

Abstract
The neutrophil gelatinase-associated lipocalin 2 (LCN2) is a critical inflammatory mediator persistently induced during endotoxemia, contributing to tubular damage and kidney failure. The intracellular process responsible for persistent induction of LCN2 by bacterial endotoxin Lipopolysaccharide (LPS) is not well understood. Using primary kidney fibroblasts, we observed that LPS-induced LCN2 expression requires a coupled circuit involving an early transient phase of AP-1 path and a late persistent phase of C/EBPδ path, both of which are dependent upon the interleukin 1 receptor associated kinase 1 (IRAK-1). Using immunoprecipitation analysis we observed transient binding of AP-1 to the promoters of both TNFα and C/ebpδ. On the other hand, we only observed persistent binding of C/EBPδ to its own promoter but not on TNFα. Blockage of new protein synthesis using cyclohexamide significantly reduced the expression of C/EBPδ as well as LCN2. By chromatin immunoprecipitation analyses, we demonstrated that LPS recruited C/EBPδ to the Lcn2 promoter in WT, but not IRAK-1 deficient fibroblasts. A differential equation-based computational model captured the dynamic circuit leading to the persistent induction of LCN2. In vivo, we observed elevated levels of LCN2 in kidneys harvested from LPS-injected WT mice as compared to IRAK-1 deficient mice. Taken together, this study has identified an integrated intracellular network involved in the persistent induction of LCN2 by LPS.
Introduction
Initially identified in neutrophils as a gelatinase-associated small protein [1], LCN2 was recognized as an innate defense molecule by sequestering and depleting iron-containing siderophores and curbing bacterial growth [2]. Recent studies reveal that LCN2 can be potently induced by inflammatory stimulants and widely expressed in vital organs and tissues such as kidney, heart, and brain [3], [4], [5], [6]. Elevated level of LCN2 in kidney is a well-recognized marker for both chronic kidney diseases, reflecting the extent of kidney damage [7], [8]. LCN2 is not only a marker, but also a key contributor of kidney disease, as reflected by the alleviation of chronic kidney damage in mice with Lcn2 gene deletion [6].
Bacterial endotoxin (lipopolysaccharide-LPS) is a potent inducer of LCN2 [9], [10], [11]. Elevated levels of endotoxin are seen in both acute and chronic conditions. Acute endotoxemia leads to septic shock and multi-organ damages including acute kidney failure [12]. In contrast, chronic endotoxemia associated with obesity, aging, and other adverse health conditions can lead to persistent inflammatory complications and chronic diseases [11], [13], [14]. LCN2 levels persist during both acute and chronic phases of kidney damage, and may serve as a key propagating factor for chronic kidney disease [6]. However, the molecular mechanism underlying the persistent expression of LCN2 is not well understood.
Studies using other inflammatory stimulants such as IL-17 and IL-1β indicate that the induction of LCN2 requires multiple transcription factors including AP-1 and C/EBPδ [9], [15]. AP-1 activation by LPS is transient and responsible for the transient induction of pro-inflammatory mediators [16]. On a separate report, C/EBPδ was implicated in propagating the persistent activation of TLR4 pathway [17].
Based on these observations, we examined the molecular circuit underlying the persistent induction of LCN2 by LPS in kidney fibroblasts. We identified that IRAK-1, a key TLR4 intracellular signaling component, is involved in coordinating both the transient activation of AP-1, as well as the persistent activation of C/EBPδ, which are collectively responsible for the sustained expression of LCN2. We further performed a differential equation-based computational modeling, which complemented our experimental data and revealed a dynamic intracellular signaling circuit responsible for the persistent induction of LCN2 by LPS.
Materials and Methods
Reagents
LPS (Escherichia coli 0111:B4) and Cycloheximide were obtained from Sigma Aldrich. Anti-LCN2 antibody was purchased from R&D Systems. Anti- IkBα antibody was purchased from Cell Signaling. Anti-Lamin B (ab-16048) was purchased from Abcam. Anti-C/EBPδ (M-17), anti-cJun (H-79), anti-P65 (F-6), anti-GAPDH (FL-335), anti-IRAK-1 (F-4), and anti-β-actin (C-4) antibodies were from Santa Cruz Biotechnology.
Kidney Fibroblast cultures
Both WT and IRAK-1−/− C57/BL/6 mice were housed and bred in the Derring Hall Animal Facility in compliance with approved Animal Care and Use Committee protocols at Virginia Polytechnic Institute and State University. Murine kidney fibroblasts were isolated as previously described [18]. Fibroblast cells were grown in 50∶50 DMEM/F12 #10-092-CV (Mediatech™, Inc., VA) supplemented with 10% FBS #SH30071.03 (Hyclone™) and 1% Penicillin/Streptomycin at 37°C with 5% CO2.
Real time RT-PCR (qRT-PCR)
Total RNAs were harvested from murine kidney fibroblasts treated with or without LPS using Trizol® Reagent (Invitrogen) according to the manufacturer's protocol. cDNAs were generated using the High Capacity cDNA Reverse Transcription Kit (Applied BioSystems™, Foster, CA). Real-time PCR analyses were performed using the iQ Sybr® Green Supermix (BioRad™ Laboratories, Hercules, CA) on an IQ5 thermocycler (BioRad). The relative levels of transcripts were calculated using the ΔΔCt method using GAPDH as the internal control. The relative levels of mRNA from the untreated samples were adjusted to 1 and served as the basal control value. The following primers were used perform qRT-PCR: mouse LCN2 forward: 5′- TTT CAC CCG CTT TGC CAA GT-3′, reverse: 5′-GTC TCT GCG CAT CCC AGT CA-3′; mouse C/ebpδ forward: 5′-ACT TCA GCG CCT ACA TTG ACT CCA-3′, reverse: 5′-TGT TGA AGA GGT CGG CGA AGA GTT-3′; mouse GAPDH forward: 5′-AAC TTT GGC ATT GTG GAA GGG CTC-3′, reverse: 5′- GGA AGA GTG GGA GTT GCT GTT GA-3′; mouse TNFα forward: 5′-AGC CGA TGG GTT GTA CCT TGT CTA-3′, reverse: 5′-TGA GAT AGC AAA TCG GCT GAC GGT-3′; mouse Il-6 forward: 5′-ATC CAG TTG CCT TCT TGG GAC TGA-3′, reverse: 5′- TAA GCC TCC GAC TTG TGA AGT GGT-3′; mouse SOCS1 forward: 5′-AGT CGC CAA CGG AAC TGC TTC TT-3′, reverse: 5′-ACG TAG TGC TCC AGC AGC TCG AAA-3′; and mouse ATF3 forward: 5′-TCA AGG AAG AGC TGA GAT TCG CCA-3′, reverse: 5′-GTT TCG ACA CTT GGC AGC AGC AAT-3′.
Western Blot Analysis
Isolation of whole cell, cytoplasmic, and nuclear lysates were performed as described previously [19]. Briefly, fibroblasts were lysed on ice using lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10% Triton X-100, 10 mM KCl, 0.5 mM EDTA, 0.5 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1 ug/mL leupeptin, 1 ug/mL pepstatin) for 30 minutes. Samples were centrifuged at 5000 rpm for 10 minutes at 4°C. The supernatants were saved as the cytoplasmic extract and the pelleted nuclei were re-suspend and lysed in a high salt buffer (20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.4 M NaCl, 0.2 mM EDTA, 0.5 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride) on ice for 30 minutes. The samples were then centrifuged at 12,000 RPM for 20 minutes at 4°C and supernatant kept as the nuclear extract. Protein samples were analyzed by Western Blot as described before. Images were quantified with Fujifilm Multi Gauge software normalized against β actin, GAPDH, or Lamin B.
Chromatin Immunoprecipitation (ChIP) assays
Protein and chromatin from treated cells were cross-linked using 1% formaldehyde solution in complete media for 15 minutes with gentle rocking every 3 minutes at room temperature. Cells were washed twice with cold PBS and treated with a glycine solution for 5 minutes to stop the cross-linking reaction. The cells were then lysed in an ice cold buffer containing SDS and protease inhibitor cocktail. The samples were then subject to sonication in an ice water bath to shear the chromatin for 6 minutes (30 seconds on and 30 seconds off). The sheared chromatin was processed using Chip-IT Express Kit #53008 from Active Motif™. The immune-precipitated chromatin was analyzed by PCR using primer pairs that span the promoter regions of either Lcn2, C/ebpδ, and Tnfα. The primer sequences are as follows: mouse pLcn2 forward: 5′-TGA CCC ACA AGC AGT GCC CTG T-3′, reverse: 5′-ACT TGG CAA GAT TTC TGT CCC-3′; mouse pC/ebpδ forward: 5′-ACA AAC AGG AAG GAG GGA AG-3′, reverse: 5′-TCC AAG TTG GGC TGT CA-3′; and mouse pTnfα forward: 5′-AGG AGA TTC CTT GAT GCC TGG GT-3′, reverse: 5′-TTT CTG TTC TCC CTC CTG GCT AGT-3′.
In vivo LPS injection
Female IRAK-1−/− and WT C57/BL/6 mice of 12 weeks old received intraperitoneal injections with 25 mg/kg of LPS. Six hours after the injection, mice were sacrificed and the whole kidney tissue was harvested for subsequent mRNA analysis by real time RT-PCR and protein analyses by Western blot.
Computational analysis
A differential-equation based computational modeling is performed as previously described [20]. A detailed description of the modeling approach is provided in the supplementary document accompanying this manuscript.
Statistical Analysis
Data significance was determined using the student t-test. P-values less than 0.05 were considered statistically significant as indicated by an asterisk (*).
Results
LPS induces persistent expression of LCN2 in kidney fibroblasts in an IRAK-1 dependent fashion
We first compared the expression profiles of selected inflammatory genes in fibroblasts stimulated with LPS. As shown in Figure 1A, the induction of Tnfα message by LPS was transient, peaked at the 2 hour time point post challenge, and returned to basal values at the 16 hour time point. In contrast, the expression of Lcn2 mRNA persisted throughout the course of LPS treatment (Fig. 1B). Because IRAK-1 is one of the key downstream components of TLR4 pathway, we evaluated whether IRAK-1 is involved in both the transient induction of Tnfα and persistent induction of Lcn2 message. As shown in Figure 1A and 1B, the inductions of both Tnfα and Lcn2 were significantly reduced in IRAK-1 deficient kidney fibroblasts. We also confirmed the induction of LCN2 protein in wild type (WT) cells by LPS (Fig. 1C).
Figure 1. LPS stimulation induces a persistent induction of LCN2 in kidney fibroblasts.

(A) LPS (100 ng/ml) induces a transient induction of Tnfα mRNA. (B) LPS (100 ng/ml) induces a persistent induction of Lcn2 mRNA Transcript levels were measured by qRT-PCR as described above. The results are expressed as means +/− standard deviation performed in triplicate. (C) LCN2 protein levels persist after 24 hours of LPS stimulation. The levels of LCN2 were visualized by western blot.
LPS induces transient activation of AP-1 and persistent induction of C/EBPδ in kidney fibroblasts
To determine the potential mechanism for the persistent induction of LCN2 in kidney fibroblasts by LPS, we examined the activation status of relevant transcription factors including NFκB, AP-1 and C/EBPδ. As shown in Figure 2A, LPS treatment caused no apparent decrease in the levels of Iκbα in WT fibroblasts, indicating a lack of robust NFκB activation by LPS in kidney fibroblasts. Further supporting this claim, we observed that the residue nuclear p65/RelA remained constant following LPS treatment (Fig. 2B). In contrast, we observed a rapid yet transient induction of AP-1/c-Jun. Nuclear levels of AP-1/c-Jun was induced by LPS, reached its peak level by 60 to 120 minutes, and decreased at 240 minutes after LPS challenge (Fig. 2C). C/EBPδ, on the other hand, was elevated after 24 hr of LPS treatment (Fig. 2D and 2E). Given the fact that Western blot may not give rise to quantitative result, we performed RT-PCR analysis of C/ebpδ expression induced by LPS. As shown in Figure 2F, LPS significantly induced the expression of C/ebpδ, and such induction is dependent on IRAK-1.
Figure 2. LPS induces transient activation of AP-1 and persistent induction of C/EBPδ in kidney fibroblasts.

WT fibroblasts were treated with LPS (100 ng/ml) for the indicated time periods. (A) LPS fails to significantly induce IκBα degradation. Whole cell lysates were harvested and analyzed by Western blot using an IκBα specific antibody. (B) LPS fails to significantly induce p65 nuclear translocation in kidney fibroblasts. Nuclear cell lysates were harvested and analyzed by Western blot using an anti-p65 antibody. (C) LPS induces the active translocation of AP-1/c-Jun. Nuclear lysates were harvested and analyzed by Western blot. (D&E) LPS induces a persistent increase of C/EBPδ protein. Whole cell lysates were harvested and analyzed by Western blot. (F) LPS significantly induces the expression of C/ebpδ dependent upon IRAK-1. WT and IRAK-1 deficient cells were treated with LPS for indicated time period. Message levels of C/ebpδ were determined by RT-PCR. n = 3; *P<0.05.
Based on above observation and previous studies, we propose that a coupled step-wise process may lead to the persistent activation and induction of C/EBPδ. LPS may first induce an initial C/EPBδ expression primarily through a transient activation of AP-1 and residue NFκB in the nucleus [21]. Subsequently, newly synthesized C/EBPδ can auto-induce its own expression and contribute to a steady induction of C/EBPδ [17] and its downstream target gene LCN2. To test this hypothesis, we performed Chromatin immunoprecipitation (ChIP) analysis to determine the recruitment of c-Jun and C/EBPδ to the promoter of C/ebpδ. As shown in Figure 3, LPS transiently increased the recruitment of c-Jun to the promoter of C/ebpδ (Figure 3A) and Tnfα (Figure 3B), peaking at 4 hr for C/ebpδ and 1 hr for Tnfα promoter, and dropping off at the 8 hr time point. In contrast, increased recruitment of C/EBPδ at the 8 hr time point was evident on the promoter of C/ebpδ, but not Tnfα.
Figure 3. LPS induces transient recruitment of c-Jun to the promoters of Tnfα and C/ebpδ, and induces prolonged recruitment of C/EBPδ to the promoter of C/ebpδ.

WT kidney fibroblasts were treated with or without LPS for a time course. Nuclear lysates were then subjected to ChIP analysis to examine relative binding to the promoter of C/ebpδ (A) or Tnfα (B).
Both De novo synthesis and subsequent activation of C/EBPδ is required for the expression of LCN2
To further confirm that de novo synthesis of C/EBPδ is involved in LPS-induced LCN2 expression, we applied cycloheximide (CHX), a known protein synthesis inhibitor, to the culture medium. As shown in Figure 4A, CHX significantly reduced LPS-mediated induction of Lcn2 message, suggesting that new protein synthesis is required for the induction of LCN2 gene expression. In contrast, CHX has no inhibitory effect on LPS-mediated expression of C/ebpδ message (Figure 4B). This is in agreement with a previous finding that the expression of C/EBPδ and its subsequent transcriptional activation are two separate events controlled by two distinct upstream processes [21]. To further confirm the role of C/EBPδ in LPS induced expression of Lcn2, we examined the recruitment of C/EBPδ to the promoter of Lcn2 following LPS challenge. As shown in Figure 4C, LPS dramatically prolonged the elevated recruitment of C/EBPδ to the Lcn2 promoter in WT cells.
Figure 4. Lcn2 transcription requires new protein synthesis, but C/ebpδ does not. C/EBPδ is recruited to the promoter of LCN2 upon LPS stimulation in an IRAK-1 dependent manner.

(A) Cyclohexamide, a protein synthesis inhibitor, blocks Lcn2 transcription upon LPS stimulation. (B) The induction of C/ebpδ does not require new protein synthesis. (B) Wild-type kidney fibroblasts were untreated (DMSO) or pretreated with cyclohexamide 5 ug/mL for 1 hour. Immediately following pretreatment, the cells were stimulated with or without LPS for 4 hours. Transcripts were quantitated using qRT-PCR and standardized using Gapdh as the internal loading control. Each experiment was performed in triplicate. *P<0.05 (C) The recruitment of C/EBPδ to the promoter of LCN2 upon LPS stimulation depends upon IRAK-1. Nuclear lysates were subject to ChIP analysis using a C/EBPδ specific antibody. The arrow points to the specific amplification signal for C/ebpδ promoter. The * sign indicates a non-specific amplification.
The lack of inducible negative regulators contributes to prolonged expression of LCN2 in kidney fibroblasts upon LPS stimulation
Through the classical NFκB pathway, LPS is known to induce negative regulators that subsequently turn off the expression of pro-inflammatory genes. Among the negative regulators, SOCS1 suppresses the upstream LPS signaling pathway by inhibiting IRAK-1 [22]. On the other hand, ATF3 is shown to compete and suppress the activity of C/EBPδ [17], [23]. Given the fact that we did not observe noticeable activation of the NFκB pathway, we hypothesize that LPS may not be able to induce related negative regulators in kidney fibroblasts. Indeed, we demonstrated that LPS failed to induce the expression of Socs 1 and Atf 3 (Fig. 5A). As a consequence, we observed that there was no apparent degradation of IRAK-1 protein in kidney fibroblasts following a 4 hr LPS treatment (Fig. 5B). Taken together, our data indicate that LPS treatment in kidney fibroblasts failed to induce negative regulators that modulate the persistent activation of C/EBPδ, leading to a sustained induction of its downstream gene Lcn2.
Figure 5. Socs1 and Atf3, negative regulators of TLR4 signaling, are not induced and IRAK-1 remains intact upon LPS stimulation in kidney fibroblasts.

(A) The expressions of Socs1 and Atf3 were not induced after stimulation with LPS. Wild-type kidney fibroblasts were either untreated or treated with 100 ng/mL LPS for 4, 6, or 10 hours. Socs1, Atf3, and Il-6 transcripts were measured by real time RT-PCR assays and standardized against Gapdh levels. Each experiment was performed in triplicate. Data is depicted as means +/− standard deviation. (B) LPS does not cause the degradation of IRAK-1. Wild-type kidney fibroblasts were either untreated or treated with 100 ng/mL LPS for either 2 or 4 hours. Whole cell lysates were harvested and analyzed by Western blot with IRAK-1 specific antibodies.
In vivo induction of LCN2 expression in an IRAK-1 dependent fashion
To test the in vivo relevance of our finding, we examined the expression of LCN2 in kidney tissues from WT and IRAK-1 deficient mice following LPS challenge. As shown in Figure 6, LPS injection induced significant induction of Lcn2 messages as well as LCN2 proteins in kidney tissues harvested from LPS-injected WT mice. In contrast, the levels of Lcn2 messages were significantly lower in kidneys harvested from IRAK-1 deficient mice injected with LPS.
Figure 6. Loss of IRAK-1 causes decreased expression of Lcn2 mRNA and protein in vivo.

WT and IRAK-1−/− C57/BL/6 female mice of 12 weeks old (6 each) were intraperitoneally injected with either 30 mg/kg of LPS or PBS for 6 hours. (A) Kidney tissues were extracted and subject to RNA extraction. Lcn2 transcripts were measured by qRT-PCR assays and standardized against their respective controls (mice receiving PBS injections). Data is depicted as three separate mice (both WT and IRAK-1−/−). *P<0.05 (B) Protein lysates were extracted from the kidney tissues and subjected to Western blot. Blots were analyzed using LCN2 specific antibodies. The same blot was probed with β-actin as a loading control.
Computational analyses of the molecular circuit responsible for the persistent induction of LCN2 in kidney fibroblasts
We further fit our experimental data into a differential equation based computation model (Fig. 7). This model takes into consideration of the positive feedback loop involving C/EBPδ that helps to maintain a sustained expression of LCN2.
Figure 7. Computational simulation captures the dynamic features of the experimental data.

(A) The network motif responsible for the transient induction of Tnfα and the persistent induction of Lcn2. (B) Computational simulation of the transient induction of Tnfα and persistent induction of Lcn2. Circles denote actual experimental data. Lines represent computational simulations.
This model is based on a coarse-grained TLR4-signaling network that highlights the core pathways controlling the induction of LCN2. As is shown in Figure 7A, this signaling circuit is composed of both an early-phase module with transient response and a late-phase module with persistent response. Each module can be viewed as an information processor that receives inputs from upstream signals, and generate outputs that regulate downstream response. The two modules are connected. The transcription of C/EBPδ is dependent on AP-1/c-Jun, and serves as the core of the late-phase module.
Table 1 lists the equations in the computational model. Each ODE describes the change of a molecular species in the relative level as a function of time. Parameters such as mRNA degradation rates are estimated based on our data or from existing literatures. Other parameters such as activating threshold k and relative weights K are estimated using genetic algorithm of least square regression (MATLAB® 2009b, the MathWorks, Natick, MI). All parameters are listed in Table 2.
Table 1. Ordinary Differential Equations used in the model.
| Variable | Name | Differential Equations |
| x 1 | c-Jun |
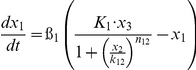
|
| x 2 | MKP-1 |
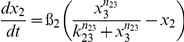
|
| x 3 | MAPK |
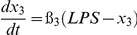
|
| x 4 | TNFα |
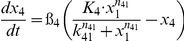
|
| x 5 | IKKε |
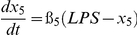
|
| x 6 | C/EBPδ |
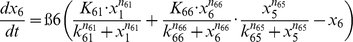
|
| x 7 | LCN2 |
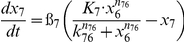
|
Subscripts of parameters denote the direction of regulations. For example, k 61 represents the threshold for c-Jun (x1) to activate C/EBPδ (x6).
Table 2. List of the estimated parameter values.
| Parameter | Value | Description |
| β 1 |
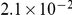
|
Degradation rate of x 1 (based on Western Blot data) |
| β 2 |
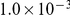
|
Degradation rate of x 2 |
| β 3 |
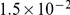
|
Degradation rate of x 3 |
| β 4 |
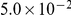
|
Degradation rate of x 4 (based on RT-PCR data) |
| β 5 |

|
Degradation rate of x 5 |
| β 6 |
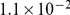
|
Degradation rate of x 6, based on (2) |
| β 7 |
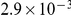
|
Degradation rate of x 7 (based on RT-PCR data) |
| K 1 | 2.1 | Weighted factor |
| K 4 | 11.0 | Weighted factor |
| K 61 | 8.6 | Weighted factor |
| K 66 | 1.3 | Weighted factor |
| K 7 | 1.2 | Weighted factor |
| k 12 |
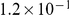
|
Threshold of x 2 to inhibit x 1 |
| k 23 |
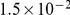
|
Threshold of x 3 to activate x 2 |
| k 41 | 1.7 | Threshold of x 1 to activate x 4 |
| k 61 | 1.4 | Threshold of x 1 to activate x 6 |
| k 66 |
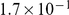
|
Threshold of x 6 to activate x 6 (auto-regulation) |
| k 65 |
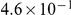
|
Threshold of x 5 to activate x 6 |
| k 76 |
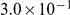
|
Threshold of x 6 to activate x 7 |
| n 12 | 4 | Coefficient of nonlinearity for x 2 to inhibit x 1 |
| n 23 | 4 | Coefficient of nonlinearity for x 3 to activate x 2 |
| n 41 | 4 | Coefficient of nonlinearity for x 1 to activate x 4 |
| n 61 | 4 | Coefficient of nonlinearity for x 1 to activate x 6 |
| n 66 | 4 | Coefficient of nonlinearity for x 6 to activate x 6 |
| n 65 | 4 | Coefficient of nonlinearity for x 5 to activate x 6 |
| n 76 | 4 | Coefficient of nonlinearity for x 6 to activate x 7 |
Parameters such as mRNA degradation rates are estimated based on our data or from existing literatures. Values of other parameters such as activation/inhibition threshold k and relative weights K are estimated to fit in the time course data by genetic algorithm of least square regression. Degradation rates are in the unit of/min. Activation/inhibition thresholds, nonlinearity coefficients and relative weights for multiple inputs are non-dimensionalized.
As is shown in Figure 7B, our computational model based on the proposed signaling circuitry fits well with the experimental data. The model reveals that the essential requirement of the biphasic dynamics of C/EBPδ and LCN2 is the proper timing of the two consecutive modules. The immediate yet transient induction in c-Jun explains the first pulse in C/EBPδ, when the early-phase module dominates the dynamics of the system. As soon as c-Jun decays, the late-phase module relays the signal and an auto-regulation of C/EBPδ gives rise to the persistent induction of both C/EBPδ and LCN2. IKKε plays an important role in ensuring the correct timing of the second module. Experimentally, it was shown that IKKε activates the activity of C/EBPδ, therefore the auto-regulation of C/EBPδ is IKKε-dependent (21).
Taken together, our computational simulation based on the intracellular signaling circuitry identified in this report fitted well with the experimental data, capturing the dynamic intracellular circuits that give rise to transient induction of TNFα and persistent induction of LCN2.
Discussion
In this report, we have identified that LPS can induce a persistent expression of LCN2 in kidney fibroblasts. The intracellular signaling circuit responsible for the persistent induction of LCN2 involves dynamic interaction among AP-1 and C/EBPδ. An early phase of a transient AP-1 activation helps to usher in a late phase of C/EBPδ activation. IRAK-1 is responsible for the propagation of both phases necessary for the persistent induction of LCN2.
Our study shed light on the dynamic and functional circuitry within cells when challenged with bacterial endotoxin. Although empirical knowledge reckons that integrated intracellular circuits instead of linear cascades are more capable of interpreting and responding properly to environmental cues including LPS, almost all existing studies have adopted conventional approaches in defining the signaling cascades downstream of LPS and TLR4 receptors [24]. To this regard, a large number of signaling molecules and pathways have been discovered that can be activated by LPS [25]. However, how these signals are properly integrated and give rise to distinct cellular responses is not clear. This work builds upon conventional information uncovered in the area of LPS signaling within host cells, and reveals an integrated circuit that couples a transient activation of AP-1 with a sustained activation of C/EBPδ.
Our effort is made possible by the integrated approach combining experimental tests and computational analyses. Taken into consideration of the experimental data, we have employed a differential equation-based approach to analyze time-dependent activation of AP-1 and C/EBPδ, as well as expression of Tnfα and Lcn2. As a consequence, genes primarily under the control of AP-1 such as Tnfα experience a transient pattern of expression. The expression of C/ebpδ, on the other hand, is initially activated by AP-1, and subsequently sustained by its own auto-activation [17], [21]. Due to the transient nature of AP-1 activation, we observed a transient dip in the expression of C/ebpδ (around the 4 hr time point). Subsequent self-activation of C/EBPδ then sustains the continued expression of C/ebpδ and its downstream gene Lcn2. A detailed computational analysis can be seen in the supplementary material.
It is likely that there will be significant perturbations as well as additional connections surrounding this circuit in different cell types. Consequently, this may allow distinct outcomes in different cell types in response to bacterial endotoxin. For example, the expression of C/EBPδ in macrophages induced by LPS eventually subsides, due to a novel negative feedback initiated by ATF3 [17]. In contrast, we observed that LPS failed to induce Atf3 in kidney fibroblasts, which may explain the sustained expression and activation of C/EBPδ. This may due to the fact that fibroblasts are less sensitive to the LPS challenge as compared to macrophages. Indeed, our recent study indicates that a much lower LPS dose (∼50 pg/ml) induces a persistent activation of C/EBPδ in macrophages [26], similar to the effect seen in this study using fibroblasts with a high dose LPS. Further supporting this claim, we observed that IRAK-1 is relatively stable in kidney fibroblasts treated with 100 ng/ml LPS. In contrast, it was observed that 100 ng/ml LPS causes IRAK-1 degradation in macrophages, which can dampen the signals leading to C/EBPδ activation, leading to LPS tolerance [27]. Further studies employing systems biology approach are clearly warranted to clarify the differential sensing mechanisms of LPS signal strength in different cell types. On a separate note, the integrated systems approach will likely explain the complex phenotypes such as LPS priming and tolerance exhibited in cells treated with either a low or a high dose LPS.
Our findings bear significant clinical implication during the pathogenesis of chronic and persistent inflammatory diseases. Local tissues and fibroblasts encountering bacterial endotoxin may exhibit persistent activation of the C/EBPδ circuit without a dampening mechanism. As a consequence, inflammatory mediators such as LCN2 may accumulate and lead to chronic tissue damages including kidney failure. Strategies targeting at this functional circuit may hold promise in designing therapeutic strategies to combat chronic inflammatory diseases.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work is supported by an internal institutional fund from the Institute of Critical Technology and Applied Sciences from Virginia Tech. No additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268:10425–10432. [PubMed] [Google Scholar]
- 2.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 3.Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. 1997;45:17–23. doi: 10.1006/geno.1997.4896. [DOI] [PubMed] [Google Scholar]
- 4.Lu YC, Kim I, Lye E, Shen F, Suzuki N, et al. Differential role for c-Rel and C/EBPbeta/delta in TLR-mediated induction of proinflammatory cytokines. J Immunol. 2009;182:7212–7221. doi: 10.4049/jimmunol.0802971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding L, Hanawa H, Ota Y, Hasegawa G, Hao K, et al. Lipocalin-2/neutrophil gelatinase-B associated lipocalin is strongly induced in hearts of rats with autoimmune myocarditis and in human myocarditis. Circ J. 2010;74:523–530. doi: 10.1253/circj.cj-09-0485. [DOI] [PubMed] [Google Scholar]
- 6.Viau A, El Karoui K, Laouari D, Burtin M, Nguyen C, et al. Lipocalin 2 is essential for chronic kidney disease progression in mice and humans. J Clin Invest. 2010;120:4065–4076. doi: 10.1172/JCI42004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolignano D, Coppolino G, Lacquaniti A, Buemi M. From kidney to cardiovascular diseases: NGAL as a biomarker beyond the confines of nephrology. Eur J Clin Invest. 2010;40:273–276. doi: 10.1111/j.1365-2362.2010.02258.x. [DOI] [PubMed] [Google Scholar]
- 8.Bonomini F, Foglio E, Rodella LF, Rezzani R. Clinical biomarkers in kidney diseases. Front Biosci (Schol Ed) 2010;2:591–615. doi: 10.2741/s88. [DOI] [PubMed] [Google Scholar]
- 9.Cowland JB, Sorensen OE, Sehested M, Borregaard N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol. 2003;171:6630–6639. doi: 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 10.Sunil VR, Patel KJ, Nilsen-Hamilton M, Heck DE, Laskin JD, et al. Acute endotoxemia is associated with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp Mol Pathol. 2007;83:177–187. doi: 10.1016/j.yexmp.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smolens P, Stein JH. Pathophysiology of acute renal failure. Am J Med. 1981;70:479–482. doi: 10.1016/0002-9343(81)90568-4. [DOI] [PubMed] [Google Scholar]
- 13.Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59:172–181. doi: 10.2337/db09-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 15.Shen F, Hu Z, Goswami J, Gaffen SL. Identification of common transcriptional regulatory elements in interleukin-17 target genes. J Biol Chem. 2006;281:24138–24148. doi: 10.1074/jbc.M604597200. [DOI] [PubMed] [Google Scholar]
- 16.Su JZT, Tyson J, Li L. The Interleukin-1 Receptor-Associated Kinase M (IRAK-M) selectively inhibits the alternative, instead of the classical NFkB pathway. Journal of Innate Immunity. 2009;1:164–174. doi: 10.1159/000158541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litvak V, Ramsey SA, Rust AG, Zak DE, Kennedy KA, et al. Function of C/EBPdelta in a regulatory circuit that discriminates between transient and persistent TLR4-induced signals. Nat Immunol. 2009;10:437–443. doi: 10.1038/ni.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peairs A, Dai R, Gan L, Shimp S, Rylander MN, et al. Epigallocatechin-3-gallate (EGCG) attenuates inflammation in MRL/lpr mouse mesangial cells. Cell Mol Immunol. 2010;7:123–132. doi: 10.1038/cmi.2010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maitra U, Parks JS, Li L. An innate immunity signaling process suppresses macrophage ABCA1 expression through IRAK-1-mediated downregulation of retinoic acid receptor alpha and NFATc2. Mol Cell Biol. 2009;29:5989–5997. doi: 10.1128/MCB.00541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tyson JJ, Novak B. Functional motifs in biochemical reaction networks. Annu Rev Phys Chem. 2010;61:219–240. doi: 10.1146/annurev.physchem.012809.103457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kravchenko VV, Mathison JC, Schwamborn K, Mercurio F, Ulevitch RJ. IKKi/IKKepsilon plays a key role in integrating signals induced by pro-inflammatory stimuli. J Biol Chem. 2003;278:26612–26619. doi: 10.1074/jbc.M303001200. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 23.Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, et al. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto M, Akira S. Lipid A receptor TLR4-mediated signaling pathways. Adv Exp Med Biol. 2009;667:59–68. doi: 10.1007/978-1-4419-1603-7_6. [DOI] [PubMed] [Google Scholar]
- 26.Maitra U, Gan L, Chang S, Li L. Low-Dose Endotoxin Induces Inflammation by Selectively Removing Nuclear Receptors and Activating CCAAT/Enhancer-Binding Protein {delta}. J Immunol. 2011;186:4467–4473. doi: 10.4049/jimmunol.1003300. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Cousart S, Hu J, McCall CE. Characterization of interleukin-1 receptor-associated kinase in normal and endotoxin-tolerant cells. J Biol Chem. 2000;275:23340–23345. doi: 10.1074/jbc.M001950200. [DOI] [PubMed] [Google Scholar]