

Abstract
The Anatomical Therapeutic Chemical (ATC) classification system, recommended by the World Health Organization, categories drugs into different classes according to their therapeutic and chemical characteristics. For a set of query compounds, how can we identify which ATC-class (or classes) they belong to? It is an important and challenging problem because the information thus obtained would be quite useful for drug development and utilization. By hybridizing the informations of chemical-chemical interactions and chemical-chemical similarities, a novel method was developed for such purpose. It was observed by the jackknife test on a benchmark dataset of 3,883 drug compounds that the overall success rate achieved by the prediction method was about 73% in identifying the drugs among the following 14 main ATC-classes: (1) alimentary tract and metabolism; (2) blood and blood forming organs; (3) cardiovascular system; (4) dermatologicals; (5) genitourinary system and sex hormones; (6) systemic hormonal preparations, excluding sex hormones and insulins; (7) anti-infectives for systemic use; (8) antineoplastic and immunomodulating agents; (9) musculoskeletal system; (10) nervous system; (11) antiparasitic products, insecticides and repellents; (12) respiratory system; (13) sensory organs; (14) various. Such a success rate is substantially higher than 7% by the random guess. It has not escaped our notice that the current method can be straightforwardly extended to identify the drugs for their 2nd-level, 3rd-level, 4th-level, and 5th-level ATC-classifications once the statistically significant benchmark data are available for these lower levels.
Introduction
Nowadays, the Anatomical Therapeutic Chemical (ATC) classification system, recommended by the World Health Organization (WHO), is the most widely recognized classification system for drugs. This classification system divides drugs into different groups according to the organ or system on which they act and/or their therapeutic and chemical characteristics. Accordingly, the ATC classification is very helpful for studying utilization of drugs and categorizing them according to different purposes, therapeutic properties, chemical and pharmacological properties (see Report of the WHO Expert Committee, 2005; World Health Organ Tech Rep, Ser:1–119). In the ATC classification system, drugs are classified into 14 main classes (http://www.whocc.no/atc/structure_and_principles/). In order to understand this kind of complicated classification system, some efforts have been made [1], [2]. In a pioneer study, Gurulingappa et al. [2] proposed a method to study the ATC-classification system by combining the information extraction and machine learning techniques. However, their method can be used to identify the drug compounds only within the class of “Cardiovascular System”, one of the 14 main ATC classes.
During the past decade, many compound databases, such as KEGG (Kyoto Encyclopedia of Genes and Genomes) [3], [4], have been established. From these databases many compounds and their properties can be acquired. Such abundant informations provide an opportunity to analyze ATC classification system in greater detail. Encouraged by the successes of using machine learning and data mining methods to investigate complicated problems in a variety of biological areas [5], [6], [7], [8], [9], the present study was initiated in an attempt to develop a powerful method by which one can identify query drugs compound among all their 14 posible main classes.
According to a recent comprehensive review [10], to establish a really useful statistical predictor for a biological system, we need to consider the following procedures: (i) construct or select a valid benchmark dataset to train and test the predictor; (ii) formulate the samples concerned with an effective mathematical expression that can truly reflect their intrinsic correlation with the target to be predicted; (iii) introduce or develop a powerful algorithm (or engine) to operate the prediction; (iv) properly perform cross-validation tests to objectively evaluate the anticipated accuracy of the predictor. Below, let us describe how to deal with these steps one by one.
Materials and Methods
Recently, the information of protein-protein interactions have been used for predicting various attributes of proteins (see, e.g., [11], [12], [13]), implying that interactive proteins are more likely to share common biological functions [11] than non-interactive ones [14]. Likewise, it is more likely that two interactive drug compounds may have the similar biological function. Actually, it is generally accepted that compounds with similar physicochemical properties often involve in similar biological activities [1]. Accordingly, it is reasonable to assume that the interactive drugs may likely belong to the same ATC-class, and so do those drugs with similar structures. Based on such rational, let us construct the following benchmark to develop a new method for identifying the ATC-classes of drugs.
Benchmark Dataset
The dataset for drugs was obtained from the public available database KEGG [3], [4] at ftp://ftp.genome.jp/pub/kegg/medicus/drug/drug (June, 2011). There are totally 9,758 drugs. After excluding those without the information of ATC-codes, the remaining are 4,376 drug samples, from which further screening was performed to remove those without the information of both chemical-chemical interactions and chemical-chemical similarities. After the above winnowing procedures, we finally obtained the benchmark dataset 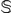 containing 3,883 drugs classified into 14 main ATC-classes, as can be formulated by
containing 3,883 drugs classified into 14 main ATC-classes, as can be formulated by
| (1) |
where 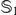 represents the subset for the 1st main ATC class called “Alimentary tract and metabolism”,
represents the subset for the 1st main ATC class called “Alimentary tract and metabolism”, 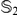 the subset for the 2nd main ATC class “Blood and blood forming organs”,
the subset for the 2nd main ATC class “Blood and blood forming organs”, 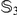 the subset for the 3rd main ATC class “Cardiovascular system”, and so forth (cf.
Table 1
); while
the subset for the 3rd main ATC class “Cardiovascular system”, and so forth (cf.
Table 1
); while 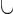 represents the symbol for “union” in the set theory. For convenience, hereafter let us just use C
1, C
2, C
3, …, C
14 as the tags of the 14 classes. A breakdown of the 3,883 drugs into the 14 main ATC-classes is given in
Table 1
. For the codes of these drugs in each of the 14 classes, see Supporting Information S1. During the course of constructing the benchmark dataset, the information from http://www.genome.jp/kegg-bin/get_htext?br08303.keg was used that collected the drug compounds and their ATC classification information from http://www.whocc.no/atc_ddd_index/ and provided the ATC code for each drug.
represents the symbol for “union” in the set theory. For convenience, hereafter let us just use C
1, C
2, C
3, …, C
14 as the tags of the 14 classes. A breakdown of the 3,883 drugs into the 14 main ATC-classes is given in
Table 1
. For the codes of these drugs in each of the 14 classes, see Supporting Information S1. During the course of constructing the benchmark dataset, the information from http://www.genome.jp/kegg-bin/get_htext?br08303.keg was used that collected the drug compounds and their ATC classification information from http://www.whocc.no/atc_ddd_index/ and provided the ATC code for each drug.
Table 1. Breakdown of the benchmark dataset  according to the 14 main ATC classes.
according to the 14 main ATC classes.
| Tag | The 1st-level ATC class | Number of drugs |
| C 1 | Alimentary tract and metabolism | 540 |
| C 2 | Blood and blood forming organs | 133 |
| C 3 | Cardiovascular system | 591 |
| C 4 | Dermatologicals | 421 |
| C 5 | Genito-urinary system and sex hormones | 248 |
| C 6 | Systemic hormonal preparations, excluding sex hormones and insulins | 126 |
| C 7 | Antiinfectives for systemic use | 521 |
| C 8 | Antineoplastic and immunomodulating agents | 232 |
| C 9 | Musculo-skeletal system | 208 |
| C 10 | Nervous system | 737 |
| C 11 | Antiparasitic products, insecticides and repellents | 127 |
| C 12 | Respiratory system | 427 |
| C 13 | Sensory organs | 390 |
| C 14 | Various | 211 |
Number of total virtual drugs 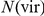
|
4,912a | |
Number of total structural different drugs 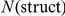
|
3,883b | |
See Eqs.2–3 for the definition about the number of virtual drugs, and its relation with the number of structural different drugs.
Of the 3,883 structural different drugs, 3,295 belong to one class, 370 to two classes, 110 to three classes, 37 to four classes, 27 to five classes, and 44 to six classes. See Supporting Information S1 for the detailed drug codes listed in each of 14 ATC-classes.
Because some drugs may belong to more than one main ATC-class, like the case in dealing with proteins with multiple location sites [15], [16], [17], it is instructive to introduce the concept of the “virtual drugs” as illustrated as follows. A drug compound belonging to two different ATC-classes will be counted as 2 virtual samples even though they have an identical chemical structure; if belonging to three different classes, 3 virtual samples; and so forth. Accordingly, the total number of the different virtual drug samples is generally greater than that of the total different structural drug samples. Their relationship can be formulated as follows [18]
| (2) |
where 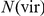 is the number of total different virtual drug samples in
is the number of total different virtual drug samples in 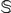 ,
, 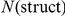 the number of total different structural drugs,
the number of total different structural drugs, 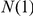 the number of drugs belonging to one ACT-class,
the number of drugs belonging to one ACT-class, 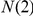 the number of drugs belonging to two ATC-classes, and so forth; while
the number of drugs belonging to two ATC-classes, and so forth; while 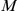 is the number of total main ACT-classes (for the current case,
is the number of total main ACT-classes (for the current case, 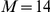 (cf.
Table 1
).
(cf.
Table 1
).
For the current 3,883 drugs in 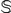 , 3,295 occur in one class, 370 in two classes, 110 in three classes, 37 in four classes, 27 in five classes, 44 in six classes, and none in seven or more classes (
Figure 1
). Substituting these data into Eq.1, we have
, 3,295 occur in one class, 370 in two classes, 110 in three classes, 37 in four classes, 27 in five classes, 44 in six classes, and none in seven or more classes (
Figure 1
). Substituting these data into Eq.1, we have
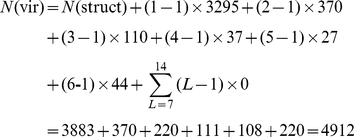 |
(3) |
which is fully consistent with the figures in Table 1 and the data in Supporting Information S1.
Figure 1. An illustration to show the distribution about the numbers of ATC-classes a same drug may belong to.

For the 3,883 drugs in 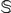 , 3,295 belong to one class, 370 to two classes, 110 to three classes, 37 to four classes, 27 to five classes, 44 to six classes, and none to seven or more classes.
, 3,295 belong to one class, 370 to two classes, 110 to three classes, 37 to four classes, 27 to five classes, 44 to six classes, and none to seven or more classes.
Prediction Based on Chemical-Chemical Interactions
Based on the fact that the interactive compounds often involve in similar biological activities [11], it is feasible to predict the ATC-class of a query drug using the information of chemical-chemical interactions, as described below.
STITCH (Search tool for interactions of chemicals) [19] is a large database containing known and predicted interactions between chemicals and between proteins derived from experiments, literature and other databases. We downloaded the information of chemical-chemical interactions from http://stitch.embl.de:8080/download/chemical_chemical.links.v2.0.tsv.gz. Each of these interactions was evaluated by a confidence score, ranging from 1 to 1000, to reflect the likelihood of its occurrence. For any two drugs d
1 and d
2, their interaction confidence score was denoted by 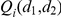 . Particularly, if the interaction between d
1 and d
2 does not exist in STITCH, their interaction confidence score was set as zero, i.e.,
. Particularly, if the interaction between d
1 and d
2 does not exist in STITCH, their interaction confidence score was set as zero, i.e., 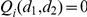 .
.
Suppose that a training dataset 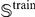 consists of n drugs
consists of n drugs 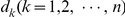 , and that the 14 main ATC-classes are denoted by
, and that the 14 main ATC-classes are denoted by 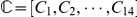 , where C
1 represents “Alimentary tract and metabolism”, C
2 “Blood and blood forming organs”, and so forth (see
Table 1
). The ATC-classes of any drug di can be formulated as
, where C
1 represents “Alimentary tract and metabolism”, C
2 “Blood and blood forming organs”, and so forth (see
Table 1
). The ATC-classes of any drug di can be formulated as
| (4) |
where
| (5) |
According to the chemical-chemical interaction approach, the likelihood for a query drug 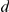 belonging to Cj, denoted as
belonging to Cj, denoted as 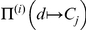 , can be calculated by
, can be calculated by
| (6) |
where 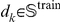 means that
means that 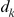 is an element of the training dataset
is an element of the training dataset 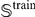 . According Eq.6, the likelihood that
. According Eq.6, the likelihood that 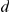 belongs to Cj can be formulated as the maximum of the interaction confidence scores between
belongs to Cj can be formulated as the maximum of the interaction confidence scores between 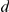 and those drugs that belong to Cj in the training dataset
and those drugs that belong to Cj in the training dataset 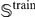 . Obviously, the larger the score is, the more likely that
. Obviously, the larger the score is, the more likely that 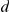 belongs to
belongs to 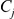 . When
. When 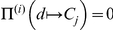 , it means that the probability for the drug
, it means that the probability for the drug 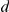 belonging to the class Cj is zero. Given a query drug compound
belonging to the class Cj is zero. Given a query drug compound 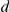 , suppose the outcome derived from Eq.6 is
, suppose the outcome derived from Eq.6 is
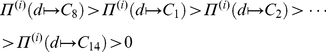 |
(7) |
which means that the highest probability for the drug 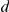 belonging to the ATC-class is
belonging to the ATC-class is 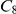 (“Antineoplastic and immunomodulating agents”), followed by
(“Antineoplastic and immunomodulating agents”), followed by 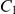 (“Alimentary tract and metabolism”), and so forth (cf.
Table 1
). If there is a tie between two terms in Eq.7, then the probabilities for the drug belonging to the two corresponding classes are the same. But this kind of tie case rarely happened.
(“Alimentary tract and metabolism”), and so forth (cf.
Table 1
). If there is a tie between two terms in Eq.7, then the probabilities for the drug belonging to the two corresponding classes are the same. But this kind of tie case rarely happened.
Note that the outcome of Eq.6 might turn out to be trivial, i.e.,
| (8) |
indicating that no chemical-chemical interaction exists for the query drug 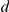 in the training dataset
in the training dataset 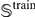 ; i.e.,
; i.e.,
| (9) |
Under such a circumstance, no meaningful result would be obtained by the “interaction-based” method, and we should instead use the “similarity-based method as described in the next section.
Prediction Based on Chemical-Chemical Similarities
Likewise, based on the fact that the compounds with similar physicochemical properties often have the same biological activities [1], we can also use the information of chemical-chemical similarities as another feasible avenue to predict the ATC-class for a query drug. To realize this, let us first introduce how to use graphical representation to measure the similarity between two drug compounds.
Graphical approaches can provide intuitive pictures and useful insights for studying and analyzing complicated biological systems, as demonstrated by many studies on a series of important biological topics (see, e.g., [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]). Here, a special graphic approach was utilized to estimate the similarity of two compounds. Hattori et al.
[31] first proposed a means to measure the similarity of two compounds via their graph representations. Since each chemical structure can be easily represented by a 2D (two-dimensional) graph where vertices stand for atoms and edges for bonds between them, the similarity of two compounds can be estimated by the Jaccard coefficient [32], [33] based on their maximum common subgraph. The similarity scores between compounds by this method can be obtained from the website at http://www.genome.jp/ligand-bin/search_compound. According to the graphical method by Hattori et al.
[31], given two drug compounds d
1 and d
2, their similarity score was denoted by 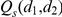 . When the similarity score between d
1 and d
2 does not exist in http://www.genome.jp/ligand-bin/search_compound, their similarity was set as zero; i.e.,
. When the similarity score between d
1 and d
2 does not exist in http://www.genome.jp/ligand-bin/search_compound, their similarity was set as zero; i.e., 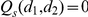 .
.
Thus, the prediction method based on the chemical-chemical similarities can be formulated in a way almost completely parallel to that of the chemical-chemical interactions as done in the preceding section.
Now, instead of Eq.6, we have
| (10) |
where the superscript and subscript “s” stands for the 1st letter of “similarity”, implying that the calculation is now based on “chemical-chemical similarity” instead of “chemical-chemical interaction” as done in Eq.6.
Prediction by Integrating the Interaction-Based and Similarity-Based Methods
Given a query drug compound 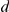 , when the integrated method was used to identify its ATC-class, the prediction involved the following two steps.
, when the integrated method was used to identify its ATC-class, the prediction involved the following two steps.
Step 1
The interaction-based method (cf. Eq.6) was first applied to identify its ATC-class.
Step 2
If the probabilities thus obtained for the drug belonging to all the 14 ATC-classes were zero as indicated in Eq.8, meaning no meaningful results were obtained at all, then the prediction would continue using the similarity-based method (cf. Eq.10).
Jackknife Cross-Validation
In statistical prediction, the following three cross-validation methods are often used to examine the quality of a predictor: independent dataset test, subsampling (or k-fold crossover) test, and jackknife test [34]. However, of the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark dataset [35]. The reasons are as follows. (i) For the independent dataset test, although all the samples used to test a predictor are outside the training dataset used to train the prediction engine so as to exclude the “memory” effect or bias, the way of how to select the independent samples for testing the predictor could be quite arbitrary unless the number of independent samples is sufficiently large. This kind of arbitrariness might lead to completely different conclusions. For instance, a predictor achieving a higher success rate than the other for a given independent testing dataset might not able to keep so when tested by another independent testing dataset [34]. (ii) For the subsampling (or k-fold crossover) test, the concrete procedure usually used in literatures was the 5-fold, 7-fold or 10-fold cross-validation. The problem with this kind of subsampling test was that the number of possible selections in dividing a benchmark dataset would be an astronomical figure even for a very simple dataset, as elucidated in [35] and demonstrated by Eqs.28–30 in [10]. Therefore, in any practical subsampling cross-validation tests, only an extremely small fraction of the possible selections were taken into account. Since different selections would always yield different results even for a same benchmark dataset and a same predictor, the subsampling test could not avoid the arbitrariness either. A test method unable to generate a unique outcome should not be deemed as a good one. (iii) In the jackknife test, all the samples in the benchmark dataset will be singled out one-by-one and tested by the predictor trained by the remaining samples. During the process of jackknifing, both the training dataset and testing dataset are actually open, and each sample will be in turn moved between the two. The jackknife test can exclude the “memory” effect. Also, the arbitrariness problem as mentioned above for the independent dataset test and subsampling (or k-fold crossover) test can be avoided because the outcome obtained by the jackknife cross-validation is always unique for a given benchmark dataset. Accordingly, the jackknife test has been widely recognized and increasingly adopted by many investigators to examine the quality of various predictors (see, e.g., [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47]). Accordingly, in this study we are to use the jackknife test to examine the prediction quality as well.
Accuracy Measurement
For any given set of query drugs, we can obtain a series of candidate ATC-classes using the aforementioned prediction methods. Ranked by the likelihood according to their descending order, the prediction accuracy can be defined as
| (11) |
where CPj denotes the number of drugs whose j-th order predicted ATC-class is one of the true ATC-class, and N denotes the total number of query drugs whose ATC-classes are to be identified. According to such a definition, the result of higher ACj with smaller j or lower ACj with larger j indicates that the predicted hits are more concentrated meaning a better prediction. Obviously, the result with high 1st-order prediction accuracy AC 1 always represents a good quality of prediction.
The average number of ATC-classes for the N query drugs is defined as
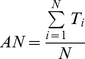 |
(12) |
where Ti is the number of ATC-classes for the i-th query drug. Thus, another parameter for measuring the proportion of the true classes successfully identified by the first m-order prediction hits can be calculated as [13]
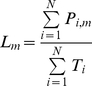 |
(13) |
where Pi,m denotes the number of the first m predicted candidate ATC-classes that are the true ATC-classes for the i-th drug in the dataset. Usually, m could take the smallest integer that is equal to or greater than AN; i.e.,
| (14) |
where the operator Int means taking the integer part of the quantity right after it. Again, the result of larger Lm with smaller m implies a better prediction with less uncertainty.
Results and Discussion
For clarity, the original benchmark dataset 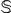 of 3,883 drugs (cf. Supporting Information S1) can be separated into two subsets; i.e.,
of 3,883 drugs (cf. Supporting Information S1) can be separated into two subsets; i.e.,
| (15) |
where 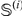 contains 2,144 drugs that had the chemical-chemical interaction information, while
contains 2,144 drugs that had the chemical-chemical interaction information, while 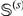 contains
contains 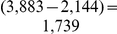 drugs that had no chemical-chemical interaction information. Listed in
Table 2
are the results obtained by the aforementioned three different prediction methods in identifying the 14 main ATC classes for the drugs investigated. By examining the table, we can observe the following.
drugs that had no chemical-chemical interaction information. Listed in
Table 2
are the results obtained by the aforementioned three different prediction methods in identifying the 14 main ATC classes for the drugs investigated. By examining the table, we can observe the following.
Table 2. The jackknife success rates by three different methods in identifying the drugs among the 14 main ATC-classes.
| Prediction order | Interaction-baseda | Similarity-basedb | Integratedc |
| 1 | 67.72% | 78.49% | 72.55% |
| 2 | 21.13% | 18.86% | 20.11% |
| 3 | 13.43% | 8.63% | 11.28% |
| 4 | 7.18% | 5.23% | 6.31% |
| 5 | 4.76% | 2.88% | 3.91% |
| 6 | 3.54% | 1.73% | 2.73% |
| 7 | 1.63% | 0.12% | 0.95% |
| 8 | 0.75% | 0.35% | 0.57% |
| 9 | 0.75% | 0.12% | 0.46% |
| 10 | 0.56% | 0.06% | 0.33% |
| 11 | 0.09% | 0.00% | 0.05% |
| 12 | 0.28% | 0.00% | 0.15% |
| 13 | 0.09% | 0.00% | 0.05% |
| 14 | 0.05% | 0.00% | 0.03% |
Using Eq.6 on the 2,144 drugs in the benchmark dataset 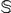 that had the chemical-chemical interaction information.
that had the chemical-chemical interaction information.
Using Eq.10 on the 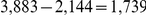 drugs in the benchmark dataset
drugs in the benchmark dataset  that had no chemical-chemical interaction information.
that had no chemical-chemical interaction information.
Using the integrated method by hybridizing Eq.6 and Eq.10 on the 3,883 drugs in the benchmark dataset  as given in Supporting Information S1.
as given in Supporting Information S1.
Performance of the Interaction-Based Method
For the 2,144 drugs in 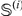 we could use Eq.6 to conduct the prediction. The results thus obtained are listed in column 2 of
Table 2
, from which we can see that the 1st-order prediction by the jackknife test on the 2,114 drugs was 67.72%. The success rates generally followed a descending trend with increasing of the order number, indicating that the predicted ATC-classes were well sorted for each of the samples investigated. The average number of the ATC-classes in
we could use Eq.6 to conduct the prediction. The results thus obtained are listed in column 2 of
Table 2
, from which we can see that the 1st-order prediction by the jackknife test on the 2,114 drugs was 67.72%. The success rates generally followed a descending trend with increasing of the order number, indicating that the predicted ATC-classes were well sorted for each of the samples investigated. The average number of the ATC-classes in 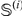 was
was 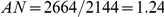 (see Eq.12). Thus, it follows according to Eq.14 that
(see Eq.12). Thus, it follows according to Eq.14 that  , meaning that the first 2-order predictions should be taken into consideration. Substituting these data into Eq.13, we obtained the overall success rate by the predictions of the first two orders for the 2,144 drugs in
, meaning that the first 2-order predictions should be taken into consideration. Substituting these data into Eq.13, we obtained the overall success rate by the predictions of the first two orders for the 2,144 drugs in  was
was 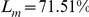 , indicating that the interaction-based method is quite promising in identifying the ATC-classed of drugs. However, this method could only be used to deal with those drugs that had the chemical-chemical interaction information.
, indicating that the interaction-based method is quite promising in identifying the ATC-classed of drugs. However, this method could only be used to deal with those drugs that had the chemical-chemical interaction information.
Performance of Similarity-Based Method
For the remaining 1,739 drugs in the dataset 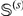 (cf. Eq.15) that did not have the chemical-chemical information, the similarity-based method (cf. Eq.10) was used as a backup, and the results thus obtained are shown in column 3 of
Table 2
. It can be seen from there that the 1st-order prediction by the jackknife test on the 1,739 drugs was 78.49%. The average number of ATC-classes for the drugs in
(cf. Eq.15) that did not have the chemical-chemical information, the similarity-based method (cf. Eq.10) was used as a backup, and the results thus obtained are shown in column 3 of
Table 2
. It can be seen from there that the 1st-order prediction by the jackknife test on the 1,739 drugs was 78.49%. The average number of ATC-classes for the drugs in 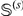 was
was 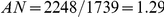 (see Eq.12), and hence we have
(see Eq.12), and hence we have 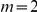 (Eq.14), meaning that the first 2-order predictions should be taken into account. Substituting these data into Eq.13, we obtained the overall success rate by the first two orders predictions for the 1,739 drugs without the chemical-chemical interaction information was 75.31%, indicating that the similarity-based method was quite promising as well.
(Eq.14), meaning that the first 2-order predictions should be taken into account. Substituting these data into Eq.13, we obtained the overall success rate by the first two orders predictions for the 1,739 drugs without the chemical-chemical interaction information was 75.31%, indicating that the similarity-based method was quite promising as well.
At a first glance at
Table 2
, it looks like that the success rates by the similarity-based method (Eq.10) are higher than those by the interaction-based method (Eq.6). However, since the success rates by the two methods as reported in
Table 2
were derived from two different datasets, 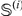 and
and 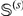 (cf. Eq.15) respectively, they might not able to reflect the true superiority between the two methods. To make a comparison between them in a more fair manner, let us construct a new dataset, denoted as
(cf. Eq.15) respectively, they might not able to reflect the true superiority between the two methods. To make a comparison between them in a more fair manner, let us construct a new dataset, denoted as 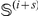 . It consists of 2,138 drugs with each containing both chemical-chemical interaction and chemical-chemical similarity informations. The details of such a dataset is given in Supporting Information S2.
. It consists of 2,138 drugs with each containing both chemical-chemical interaction and chemical-chemical similarity informations. The details of such a dataset is given in Supporting Information S2.
Listed in
Table 3
are the results obtained by the methods in identifying the 14 main ATC classes for the 2,138 drugs in the 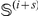 dataset. As we can see from the table, the 1st-order prediction accuracy by the interaction-based method was 67.40%, while that by the similarity-based method was 40.36%.
dataset. As we can see from the table, the 1st-order prediction accuracy by the interaction-based method was 67.40%, while that by the similarity-based method was 40.36%.
Table 3. A comparison between the similarity-based method (Eq.10) and the interaction-based method (Eq.6) in identifying the 2,138 drugs in the 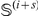 dataset (cf. Supporting Information S2).
dataset (cf. Supporting Information S2).
| Prediction order | Similarity-based | Interaction-based | Difference |
| 1 | 40.36% | 67.40% | 27.04% |
| 2 | 13.89% | 21.09% | 7.20% |
| 3 | 9.17% | 13.47% | 4.30% |
| 4 | 5.99% | 7.16% | 1.17% |
| 5 | 3.32% | 4.91% | 1.59% |
| 6 | 2.76% | 3.46% | 0.70% |
| 7 | 0.65% | 1.54% | 0.89% |
| 8 | 0.23% | 0.75% | 0.52% |
| 9 | 0.09% | 0.75% | 0.66% |
| 10 | 0.05% | 0.56% | 0.51% |
| 11 | 0.05% | 0.09% | 0.04% |
| 12 | 0.00% | 0.33% | 0.33% |
| 13 | 0.09% | 0.09% | 0.00% |
| 14 | 0.05% | 0.05% | 0.05% |
The average number of ATC-classes for the drugs in 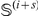 was 1.24 (see Eq.12), and hence we have
was 1.24 (see Eq.12), and hence we have  (Eq.14), meaning that the first 2-order predictions should be taken into account. Substituting these data into Eq.13, we obtained the overall success rate by the 1st two orders predictions for the 2,138 drugs in
(Eq.14), meaning that the first 2-order predictions should be taken into account. Substituting these data into Eq.13, we obtained the overall success rate by the 1st two orders predictions for the 2,138 drugs in 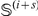 by the interaction-based method (Eq.6) was 71.26%, while that by the similarity-based method (Eq.10) was only 43.69%, indicating that the interaction-based method is superior to the similarity-based method in identifying the ATC-classes of drugs. That is why in the integrated method the first step was to use the interaction method (Eq.6) to identify the ATC-classes for any query drugs. When, and only when no meaningful result was obtained by the interaction-based method, was the similarity-based method (Eq.10) used as a backup to continue the prediction (see the Section of “Prediction by Integrating the Interaction-Based and Similarity-Based Methods”).
by the interaction-based method (Eq.6) was 71.26%, while that by the similarity-based method (Eq.10) was only 43.69%, indicating that the interaction-based method is superior to the similarity-based method in identifying the ATC-classes of drugs. That is why in the integrated method the first step was to use the interaction method (Eq.6) to identify the ATC-classes for any query drugs. When, and only when no meaningful result was obtained by the interaction-based method, was the similarity-based method (Eq.10) used as a backup to continue the prediction (see the Section of “Prediction by Integrating the Interaction-Based and Similarity-Based Methods”).
Performance of Integrated Prediction Method
Shown in the 4th column of
Table 2
are the results obtained by the integrated method in identifying the 14 main ATC classes for the 3,883 drugs in the benchmark dataset 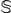 . As we can see there, the 1st-order prediction accuracy was 72.55%. The average numbers of ATC-classes for the drugs in
. As we can see there, the 1st-order prediction accuracy was 72.55%. The average numbers of ATC-classes for the drugs in 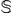 was
was 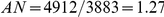 (see Eq.12). Thus, it follows according to Eq.14 that
(see Eq.12). Thus, it follows according to Eq.14 that 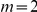 , meaning that the first 2-order predictions should be taken into consideration. Substituting these data into Eq.13, we obtained the overall success rate by the first two orders predictions for the drugs in
, meaning that the first 2-order predictions should be taken into consideration. Substituting these data into Eq.13, we obtained the overall success rate by the first two orders predictions for the drugs in 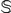 was 73.25%.
was 73.25%.
These results indicate that the integrated method performed quite well in identifying drugs among their 14 main ATC-classes, and that more attention should be paid to the results hit by the first two order predictions because they covered more than 70% of the true ATC-classes.
Finally, it is instructive to point out that although the above demonstrations were given for identifying query drug compounds among their main (or 1st level) classification, the method developed here can be straightforwardly extended to cover the 2nd, 3rd, 4th, 5th or any lower-level classification as long as the corresponding statistically significant datasets for training the predictor are available.
Supporting Information
List of the 4,376 drugs in the ATC classification system extracted from KEGG.
(PDF)
This dataset 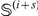 contains 2,138 drugs classified into 14 main ATC classes. Each of the drugs listed here contains both chemical-chemical interaction and chemical-chemical similarity informations. Among the 2,138 different drugs (2,655 virtual drugs), 1,838 belong to one class; 190 to two classes; 57 to three classes, 19 to four classes, 14 to five classes, and 20 to six classes. None of the drugs listed here belongs to seven and more classes.
contains 2,138 drugs classified into 14 main ATC classes. Each of the drugs listed here contains both chemical-chemical interaction and chemical-chemical similarity informations. Among the 2,138 different drugs (2,655 virtual drugs), 1,838 belong to one class; 190 to two classes; 57 to three classes, 19 to four classes, 14 to five classes, and 20 to six classes. None of the drugs listed here belongs to seven and more classes.
(PDF)
Acknowledgments
The authors are very much indebted to the Academic Editor for taking time from her busy schedule to edit our paper. Many thanks are also due to the two anonymous experts for their constructive comments, which were very helpful for strengthening the presentation of this paper.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This contribution is supported by National Basic Research Program of China (2011CB510102, 2011CB510101), National Natural Science Foundation of China (No. 31170952), Innovation Program of Shanghai Municipal Education Commission (No. 11ZZ143, No. 12YZ120, No. 12ZZ087) and Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dunkel M, Günther S, Ahmed J, Wittig B, Preissner R. SuperPred: drug classification and target prediction. Nucleic acids research. 2008;36:W55–W59. doi: 10.1093/nar/gkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurulingappa H, Kolářik C, Hofmann-Apitius M, Fluck J. Concept-based semi-automatic classification of drugs. Journal of chemical information and modeling. 2009;49:1986–1992. doi: 10.1021/ci9000844. [DOI] [PubMed] [Google Scholar]
- 3.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic acids research. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: Structure, Function, and Genetics (Erratum: ibid, 2001, Vol44, 60) 2001;43:246–255. doi: 10.1002/prot.1035. [DOI] [PubMed] [Google Scholar]
- 6.Cai YD, Lu L, Chen L, He JF. Predicting subcellular location of proteins using integrated-algorithm method. Molecular Diversity. 2010;14:551–558. doi: 10.1007/s11030-009-9182-4. [DOI] [PubMed] [Google Scholar]
- 7.Chou KC, Shen HB. ProtIdent: A web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem Biophys Res Comm. 2008;376:321–325. doi: 10.1016/j.bbrc.2008.08.125. [DOI] [PubMed] [Google Scholar]
- 8.Cai YD, Liu XJ, Xu X, Zhou GP. Support vector machines for predicting protein structural class. BMC bioinformatics. 2001;2:3. doi: 10.1186/1471-2105-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chou KC, Shen HB. Signal-CF: a subsite-coupled and window-fusing approach for predicting signal peptides. Biochem Biophys Res Comm. 2007;357:633–640. doi: 10.1016/j.bbrc.2007.03.162. [DOI] [PubMed] [Google Scholar]
- 10.Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). Journal of Theoretical Biology. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Molecular systems biology. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang T, Shi XH, Wang P, He Z, Feng KY, et al. Analysis and prediction of the metabolic stability of proteins based on their sequential features, subcellular locations and interaction networks. PLoS ONE. 2010;5:e10972. doi: 10.1371/journal.pone.0010972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu L, Huang T, Shi X, Lu WC, Cai YD, et al. Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PLoS ONE. 2011;6:e14556. doi: 10.1371/journal.pone.0014556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karaoz U, Murali TM, Letovsky S, Zheng Y, Ding C, et al. Whole-genome annotation by using evidence integration in functional-linkage networks. Proc Natl Acad Sci U S A. 2004;101:2888–2893. doi: 10.1073/pnas.0307326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou KC, Shen HB. A new method for predicting the subcellular localization of eukaryotic proteins with both single and multiple sites: Euk-mPLoc 2.0. PLoS ONE. 2010;5:e9931. doi: 10.1371/journal.pone.0009931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu ZC, Xiao X, Chou KC. iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Molecular BioSystems. 2011;7:3287–3297. doi: 10.1039/c1mb05232b. [DOI] [PubMed] [Google Scholar]
- 17.Chou KC, Wu ZC, Xiao X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Molecular Biosystems. 2012;8:629–641. doi: 10.1039/c1mb05420a. [DOI] [PubMed] [Google Scholar]
- 18.Chou KC, Shen HB. Review: Recent progresses in protein subcellular location prediction. Analytical Biochemistry. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn M, von Mering C, Campillos M, Jensen LJ, Bork P. STITCH: interaction networks of chemicals and proteins. Nucleic Acids Res. 2008;36:D684–688. doi: 10.1093/nar/gkm795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou KC, Forsen S. Graphical rules for enzyme-catalyzed rate laws. Biochemical Journal. 1980;187:829–835. doi: 10.1042/bj1870829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou GP, Deng MH. An extension of Chou's graphic rules for deriving enzyme kinetic equations to systems involving parallel reaction pathways. Biochemical Journal. 1984;222:169–176. doi: 10.1042/bj2220169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou KC. Graphic rules in steady and non-steady enzyme kinetics. Journal of Biological Chemistry. 1989;264:12074–12079. [PubMed] [Google Scholar]
- 23.Chou KC. Review: Applications of graph theory to enzyme kinetics and protein folding kinetics. Steady and non-steady state systems. Biophysical Chemistry. 1990;35:1–24. doi: 10.1016/0301-4622(90)80056-d. [DOI] [PubMed] [Google Scholar]
- 24.Althaus IW, Gonzales AJ, Chou JJ, Diebel MR, Chou KC, et al. The quinoline U-78036 is a potent inhibitor of HIV-1 reverse transcriptase. Journal of Biological Chemistry. 1993;268:14875–14880. [PubMed] [Google Scholar]
- 25.Chou KC, Kezdy FJ, Reusser F. Review: Steady-state inhibition kinetics of processive nucleic acid polymerases and nucleases. Analytical Biochemistry. 1994;221:217–230. doi: 10.1006/abio.1994.1405. [DOI] [PubMed] [Google Scholar]
- 26.Andraos J. Kinetic plasticity and the determination of product ratios for kinetic schemes leading to multiple products without rate laws: new methods based on directed graphs. Canadian Journal of Chemistry. 2008;86:342–357. [Google Scholar]
- 27.Chou KC. Graphic rule for drug metabolism systems. Current Drug Metabolism. 2010;11:369–378. doi: 10.2174/138920010791514261. [DOI] [PubMed] [Google Scholar]
- 28.Zhou GP. The disposition of the LZCC protein residues in wenxiang diagram provides new insights into the protein-protein interaction mechanism. Journal of Theoretical Biology. 2011;284:142–148. doi: 10.1016/j.jtbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou KC, Lin WZ, Xiao X. Wenxiang: a web-server for drawing wenxiang diagrams. Natural Science. 2011;3:862–865. [Google Scholar]
- 30.Zhou GP. The Structural Determinations of the Leucine Zipper Coiled-Coil Domains of the cGMP-Dependent Protein Kinase I alpha and its Interaction with the Myosin Binding Subunit of the Myosin Light Chains Phosphase. Proteins & Peptide Letters. 2011;18:966–978. doi: 10.2174/0929866511107010966. [DOI] [PubMed] [Google Scholar]
- 31.Hattori M, Okuno Y, Goto S, Kanehisa M. Development of a chemical structure comparison method for integrated analysis of chemical and genomic information in the metabolic pathways. Journal of the American Chemical Society. 2003;125:11853–11865. doi: 10.1021/ja036030u. [DOI] [PubMed] [Google Scholar]
- 32.Jaccard P. THE The Distribution of the Flora in the Alpine Zone. 1. New Phytologist. 1912;11:37–50. [Google Scholar]
- 33.Watson GA. An algorithm for the single facility location problem using the Jaccard metric. SIAM Journal on Scientific and Statistical Computing. 1983;4:748–756. [Google Scholar]
- 34.Chou KC, Zhang CT. Review: Prediction of protein structural classes. Critical Reviews in Biochemistry and Molecular Biology. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- 35.Chou KC, Shen HB. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms (updated version: Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms, Natural Science, 2010, 2, 1090–1103). Nature Protocols. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- 36.Esmaeili M, Mohabatkar H, Mohsenzadeh S. Using the concept of Chou's pseudo amino acid composition for risk type prediction of human papillomaviruses. Journal of Theoretical Biology. 2010;263:203–209. doi: 10.1016/j.jtbi.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Georgiou DN, Karakasidis TE, Nieto JJ, Torres A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou's pseudo amino acid composition. Journal of Theoretical Biology. 2009;257:17–26. doi: 10.1016/j.jtbi.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Chou KC, Wu ZC, Xiao X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS One. 2011;6:e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mohabatkar H, Mohammad Beigi M, Esmaeili A. Prediction of GABA(A) receptor proteins using the concept of Chou's pseudo-amino acid composition and support vector machine. Journal of Theoretical Biology. 2011;281:18–23. doi: 10.1016/j.jtbi.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 40.Chou KC, Shen HB. Plant-mPLoc: A Top-Down Strategy to Augment the Power for Predicting Plant Protein Subcellular Localization. PLoS ONE. 2010;5:e11335. doi: 10.1371/journal.pone.0011335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu ZC, Xiao X, Chou KC. iLoc-Gpos: A Multi-Layer Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Gram-Positive Bacterial Proteins. Protein & Peptide Letters. 2012;19:4–14. doi: 10.2174/092986612798472839. [DOI] [PubMed] [Google Scholar]
- 42.Gu Q, Ding YS, Zhang TL. Prediction of G-Protein-Coupled Receptor Classes in Low Homology Using Chou's Pseudo Amino Acid Composition with Approximate Entropy and Hydrophobicity Patterns. Protein & Peptide Letters. 2010;17:559–567. doi: 10.2174/092986610791112693. [DOI] [PubMed] [Google Scholar]
- 43.Lin J, Wang Y. Using a novel AdaBoost algorithm and Chou's pseudo amino acid composition for predicting protein subcellular localization. Protein & Peptide Letters. 2011;18:1219–1225. doi: 10.2174/092986611797642797. [DOI] [PubMed] [Google Scholar]
- 44.Mohabatkar H. Prediction of cyclin proteins using Chou's pseudo amino acid composition. Protein & Peptide Letters. 2010;17:1207–1214. doi: 10.2174/092986610792231564. [DOI] [PubMed] [Google Scholar]
- 45.Xiao X, Wu ZC, Chou KC. iLoc-Virus: A multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. Journal of Theoretical Biology. 2011;284:42–51. doi: 10.1016/j.jtbi.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Lin WZ, Fang JA, Xiao X, Chou KC. iDNA-Prot: Identification of DNA Binding Proteins Using Random Forest with Grey Model. PLoS ONE. 2011;6:e24756. doi: 10.1371/journal.pone.0024756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang P, Xiao X, Chou KC. NR-2L: A Two-Level Predictor for Identifying Nuclear Receptor Subfamilies Based on Sequence-Derived Features. PLoS ONE. 2011;6:e23505. doi: 10.1371/journal.pone.0023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of the 4,376 drugs in the ATC classification system extracted from KEGG.
(PDF)
This dataset contains 2,138 drugs classified into 14 main ATC classes. Each of the drugs listed here contains both chemical-chemical interaction and chemical-chemical similarity informations. Among the 2,138 different drugs (2,655 virtual drugs), 1,838 belong to one class; 190 to two classes; 57 to three classes, 19 to four classes, 14 to five classes, and 20 to six classes. None of the drugs listed here belongs to seven and more classes.
(PDF)