77 (24). The primary screening was performed in triplicate at compound concentration of 15 μM (the compounds were dissolved in 0.3 % DMSO) and the selected hits were tested again at a concentration of 1.5 μM. IC50 was defined as the compound concentration that leads to 50 % of inhibition.
Kinase profiling and IC50 determination
Assays were performed on the RBC’s DiscoveryDot nanoliter screening platform (Reaction Biology Corp, USA), which combines the advantages of both radioisotopes and microarrays. A kinase profiling was performed with 36 recombinant kinases. Compound 1 was tested in duplicate, at the final concentration of 1 μM. Staurosporine was used as internal control. ATP concentration was 1 μM for all reactions. The purity of the molecule 1 was superior to 95 % and its synthesis is described in (25).
Compound 1, VX-680 and Staurosporine were tested against five selected kinases in a 10-dose serial dilution starting at 2 μM; ATP concentration was 1 μM for all reactions. Assays were run in duplicate. IC50 was defined as the compound concentration that leads to 50 % of inhibition.
Cell culture
HeLa, HCT-116, Hek-293 and LL/2 were grown on Dulbecco’s modified Eagle’s medium (Biowhittaker, Europe). H358 cells were propagated in RPMI. Media were supplemented with 10% foetal bovine serum (Biowhittaker, Europe).
HeLa (Aurora B –GFP) stable cell lines were already described elsewhere (26). HEK-293 cells were transfected with a plasmid expressing the fusion histone H2A – GFP and then a clonal fluorescent cell was selected and amplified.
Cell proliferation assays were conducted in 96 well cultures plates. Assays were run in triplicate. Serial dilution of compounds started at 2 μM and viability was estimated, at day 5, by addition of MTT cell counting (Promega).
The multicellular tumor spheroid (MTS) model
We have adapted the hanging-drop method (28) to produce HCT116 spheroids of similar diameters. 500 cells were suspended on the lid of an agar coated 24-petri dishes containing culture media. 48h later the spheroids were transferred to the culture medium. Spheroid volumes were measured before drug treatment (D0). HCT116 spheroids were treated for 5 days with different concentrations of the compounds. Control HCT116 spheroids were grown under the same conditions, but without drug treatment. The size of each spheroid was determined by measuring 2 orthogonal diameters (d1 and d2) using an inverted microscope. The volume was calculated according to the formula: V=4/3πr3 where r=1/2√d1xd2. Spheroid growth was calculated by measuring the variations in volume.
Ex vivo microscopy
Ex vivo experiments were conducted on cells grown on Lab-Tek chambered coverglass (Nalge Nunc International) and maintained under standard culture conditions as described in 26. Images were acquired on a Zeiss LSM510 system using a Planapochromat 40 X water immersion objective. GFP was excited with a 488 nm Argon 2 laser (power varying from 0.1 to 2%). Confocal slices are shown.
Immunofluorescence
Cells grown for 24 hours on glass coverslips were fixed at 37°C in 4% formaldhehyde, 2% sucrose and then immunofluorescences were then performed as described in (27). Phosphorylated Histone H3 was detected by a polyclonal rabbit antibody (Upstate). Aurora B was detected by using mouse monoclonal AIM-1 (1/100, Transduction Laboratories). DNA was visualized with 0.1 mM Hoechst 33342 (Sigma). Images were collected with a ZEISS 510 Laser Scanning Confocal microscope with a 63x immersion oil objective. Slices of 0.5 micron are shown.
Cells seeded on glass-coverslips were arrested in mitosis by the addition of paclitaxel (33 nM). Immunohistochemistry was performed on 8 nm thick frozen sections as describe above.
In vivo experiments
In vivo experiments were conducted on four-weeks old female Swiss nude mice (Iffa Credo, Marcy l’Etoile, France). After one week of adaptation in the animal facility (French agreement number A38-516-01), the mice were inoculated subcutaneously with 2 x106 exponentially growing H358 cells mixed with growth factor free matrigel (1/1, BD). Tumours were established at seven days post-injection. Then the mice from each cage were randomly divided into two groups, which allowed the equalization of the mean tumour size of each group. Tumour volumes were determined by measuring two perpendicular diameters using a clipper and then calculated as follows: V = d12 × d2/2, where d1 and d2 are the smallest and the largest diameters, respectively. One mice group (7 animals) received the treatment (compound 1, 100 μg per 20 g mouse in vehicle buffer (PEG 300/PBS/DMSO; 5/4/1) intraperitoneally, whereas the control group (6 mice) was injected with vehicle only. Mice were injected four times a week (Monday, Tuesday, Thursday and Friday). Once a week, mice were weighed and the volumes of the tumours were measured. Only one animal (from the control group) displayed a weight loss and was sacrificed. All other mice received 23 injections, then sacrificed and autopsied. For further analysis, blood was sampled and tumours were dissected and weighed. A piece of tumour was also embedded in OCT compound and frozen for immuno-histochemistry experiments. A second similar separate experiment that included five animals for each group was also carried out.
Results
High throughput screening for identification of Aurora kinase inhibitors
We have developed an Aurora kinase assay for the identification of small molecule inhibitors of Aurora kinases. The assay is based on the phosphorylation of recombinant histone H3 by a purified recombinant Aurora A kinase domain under non-saturating ATP concentrations (see Materials and Methods for detail). The calculated statistical Z’ factor was 0.77 making the assay suitable for high throughput screening. In the screen of the Institut Curie-CNRS proprietary library (http://chimiotheque-nationale.enscm.fr), which contains 6560 distinct mono- to penta-heterocyclic compounds, forty molecules were found to inhibit Aurora kinase by more than 50 % when assayed at a concentration of 15 μM.
Of the fourteen most active compounds identified, six belong to the benzo[e]pyridoindole family (Figure 1). Non-pertinent “hits” corresponding to bi- and tricyclic aromatic compounds bearing reactive (aldehyde) functionality were also found. Because of their high chemical reactivity such compounds were considered as false positive and discarded.
Figure 1. Novel Aurora kinase inhibitors identified by high throughput screening.
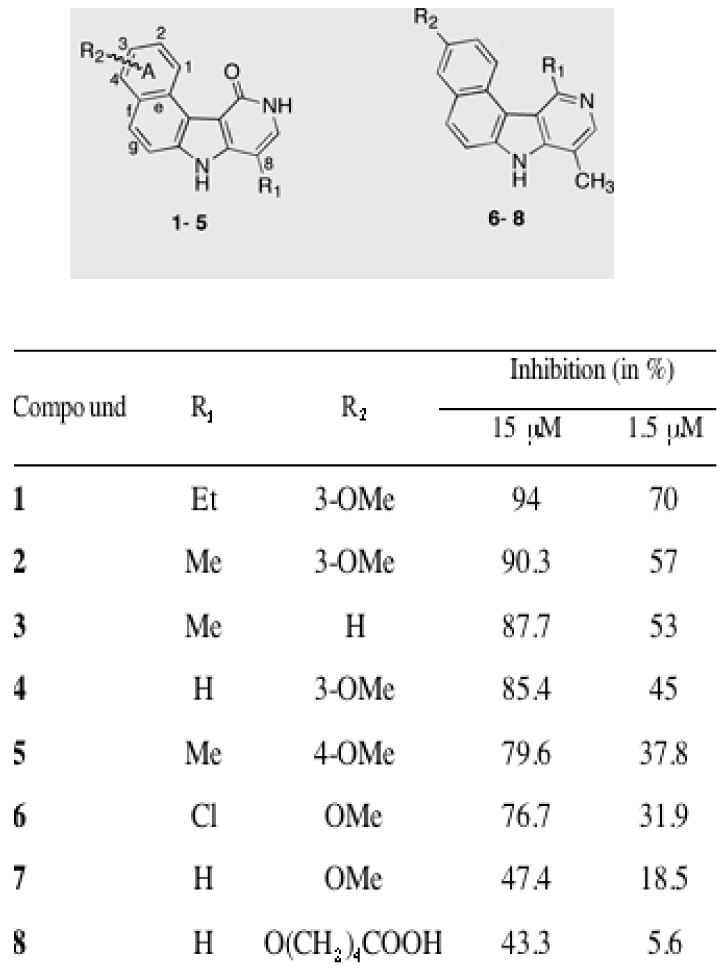
The chemical structure and the preferential conformations of the identified eight most potent hits are presented. The inhibitory efficiency (percent of inhibition) towards the kinase domain of Aurora A for each hit at 15 μm and 1.5 μM are also shown.
The most potent molecules 1 and 2 exhibit an OMe group at the 3-position of the benzo (A-ring). However, the observed high activities of compounds 3 and 5 allow the conclusion that this position of the OMe group is not a requirement for activity, and can be further moved to the 4-position (Figure 1). The presence of an alkyl chain at position 8 is, however, crucial for the inhibitory effect of these molecules, since its absence resulted in a clear loss in activity (compare 1 with 4). Replacement of the pyridinone carbonyl oxygen by a hydrogen (7) or a chlorine atom (6) resulted also in a loss of activity. Finally, the double modification in which R1 and R2 were changed, as in 8, led to almost complete loss of activity as measured at 1.5 μM.
In an independent study (C. Cochet, personal communication) compounds 1–8 were evaluated in vitro for their activity against Casein Kinase II (CKII). In this screen compound 6 was found active. The other indole compounds displayed essentially no affinity for CKII. With this in mind, we have further focused on the characterization of the biological activity of benzo[e]pyridoindole 1 (C1), the compound showing the strongest inhibitory effect (Figure 1), and we have compared its activity with that of the reference Aurora kinase inhibitor VX-680 (22).
The specificity of the molecule C1 (at 1 μM concentration) towards a panel of 36 different kinases was analysed (Figure 2). Most of the kinases tested were not affected by the presence of C1 (Figure 2A). C1 targets essentially Aurora A, MELK and FGFR1 kinases. These kinases were inhibited by 91, 96 and 92 %, respectively (Figure 2A). C1 had also some inhibitory activity towards CHK1, MEK1, RSK1, RSK2 but did not target the cell cycle kinases CDK and PLK (Figure 2A). Among the Aurora kinase family, C1 exhibited an IC50 of 61 nM, 31 nM and 124 nM for the Aurora kinases A, B and C, respectively (Figure 2B). These IC50 were similar to those determined in the same assay for VX-680, but the selectivity of the two inhibitors was slightly different. Indeed, VX-680 was more potent towards Aurora A, while the highest inhibitory effect of C1 was towards Aurora B. C1 inhibited both CHK1 (IC 50 = 262 nM) and MELK (IC50 = 42 nM), whereas VX-680 was poorly efficient towards these kinases. Both inhibitors exhibited thus different selectivity (Figure 2B), although both of them are ATP competitive Aurora inhibitors (Figure 2C and (17)).
Figure 2. Kinase inhibition selectivity of C1.
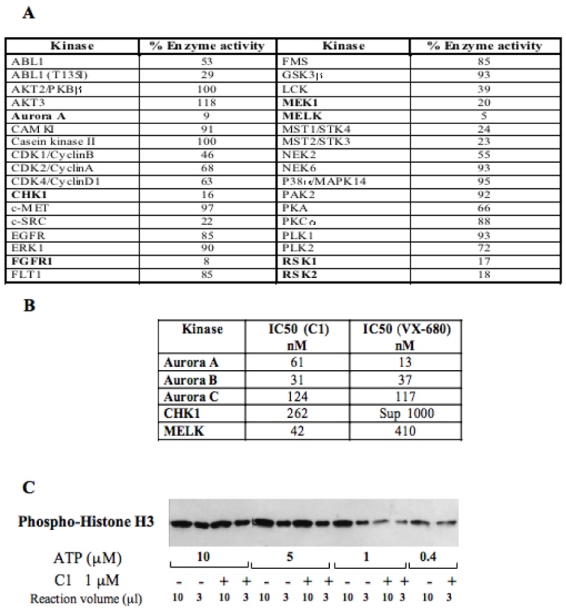
(A) C1 kinase profiling. 36 recombinant kinases were used in the study. The percentage of the remaining kinase activity measured upon treatment with 1 μM C1 in a solution containing 1 μM ATP is presented. Kinases, inhibited by more than 80 %, are indicated in bold.
(B) IC50 values for the five best kinase targets of C1. For comparison the IC50 for VX-680 for the same kinases is also shown. The measurements of IC50 for both compounds were carried out under identical conditions.
(C) Western blot analysis of the effect of C1 on the phosphorylation of histone H3 by Aurora A domain kinase. A kinase assay was carried out either in absence (−) or in the presence (+) of C1 (1μM) and at decreasing concentrations of ATP. Each reaction contained the same amount of histone H3. After completion of the reaction, 10 and 3 μl of the reaction mixtures containing either 10, or 5 or 1μM ATP were run on an SDS gel (for the reaction carried out in the presence of 0.4 μM ATP, an aliquot of only 10 μl was run). After blotting, the phosphorylation of histone H3 was revealed by a specific antibody against the phosphorylated histone H3.
Effect of C1 on HeLa cells
The Aurora kinase inhibition efficiency of C1 was also studied ex vivo in HeLa cells in culture (Figures 3 and 4). To directly demonstrate the inhibitory effect of C1 we have studied the phosphorylation status of histone H3 by Western blotting (histone H3 is one of the favoured substrate of Aurora B and the entry in mitosis correlates with a burst in histone 3 phosphorylation). (Figures 3 A and 4 A). Treatment of the cells with C1 led to a significant decrease of H3 phosphorylation in mitotic HeLa cells (Figure 3A). Moreover treatment of HeLa cells with compound 1 resulted in severe morphological perturbations of the HeLa cell nuclei (Figures 3B, 3C). In particular, cells acquired enlarged lobed nuclei. Quantification showed that treatment with either C1 (2 μM) or VX-680 (300 nM) resulted in the generation of a comparable number of cells with such irregular nuclei (21 % for C1 and 23 % for VX-680, Figure 3C). Note that under the same conditions, only ~1% of the control non-drug treated cells exhibited irregular nuclei (Figure 3C).
Figure 3. Effect of C1 in HeLa cells.
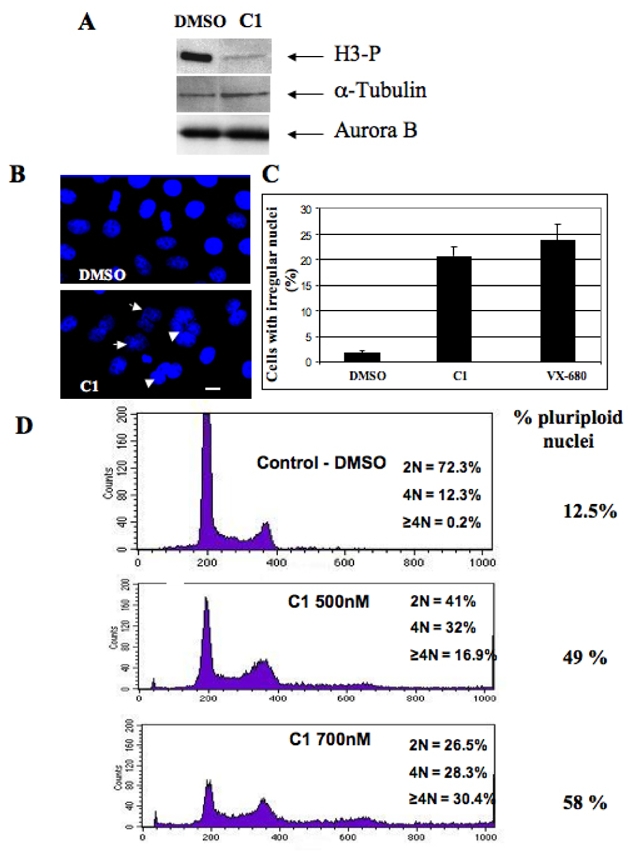
(A) Western blot analysis of histone H3 phosphorylation. HeLa cells were incubated overnight with Nocodazole (50 nM) in the presence of either DMSO or C1 (2 μM). Cells were then collected and lysed. Identical amounts of the lysed cell samples were run on 18% acrylamide gel containing SDS. After transfer, the blot was revealed using an antibody against phosphorylated histone H3. The same membrane was also revealed using antibodies against α-tubulin and Aurora B for estimation of both the amount of loaded proteins and of mitotic cells, respectively.
(B) Treatment with C1 resulted in perturbation of the structure of the cell nuclei. HeLa cells were incubated overnight in the presence of either C1 (2 μM) or DMSO (1%). Nuclei were stained with Hoechst 33 342 and visualized by fluorescence microscopy. Arrows indicate irregular nuclei. Similar perturbations in the nuclear structure were obtained upon treatment with 300 nM VX-680 (not shown). Bar, 5 microns.
(C) Quantification of the data presented in (B). The percentage of irregular (lobed and polyploid) nuclei was determined in two independent experiments; 100 cells were analyzed per experiment.
(D) FACS analysis shows that treatment with C1 results in a dramatic increase of the amount of polyploid cells. The experiments were performed with control HeLa cells (incubated only in the presence of 1% DMSO) and HeLa cells incubated with C1 (at either 500 nM or 700 nM) for 48 hours. DNA was stained with propidium iodine and the samples were analyzed by using a Beckton-Dickinson FACS analyzer. The percentage of polyploid cells (cells with a ploidy ≥2N) is indicated on the right part of the figure.
Figure 4. Effects of C1 on mitotic cells.
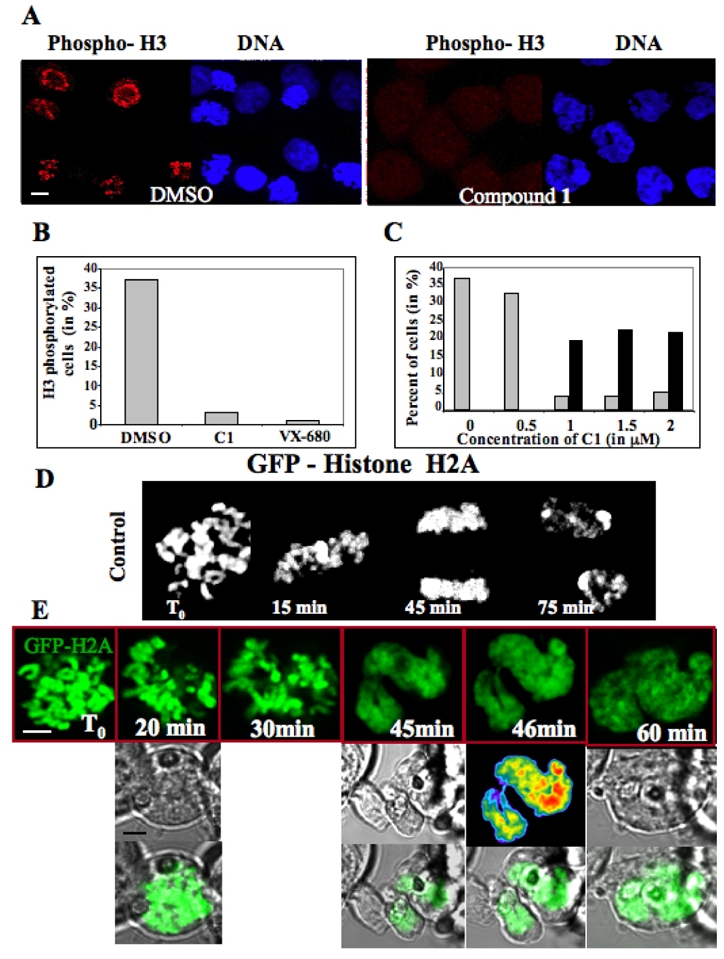
(A) Visualization of the phosphorylation status of histone H3 in paclitaxel (33 nM) treated cells in the presence of either DMSO or C1. H3 phosphorylation (red) was visualised by a specific antibody and DNA (blue) was stained by Hoechst 33342. Similar results were obtained with VX-680 (not shown). The bar, 5 μm.
(B) Quantification of the data presented in (A). 500 cells were analysed in each experiment and the data of two independent experiments are shown.
(C) Treatment with C1 induces mitotic slippage. HeLa cells were arrested in mitosis by overnight incubation in paclitaxel (33 nM). The cells were then incubated with the indicated concentrations of C1 for two additional hours and fixed. Mitotic and polyploid cells were scored. ~100 nuclei were analysed in two different experiments; grey, mitotic cells; black, cells with polylobed nuclei.
(D) Time-lapse microscopy of a stable mitotic HEK-293 cell expressing GFP-H2A histone fusion. (E) Time-lapse microscopy of C1 treated mitotic HEK-293 cells stably expressing GFP-H2A. C1 (1 μM final concentration) was added to the overnight paclitaxel (33 nM) treated cells and then the behaviour of the cell was continuously imaged. Representative photos, made at the indicated time points, are shown. The first, second and third rows present the fluorescent GFP signal, the transmission signal and the merge signal, respectively. The bar, 5 μm.
Treatment with C1 led also to cell polyploidy (Figure 3D). Mitotic (or 4N) cells represent about 12.5 % of the population in the control, whereas they increase to 30 % in the presence of compound 1 (Figure 3D). Moreover the drug-dependent generation of enlarged nuclei (Figure 3B) correlates with a significant increase of ploidy (58 % of the cells are polyploid in the presence of 700 nM compound 1, Figure 3D). These phenotypes (lobed nuclei and polyploid cells) were associated with an inactivation of Aurora-B (1), suggesting that C1 is an efficient inhibitor of the Aurora B kinase activity.
Following an overnight paclitaxel treatment, the bulk mitotic cells tested positive for phospho-histone H3 (~37% of the total cell population), whereas only 3% of the total cell population was scored positive in the presence of compound 1 (2 μM) as compared to 1% for VX-680 (300 nM), the reference Aurora B kinase inhibitor (Figure 4 A, B). Note that upon C1 treatment the 4N cells number increased ~3-fold (Figure 3D), but nonetheless a very strong inhibition of H3 phosphorylation was observed in the drug treated cells (Figure 4 A-C). The depletion of Aurora B kinase or its inactivation allowed paclitaxel treated cells to escape mitosis (mitotic slippage) (16). If C1 is really responsible for inactivating Aurora B, mitotic slippage should also be observed. With this in mind, we treated cells overnight with paclitaxel (33 nM), then added increasing C1 concentrations and pursued the incubation for two hours (Figure 4C). The percentage of lobed nuclei was negligible in the control and in the 0.5 μM C1 treated cells. The increase of C1 concentration up to 1 μM dramatically affected the cells. Indeed, while 37 % of the cells were in metaphase in the control, this percentage decreased to less than 5 % in the presence of 1 μM C1. At 0.5 μM C1 essentially no polyploid cells and no cells with irregular nuclei were observed. At 1 μM C1 a drastic increase of such “defective” cells was detected, i.e. ~22 % of the total cell population exhibited such morphological defects (Figure 4 C). Together, these data show that cells treated with both paclitaxel and C1 exhibited mitotic slippage and therefore, reinforce the conclusion that C1 is an inhibitor of the Aurora B kinase activity.
The absence of Aurora B or the inhibition of its activity affected chromosome segregation (15) and thus, treatment with C1 was expected to have the same effect, which should result in the strong polyploidy already observed (Figure 3). To study this in real time, we have used time-lapse microscopy and stable cell lines expressing GFP-H2A, cells which allow visualization of chromatin DNA (Figure 4D and E). The control cells (without drug treatment) showed a typical mitotic behaviour with properly aligned chromosomes at metaphase, which, as mitosis proceeded, were segregated and gave finally rise to the nuclei of the two daughter cells (Figure 4D). The treatment with 1μM C1 inhibited the chromosome alignment and the formation of metaphasic plate, chromosomes failed to segregate, and with time they loose their rodlike shape, decondensed and formed interphase like structure (Figure 4E), In all analyzed cells (more than 30 in three separate experiments), the interphase type chromatin was always connected by bridges (see Figure 4E, 46 min). These data demonstrate that upon treatment with C1 cells exit from mitosis without segregating chromosomes.
Interference with the function of the protein passenger complex resulted in a distribution of its members on the entire chromosome (2). Treatment with C1 inhibited the enzymatic activity of Aurora B, and thus one should expect similar effects in the presence of C1. To test this we have followed in real time by time-lapse microscopy the mitotic behaviour of Aurora B-GFP in stable HeLa cell lines in the presence or absence of C1 (Figure 5). Aurora B-GFP, in the control cells, had a typical passenger protein localisation, decorating the centromeres at metaphase and then transferring to the central spindle and the midbody as mitosis proceeded (Figure 5A). As expected for an Aurora B inhibitor, C1 prevented centromere alignment on the metaphasic plate (Figure 5A, C1 500 nM). Treatment with 1 μM C1 resulted in the partial dissociation of the GFP-tagged Aurora kinase from the centromeres (40 minutes point, Figure 5A) and with time, Aurora B-GFP decorated also the entire chromatin (100 minutes, Figure 5A). A similar behaviour of Aurora B-GFP was observed upon treatment with VX-680, the effect being observed with a concentration of 300 nM (Figure 5A). Immunofluorescense microscopy showed that the treatment with C1 of paclitaxel arrested mitotic cells resulted in a partial redistribution of the endogenous Aurora B on whole chromosomes (Figure 5B), a result in agreement with the time-lapse microscopy data.
Figure 5. C1 induces the redistribution of Aurora B.
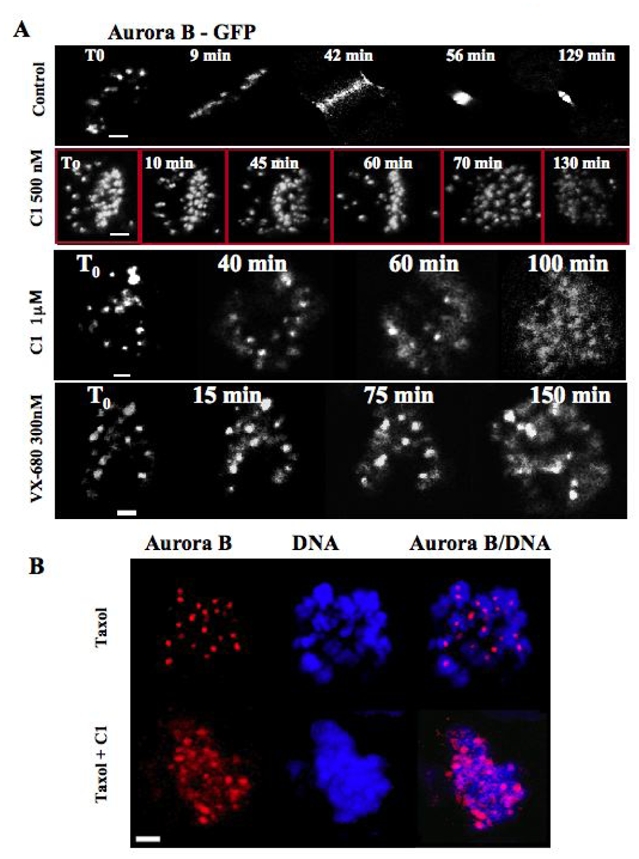
(A) Time lapse microscopy of a mitotic stable HeLa cells expressing Aurora B-GFP fusion in the absence (control) or the presence of either C1 (at 500 nM or 1 μM) or VX-680 (300 nM). The compounds were added to the cell culture and then the behaviour of the cells was continuously imaged. Representative photos, made at the times indicated, are presented. Note that both C1 and VX-680 delayed mitosis onset. In addition, the treatment with 1 μM C1 as well as with VX-680 resulted in a partial delocalization of Aurora B-GFP on the entire chromosomes.
(B) C1 treatment induces also the redistribution of endogenous Aurora B. Overnight paclitaxel (33 nM) treated HeLa cells were incubated for two hours with C1 (1 μM), fixed and analyzed by immunofluorescence microscopy. The localization of Aurora B (red) was detected by anti-Aurora B antibody. DNA (blue) was stained by Hoechst 33342. Note that the strictly punctuated pattern (exclusively centromeric localization) of Aurora B in the control (paxitaxel) cells is no longer observed in the C1 treated cells. Aurora B being partly diffused on chromatin.
In summary, these data illustrate in real time that the treatment of the cells with C1 affected similarly to VX-680 the behaviour of Aurora B during the cell cycle, but with 2–3 fold smaller efficiency (1 μM of C1 was necessary to observe effect close to those found with 300 nM VX-680, see figure 5).
Effect of C1 treatment on tumour cell line viability
The viability of tumour cell lines upon C1 treatment was investigated and compared with the effect of VX-680. Four distinct cell lines (HeLa, LL/2, H358 and HCT-116) were grown as adherent cells and the concentration that inhibits growth by two (IC50) was determined (Figure 6A). Compound 1 was found to be more potent than VX-680 to inhibit growth of HeLa cells. In the other cell lines studied it was found to be between 1.6 to 8.9 less potent than the VX-680 (Figure 6A). Hematopoietic and lung cancer cells behaved mostly like HCT116 cells, i.e. the effect of C1 was 6–7 fold less than this of VX-680 (data not shown). We have also investigated the effects of C1 on the HCT-116 carcinoma cells growing as MCTS by measuring the volumes of the spheroids. After treatment for 5 days the IC50 for HC-T116 spheroid growth inhibition was 1066 nM for compounds 1, while it was of 108 nM for VX-680 (Figure 6A).
Figure 6. Effects of C1 on cell viability in two-dimensional and three-dimensional culture conditions; Effect on mice bearing H358 tumours.
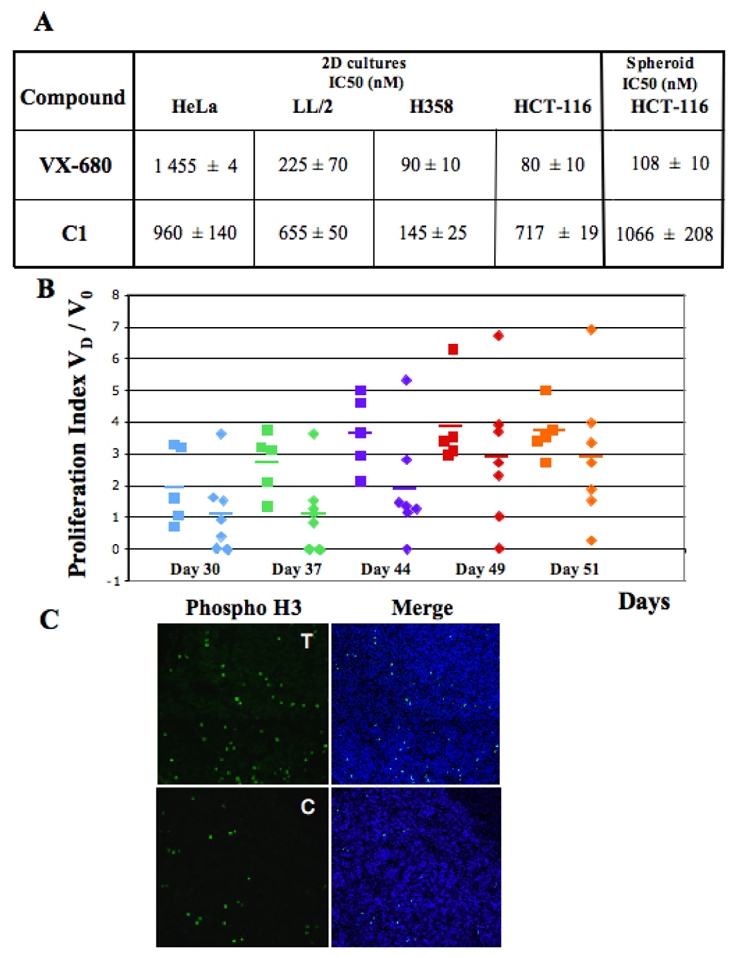
(A) C1 and VX-680 IC50 for HeLa, LL/2, H358 and HCT-116 cells in culture (two-dimensional conditions) and for HCT-116 spheroids (3D conditions). Cell growth and viability were tested under standard conditions in 96 well culture plates with MTT (Promega) cell assay. The average of three independent experiments is shown. Note that similar IC50 were determined for C1 and VX-680 towards H358 cells.
(B) Proliferation indexes of H358 tumor growth. Mice were injected with 2 ×106 cells. Tumor dimensions were measured at days 7, 8, 30, 37, 44, 49 post-injection and the 20 proliferation index (the ratio Vd/V0, where Vd is the volume of the tumour at the respective day, and V0 is the average volume of the tumour calculated at day 7 and 8). Square: control mice; diamond: treated mice. The data for day 30 (D30) post-injection are in blue, D37 data are in green, D44 data are in violet, D49 data are in red and D51 in orange. Each point presents the proliferation index of one mouse tumour and the horizontal bars represent the average index of the series.
(C) Histone H3 phosphorylation is decreased in C1 treated tumours. At day 51 post-injection the control and the C1 treated H358 tumour bearing cell mice were sacrificed and a small part of the tumours were immediately frozen. The phosphorylation status of histone H3 in frozen sections of the tumours of the control (C) or C1 treated mice (T) was visualized by using a specific anti-phosphorylated histone H3 antibody (in green). DNA (blue) was stained by Hoechst 33342. Merge (DNA plus phospo – histone H3) is shown.
Effect of C1 on tumour growth in mice
The efficiency of compound 1 was also studied in mice bearing H358 tumours. Mice bearing H358 tumours were injected with either C1 (5 mg/kg) and with the vehicle or vehicle only. The concentration of C1 was limited by its solubility in aqueous buffer. Mice were healthy after repetitive injections of the compound and grew normally. The only anomaly observed was related to the kidneys, which were slightly less coloured. Blood analysis indicated neutropenia in the mice repetitively treated with C1 (average 1 ×106 neutrophil/l) compared to the control mice (average 2.6 ×106 neutrophil/l).
In the control group, tumours grew progressively and except for one, their growing index (i.e. the ratio of the tumour volume measured at the indicated time (VD) to the initial (days 7 and 8) volume of the tumour (V0)) was ~3.5 at the end of the experiment (day 51, Figure 6B). Upon injection of C1, the tumour growth index was found to be very dispersed. Three tumours were still decreased in size at day 30, while the others behaved as control tumours. At day 37 only two tumours have regressed, while four had an index of 1 and one was growing. At day 44 two tumours increased in size (index 5.3 and 2.8) whereas two were still under control (index inferior at 1). At the end of the experiment (day 51) one tumour in the treated group has an index smaller than 1 (index 0.2). Conversely one tumour has an index superior to 6 suggesting that repetitive injections induced resistance to compound 1. Consequently the differences (mean tumour index and dispersion) between treated and control tumours were not statistically significant (Student-t and Fisher-F test). Importantly, tumours with a decreased size (growth index below 1) were always found in the treated group after 30 days. We also found that the level of H3-phosphorylation was lower in the tumours of the C1 treated mice compared to those of the untreated control mice (Figure 6C). Similar results were obtained in a second separate experiment (data not shown).
Discussion
By screening a library containing 6560 mono- to penta-heterocyclic compounds, we have identified forty molecules able to significantly inhibit Aurora A kinase. The most active compounds belong to the benzo[e]pyridoindole family. These active benzopyridoindoles exhibit a “crescent moon” shape determined by the benzo[e] ring fusion. This geometry seems to offer the best complimentary shape to the Aurora kinase ATP site, since only two amongst the two hundred tetracyclic indole compounds screened (including the related benzo[g] and benzo[f]pyridoindoles and linear pyrido[4,3-b]carbazoles (ellipticine analogues)) displayed some weak inhibitory activity at 1.5 μM. The best hit (C1) was shown to be a potent in vitro, inhibitor of the three different Aurora kinases with IC50 of 61 nM, 31 nM and 124 nM for Aurora A, B and C, respectively. The profiling results reveal that C1 also efficiently inhibits also two other kinases, FGFR and MELK. MELK, maternal embryonic leucine zipper kinase, exhibits maximal activity during mitosis but its physiological substrates are unknown (29).
Several different families of Aurora kinase inhibitors have now been identified and the structural basis for their inhibition selectivity was described (30). For example, the tight association of either VX-680 or hesperadin with the hydrophobic pocket in the active site determined the high selectivity of the inhibitors (31–33). Note that C1, as hesperadin (a member of the SU-family of indolinone type kinase inhibitors), exhibits a cyclic amide functionality (15). Two-dimensional superimposition of C1 and hesperadin via their amide functionalities showed that the three phenyl rings in hesperadin form a “disconnected” crescent moon shape type structure, suggesting that C1 interacts with Aurora kinases in a manner similar to that of hesperadin, i.e. it may form a tight complex with the hydrophobic pocket of the kinase active site.
C1 prevents histone H3 phosphorylation in mitotic cells. This strongly indicates that in vivo the main target of C1 would be Aurora B. In agreement with this, the C1 treated cells did not exhibit monopolar phenotype, which is typically observed upon efficient inhibition of Aurora A. In addition, C1 impaired chromosome alignment and segregation as well as anaphase onset. It increased significantly the ploidy of the cells and induced mitotic slippage. All these effects were observed upon ablation of Aurora B (2, 6), further suggesting that ex vivo the main target of C1 is this kinase. The two other strong inhibitors of Aurora kinases, VX-680 and hesperadin, were also reported to selectively target Aurora B ex vivo and to override the spindle checkpoint (15, 33). We have also shown that both C1 and VX-680 were able to partly delocalise Aurora B kinase all over the chromosomes.
Both C1 and VX-680 efficiently prevented tumor cell proliferation. Although C1 and VX-680 have comparable in vitro inhibition efficiency towards Aurora kinases, the C1 antiproliferative activity was found to be between 1.6 and 8.9 folds weaker in the different cell lines studied compared to that of VX-680. The multicellular tumor spheroid (MCTS) model represents an intermediate level of complexity between cell growing as in vitro monolayers and solid tumours in animals. At a micromolar concentration, C1 decreased by two the spheroid volumes of HCT116 carcinoma cells growing as MCTS. The mild difference of IC50 in monolayer cultured cells compared to the spheroid model suggests a good biodisponibility of these compounds and its absence of multicellular resistance.
Furthermore, the effect of C1 was maximal (and comparable to that of VX-680) in H358 cell line, a cell line established from a very aggressive NSCLC tumour (Non Small Cancer Lung Cell). Patients bearing this type of tumours showed very bad prognoses and only a few relatively potent therapeutic agents against this type of tumours are available (34). With this in mind, we have further studied the in vivo effect of C1 on mice bearing H358 tumours. Repetitive injections of C1 were well tolerated by the animals and affected tumour growth at least for one and a half month. Note that only in few cases a decrease of the initial volume of the tumours was detected by the repetitive injections with C1 during the whole time course of the two independent experiments. This suggests that tumours escaped and became resistant to the compound upon repetitive injections.
In summary, our studies in mice revealed encouraging results with noticeable effect on a resistant model of tumours, and with few toxic effects observed so far. Benzopyridoindoles might thus be viewed as new leads for the development of Aurora kinase inhibitors that would have interesting potentials in NSCLC treatment. Combination of therapies, especially those acting via different mechanisms are proposed for improving the survival of patients with NSCLC; Aurora inhibitors, and among them Benzo[e]pyridoindole derivatives, may be included in these treatments.
Acknowledgments
The authors thank Fabienne Sirot for help in the initial phase of this work and Céline Frangia for her skilful technical assistance. We thank Dr Dimitrios Shkoufias for reading the manuscript and suggestions. Thanks to the ChemAxon company (http://www.chemaxon.com), which has allowed the free use of the MarvinView package. T.H. was supported by a Vietnam/French program and was a recipient of an ARC fellowship. This work was supported by INSERM, ANR (Project R05075CC), ARECA and Rhônes-Alpes Canceropole CLARA (EpiProNetwork). S.D. acknowledges La Ligue Nationale contre le Cancer (Equipe labelisée La Ligue).
Bibliography
- 1.Vader G, Lens SM. The aurora kinase family in cell division and cancer. Biochim biophys Acta. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 2.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 3.Sardon T, Peset I, Petrova B, Vernos I. Dissecting the role of Aurora A during spindle assembly. EMBO J. 2008;27:2567–79. doi: 10.1038/emboj.2008.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155:1109–16. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora A cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–8. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lens SM, Medema RH. The survivin/Aurora B complex: its role in coordinating tension and attachment. Cell Cycle. 2003;2:507–10. doi: 10.4161/cc.2.6.559. [DOI] [PubMed] [Google Scholar]
- 7.Ditchfield C, Johnson VL, Tighe A, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ditchfield C, Keen N, Taylor SS. The Ipl1/Aurora kinase family: methods of inhibition and functional analysis in mammalian cells. Methods Mol Biol. 2005;296:371–81. doi: 10.1385/1-59259-857-9:371. [DOI] [PubMed] [Google Scholar]
- 9.Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- 10.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. Embo J. 1998;17:3052–65. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005;15:241–50. doi: 10.1016/j.tcb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–36. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 13.Girdler F, Gascoigne KE, Eyers PA, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006 Sep 1;119(Pt 17):3664–75. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 14.Jackson JR, Patrick DR, Dar MM, Huang PS. Targeted anti-mitotic therapies: can we improve on tubulin agents? Nature Reviews Cancer. 2007;7:107–17. doi: 10.1038/nrc2049. [DOI] [PubMed] [Google Scholar]
- 15.Hauf S, Cole RW, LaTerra S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–94. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gadea BB, Ruderman JV. Aurora kinase inhibitor ZM447439 blocks chromosome-induced spindle assembly, the completion of chromosome condensation, and the establishment of the spindle integrity checkpoint in Xenopus egg extracts. Mol Biol Cell. 2005;16:1305–18. doi: 10.1091/mbc.E04-10-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington EA, Bebbington D, Moore J, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 18.Fancelli D, Moll J, Varasi M, et al. 1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: identification of a potent aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J Med Chem. 2006;49:7247–51. doi: 10.1021/jm060897w. [DOI] [PubMed] [Google Scholar]
- 19.Soncini C, Carpinelli P, Gianellini L, et al. PHA-680632, a novel Aurora kinase inhibitor with potent antitumoral activity. Clin Cancer Res. 2006;12:4080–9. doi: 10.1158/1078-0432.CCR-05-1964. [DOI] [PubMed] [Google Scholar]
- 20.Hoar K, Chakravarty A, Rabino C, et al. MLN8054, a small-molecule inhibitor of Aurora A, causes spindle pole and chromosome congression defects leading to aneuploidy. Mol Cell Biol. 2007;27:4513–25. doi: 10.1128/MCB.02364-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–48. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 22.Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noronha G, Cao J, Chow CP, et al. Inhibitors of ABL and the ABL-T315I mutation. Curr Top Med Chem. 2008;8:905–21. doi: 10.2174/156802608784911635. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen CH, Bisagni E, Lavelle F, Bissery MC, Huel C. Synthesis and antitumor properties of new 4-methyl-substituted- pyrido[4,3-b]indoles (gamma-carbolines) Anticancer Drug Des. 1992;7:219–33. [PubMed] [Google Scholar]
- 26.Delacour-Larose MMA, Skoufias DA, Margolis RL, Dimitrov S. Distinct dynamics of Aurora B and Survivin during mitosis. Cell Cycle. 2004;3:1418–26. doi: 10.4161/cc.3.11.1203. [DOI] [PubMed] [Google Scholar]
- 27.Delacour-Larose M, Thi MN, Dimitrov S, Molla A. Role of survivin phosphorylation by aurora B in mitosis. Cell Cycle. 2007;6:1878–85. doi: 10.4161/cc.6.15.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Del Duca D, Werbowetski T, Del Maestro RF. Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J Neurooncol. 2004;67:295–303. doi: 10.1023/b:neon.0000024220.07063.70. [DOI] [PubMed] [Google Scholar]
- 29.Badouel C, Körner R, Frank-Vaillant M, Couturier A, Nigg EA, Tassan JP. M-phase MELK activity is regulated by MPF and MAPK. Cell Cycle. 2006;5:883–9. doi: 10.4161/cc.5.8.2683. [DOI] [PubMed] [Google Scholar]
- 30.Cheetham GM, Charlton PA, Golec JM, Pollard JR. Structural basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680. Cancer Lett. 2007;251:323–9. doi: 10.1016/j.canlet.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Cheetham GM, Knegtel RM, Coll JT, et al. Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J Biol Chem. 2002;277:42419–22. doi: 10.1074/jbc.C200426200. [DOI] [PubMed] [Google Scholar]
- 32.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol Cell. 2003;12:851–62. doi: 10.1016/s1097-2765(03)00392-7. [DOI] [PubMed] [Google Scholar]
- 33.Sessa F, Mapelli M, Ciferri C, et al. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–91. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 34.Alvarez M, Roman E, Santos ES, Raez LE. New targets for non-small-cell lung cancer therapy. Expert Rev Anticancer Ther. 2007;7:1423–37. doi: 10.1586/14737140.7.10.1423. [DOI] [PubMed] [Google Scholar]