

Abstract
A number of organic molecules which contain the 1,5-diaryl-3-oxo-1,4-pentadienyl group, referred to hereafter as the dienone moiety, have antineoplastic properties. Emphasis is made on the attachment of this structural moiety to several molecular scaffolds, namely piperidines, N-acylpiperidines, cycloalkanes and 3,4-dihydro-1H-napthalenes. Many of these compounds are potent cytotoxins having micromolar and nanomolar IC50 values towards a wide range of neoplastic and transformed cells. On occasions, greater toxicity towards neoplasms than normal cells has been demonstrated. A number of these compounds have in vivo anticancer properties and in general excellent tolerability in rodents is demonstrated. The way in which a number of physicochemical properties such as redox potentials, torsion angles, atomic charges and logP values govern cytotoxic potencies are presented. The importance of the shapes of different compounds as determined by molecular modeling in contributing to antineoplastic properties is outlined. Arguments are presented in favour of designing antineoplastics which have multiple sites of action in contrast to those bioactive molecules which have only one molecular target. A number of compounds which possess the dienone group have different modes of action some of which are chronicled in this review, such as inducing apoptosis, affecting respiration in mitochondria, inhibiting macro-molecular biosynthesis and both inhibiting and stimulating certain enzymes. Other important properties of these compounds are discussed including their anti-angiogenic, MDR-revertant and antioxidant properties. It is hoped that this eulogy of the importance of the dienone group will encourage researchers to consider incorporating this structural unit into candidate cytotoxins in the future.
Keywords: 1,5-Diaryl-3-oxo-1,4-pentadienes; cytotoxicity; cycloalkanones; 4-piperidones; 3,4-dihydro-1H-naphthalen-2-ones; anticancer activity; antineoplastics; diaryldienones
INTRODUCTION
The purpose of this review is to encourage consideration of the inclusion of the 1,5-diaryl-3-oxo-1,4-pentadienyl moiety into the design of novel antineoplastics. The reasons for this recommendation are based on the evidence that many compounds containing this structural unit display cytotoxic and anticancer properties, exercise their bioactivity in a number of different ways and display excellent tolerability in mammals.
A number of years ago, one of the authors prepared a lengthy review on the bioactivities of acyclic conjugated α,β-unsaturated ketones [1]. This monograph outlined some evidences that these compounds have a marked affinity for thiols whereas only minimal or non-existent interactions occur with amino or hydroxy groups. Later studies confirmed the thiol-selectivity of these compounds [2–3]. Since thiols are absent in nucleic acids, conjugated enones may avoid the problems of mutagenicity and carcinogenicity caused by certain contemporary anticancer drugs [4–5]. A subsequent review article from this laboratory on chalcones or 1,3-diaryl-2-propenones [6] provided further evidence of the potential usefulness of conjugated α,β-unsaturated ketones in cancer chemotherapy. Various series of compounds have been developed in which an aryl ring was placed adjacent to the β carbon atom in order to alter the polarity of the electrophilic centre which may correlate with cytotoxic potencies [7]. In addition, the steric and hydrophobic properties of molecules may be controlled by the nature of the aryl substituents.
However with the aim of preparing conjugated enones with selective or preferential toxicity to malignant cells rather than the corresponding normal cells, the theory of sequential cytotoxicity was proposed [8]. This hypothesis states that a chemical insult prior to a subsequent chemical attack on cellular constituents may produce greater toxicity to cancers than normal cells. The rationale for this theory is based on the observation that on a number of occasions, the lowering of cellular thiol concentrations before the administration of various anticancer agents enhances the cytotoxicity of these drugs towards tumours rather than normal cells [9–11]. In other words, greater sensitization of malignant cells may take place. In order to achieve successive interactions with cellular thiols, the 3-aryl-2-propenoyl group (Ar-CH=CH-CO-) present in various cytotoxins was expanded to a 1,5-diaryl-3-oxo-1,4-pentadienyl moiety (Ar-CH=CH-CO-CH=CH-Ar). Alkylation of thiols is predicted to occur as indicated in Scheme 1.
Scheme 1.

The sequential alkylations of cellular thiols by compounds containing the 1,5-diaryl-3-oxo-1,4-pentadienyl group.
One of the principal reasons for the pursuit of 1,5-diaryl-3-oxo-1,4-pentadienes (hereafter referred to as diaryldienones) is that these compounds interact with a variety of cellular constituents. In the past, many efforts have been made to design compounds or monoclonal antibodies to interact with one primary molecular target [12], but the results have often been disappointing [13]. Hence an alternative strategy is presented in this review, namely describing compounds which have the potential for multiple sites of action.
The reasons for incorporating groups which are documented to have different molecular targets into a candidate cytotoxin include the following considerations. (1) The problem of drug resistance will probably arise rapidly if the ligand inhibits only one biochemical pathway. Hence the design of antineoplastic agents with multiple sites of action is more likely to lead to effective anticancer drugs than a one ligand-one binding site approach. (2) When a normal cell becomes malignant, a number of dysregulated processes occur and an antineoplastic agent should possess pleotropic capabilities. (3) It is conceivable that one or more of the sites at which a multitargeted ligand interacts may assist its metabolism. For example, a diaryldienone may stimulate an isoform of glutathione S-transferase which will catalyze the conjugation of the unsaturated ketone with glutathione. Hence the elimination will ensure a lack of accumulation in the body and thus avoid the toxic symptoms caused by xenobiotics which are retained in body tissues. (4) A compound reacting at different locations in the body which contribute to the anticancer efficacy may be less toxic than an analog which has only one site of action. Thus consider a biochemical sequence W-X-Y-Z in a tumour cell while an analogous pathway is V-X-Y-Z in normal tissue. In addition, one may assume that the inhibition of Y into Z is considered to be the most damaging to the cells and above certain doses, cell death results. If the multitargeted ligand inhibits the target W but not V as well as inhibiting the conversion of Y into Z, then fewer compounds will be required than in the case of an analog which only inhibits the pathway Y into Z. Hence toxicity to normal cells will be less when the multitargeted cytotoxin is administered. (5) A multitargeted compound may reveal novel mechanisms of action. For example, in the case of the discovery that an antineoplastic agent acts an both W and inhibits the Y-Z sequence may reveal that these two loci of action are connected, i.e., the relationship W-X-Y-Z is demonstrated. (6) Various molecular targets may be expressed only in small groups of patients. Hence a multitargeted ligand may be effective towards a greater number of patients than using a compound which acts at one locus only. (7) Compounds which interact at a variety of sites in the body may demonstrate bioactivity in other clinical areas. For example, a cytotoxic sulfonamide may also display diuretic properties. In this case, the doses required for anticancer therapy and enhancement of diuresis should be similar.
However in order to obtain compounds with therapeutic potential, a balance needs to be achieved whereby the cytotoxin interacts with various molecular targets associated with carcinogenesis but spares those receptors which are required for the healthy functioning of the body. Unless this selective toxicity is achieved when interactions with multiple targets takes place, the differential between unwanted side effects and therapeutic gain will not be manifested.
Recently other authors have articulated the need to find anticancer agents which interact at multiple sites [14–16]. Thus this review will emphasize not only the antineoplastic properties of various diaryldienones but some of the different ways whereby antiproliferative properties are mediated. A further positive aspect of these compounds is that large doses of many of these diaryldienones can be tolerated in rodents which is in contrast to the marked toxicity of many contemporary anticancer drugs [17]. To the best knowledge of the authors, the antineoplastic properties of diaryldienones has not been reviewed previously with the exception of a brief monograph of 3,5-bis(benzylidene)-4-piperidones as novel anticancer agents [18].
This review aims to summarize different studies each of which illustrates the potential utility of diaryldienones in cancer chemotherapy. A major emphasis of this review is the demonstration that the diaryldienone motif is able to confer antineoplastic properties when incorporated into different ligands. Hence the cytotoxicity of these compounds is emphasized while reference will also be made to their in vivo anticancer properties, modes of action and other bioactivities.
CYTOTOXICITY
Four classes of diaryldienones will be considered. In the first three groups of compounds, the 1,5-diaryl-3-oxo-1,4-pentadienyl group was mounted on piperidines, cycloalkyl and 3,4-dihydro-1H-naphthalenyl scaffolds while the fourth cluster of molecules are acyclic in nature.
1. 3,5-bis(Benzylidene)-4-piperidones
The design and cytotoxic properties of various 3,5-bis(benzylidene)-4-piperidones will be reviewed followed by an account of the cytotoxicity of various N-acyl products which were not described by Jha and Duffield [18].
The reasons for the initial development of these compounds as cytotoxins was based on a number of considerations. A previous investigation revealed that the second order rate constants between a biomimetic thiol and a cluster of Mannich bases of acyclic ketones 1 was approximately 240 times faster than the corresponding enones 2 when the same substituents are in the aryl rings [19]. However while some of the acyclic Mannich bases have cytotoxic and anticancer properties [20], respiratory depression was noted in low doses for many of these types of compounds when they were administered to mice [21]. The acyclic enones are flexible molecules and therefore able to assume a variety of different shapes, some of which may have antineoplastic properties while others are responsible for the unwanted toxicity. Hence while a conjugated arylidene keto group along with a basic centre beta to the carbonyl function was retained, molecular flexibility was reduced by the incorporation of most of the molecule into a rigid piperidine ring as illustrated in Fig. (1). In addition, the incorporation of a second alkylating group was undertaken with the aim of increasing cytotoxic potencies. These considerations led to the syntheses of 3a (R1 = H) which possesses an IC50 value of 16 pM towards murine P388D1 cells [22].
Fig. 1.

The design of 3,5-bis(benzylidene)-4-piperidones as candidate antineoplastic agents.
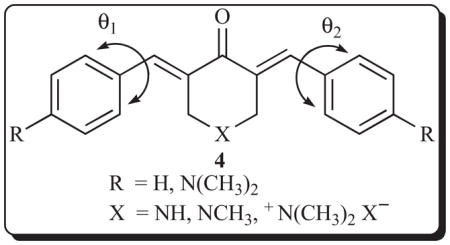
Molecular modifications of this compound by altering the nature of the basic centre and placing a 4-dimethylamino group in the aryl rings gave rise to the analogs 4 with similar high cytotoxic potencies [22]. X-ray crystallography of many of these molecules revealed that both olefinic double bonds adopted the E stereochemistry and that the aryl rings are not coplanar with the adjacent olefinic linkages. This latter phenomenon is due to nonbonded interactions between the equatorial protons at positions 2 and 6 of the piperidine ring and the ortho hydrogen atoms of the aryl rings. The torsion angles θ1 and θ2 differed in magnitude but no correlations between cytotoxic potencies and the θ1 and θ2 figures were observed.
Two experiments were undertaken with a representative compound 3a [22]. First, this dienone lowered hepatic glutathione levels in mice confirming that it is a thiol alkylator. Second, administration of doses up to and including 240 mg/kg to mice on five successive days did not cause any mortalities. An examination was undertaken in order to find if these compounds exerted their potencies in additional ways. Two related compounds 4 (R = N(CH3)2; X = +N(CH3)2 −I) and 4 (R = N(CH3)2; X = NCH3) were added to three synthetic DNAs, namely poly[d(AT)], poly da·poly dT and poly [d(TG)]·poly[d(CA)]. The quaternary ammonium compound caused a rise in the melting temperature of all three DNAs but no changes were noted with the non-quaternary ammonium compound. The behaviour of the quaternary ammonium compound indicates AT selectivity and is consistent with binding in the minor groove of DNA. Further experimentation with three unsubstituted compounds (R =H) in which X is NH.HCl, CH2 and +N(CH3)2−Br using poly[d(AT)] revealed that only binding occurred with the quaternary ammonium compound. A further study with related 3,5-bis(benzylidene)-1-methyl-4-piperidone methoiodides which are toxic to murine L1210 cells revealed that they too increased the melting temperature of poly[d(AT)] [23].
N-Acyl-3,5-bis(benzylidene)-4-piperidones
A number of N-acyl analogs of 3,5-bis(benzylidene)-4-piperidones have been prepared and the design of these compounds was based on the following considerations. First, the cytotoxicity of 3,5-bis(benzylidene)-4-piperidones may be considered to be due to the 1,5-diaryl-3-oxo-1,4-pentadienyl group interacting at a primary binding site. Hence the placement of various groups on the piperidyl nitrogen atom could enable interactions at an auxiliary binding site leading to potency increases. It is of course conceivable that groups on the nitrogen atom will impede alignment of the diaryldienone group at the primary binding site thereby lowering antineoplastic efficacy. These concepts are visualized in Fig. (2). Second, 3,5-bis(benzylidene)-4-piperidones are ionized under physiological conditions; the extent being determined by the pKa of the piperidyl nitrogen atom as well as the pH of the biological milieu [24]. The ions are likely to experience difficulty in penetrating cell membranes and hence the formation of the N-acyl analogs will prevent the nitrogen atom being charged and the permeability into malignant cells may be enhanced. The resultant amide may be bioactive per se or be a prodrug liberating the 4-piperidones by amidic hydrolysis.
Fig. 2.

The postulated interactions of N-acyl-3,5-bis(benzylidene)-4-piperidones at binding sites.
An initial study involved the preparation of N-acyl analogs of 5 in which an additional site for thiol alkylation is either present (7) or in a latentiated form (9) [25]. Examples of the compounds prepared are given in Scheme 2 and the number in parentheses refer to the potencies relative to the parent compound 5 which has an IC50 value of 2.5 μM towards murine L1210 leukemic cells. Clearly potency is elevated in both N-acylated products 7 and 9. With a view to lowering the IC50 values still further, two electron-withdrawing chloro atoms were placed in the aryl rings in order to increase the electrophilicity of the olefinic carbon atoms which gave rise to the potent cytotoxins 6, 8 and 10. The diaryldienones 5-10 along with a number of related analogs were evaluated against a panel of approximately 56 human tumour cell lines from different groups of tumours, namely leukemia, melanoma and colon, renal, small cell lung, non-small cell lung, central nervous system and ovarian cancers (referred to hereafter as the NCI screen). In particular, the average IC50 figure of 8 is 0.16 μM while the figure for melphalan is 26.3 μM revealing 8 to possess 164 times the potency of this reference drug. In particular, the average IC50 value of 8 towards five human leukemic cell lines is 36.3 nM indicating its potential as an antileukemic agent. Compound 9 and two analogs in which the dimethylamino hydrochloride group was replaced by trimethylammonium bromide and diethylamino hydrochloride substituents were incubated in a mixture of deuterated phosphate buffered saline and dimethylsulfoxide-d6 for 48 hours and 37° C. 1H NMR spectroscopy revealed the liberation of the corresponding N-acryloyl derivative 7 along with other decomposition products.
Scheme 2.

Conversion of the 3,5-bis(benzylidene)-4-piperidones 5 and 6 into the N-acylated products 7-10.
A subsequent study revealed that sodium borohydride reduction of the carbonyl group of 5 led to the corresponding alcohol 11 which has an IC50 value of 18.0 μM towards L1210 cells [26]. Thus reduction of the keto group lowered potency sevenfold suggesting that the integrity of the 1,5-diaryl-3-oxo-1,4-pentadienyl group should be maintained.
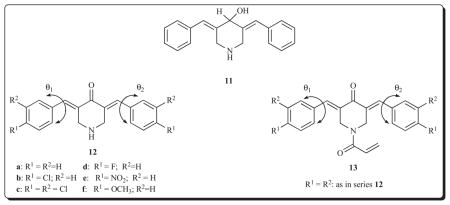
A more detailed study was undertaken subsequently in order to compare the relative cytotoxic potencies of the 3,5-bis(benzylidene)-4-piperidones 12 with the corresponding N-acryloyl derivatives 13 [27]. In addition, efforts were made to identify some of the physicochemical and biochemical parameters which contribute to the magnitude of the anti-neoplastic properties.
The average IC50 figures of the six compounds in series 12 towards human Molt 4/C8 and CEM T-lymphocytes as well as murine P388 and L1210 leukemic cells are 59.4, 33.6, 0.81 and 81.9 μM, respectively. In the case of the compounds in series 13 which have the same aryl substituents, the relative figures are 0.96, 1.13, 0.28 and 4.71 μM, respectively, i.e., the N-acryloyl derivatives 13 are, on average, 62, 30, 3 and 17 times more potent in each of these screens. Thus with the exception of the P388 cells, which are the most sensitive to these compounds, substantial increases in cytotoxic potencies were observed in creating an additional site for alkylation of cellular thiols.
The following relationships between physicochemical properties and cytotoxic potencies of the compounds in series 12 and 13 were observed. First, the Hammett σ values of the aryl substituents in series 13 correlated negatively with the IC50 figures in the P388 and L1210 bioassays. Second, the average torsion angles θ1 and θ2 in series 12 are −49.5 (± 0.05)° and 49.4 (± 0.11), respectively, while in the case of the N-acyl analogs 13, the figures are −53.2 (± 0.17) and 61.0 (± 0.24), respectively. Third, correlations have been established between the redox potentials of various compounds and different bioactivities [28], including chalcones which contain the related 3-aryl-2-propenoyl group, and cytotoxicity [29]. When the same substituents are present in the aryl rings, the redox potentials are lower in series 13 indicating their greater reducing properties. These three observations have value in the design of further analogs with a view to increasing cytotoxic potencies. Thus the placement of one or more strongly electron-withdrawing groups in the aryl ring, the insertion of substituents of different sizes in the ortho position of the aryl rings in order to increase the θ values and employing groups which have lower redox potentials should be considered.
The possible modes of action of representative compounds in series 12 and 13 were investigated. Both 12e and 13e inhibited DNA, RNA and protein biosyntheses in murine L1210 cells. The average IC50 values to accomplish these results for 12e and 13e are 239 μM and 3.90 μM, respectively. Thus 61-fold difference in potency is similar to the 78-fold difference in the IC50 values to L1210 cells. Both 12c and 13c inhibited RNA and protein biosyntheses as well as inducing apoptosis in human Jurkat T cells. In summary, the cytotoxicity of the compounds in series 12 and 13 likely includes interference with the syntheses of important biomacromolecules and by causing apoptosis.
A recent investigation also addressed the issue of placing different acyl groups on the piperidyl nitrogen of the diaryldienones 14 in order to glean further evidence of the structural requirements at the putative auxiliary binding sites [30]. Benzoylation of 14a give the corresponding amide 15 which has a similar toxicity profile as the parent compound. In order to ascertain if groups on the amidic aryl ring of 15 which have a mixture of hydrophilic and hydrophobic properties increased cytotoxic potencies, 16a was synthesized which possesses three times the potency of 14a and 15 in the Molt 4/C8 bioassay. With a view to investigating the generality of whether the N-[4-(2-alkylaminoethoxy)phenylcarbonyl] group enhances the cytotoxicity of the 3,5-bis(phenylmethylene)-4-piperidones, the following structural alterations of 16a were made. First, substituents were placed in the arylidene aryl ring which have substantially divergent σ and π values. Second, the dimethylamino group was replaced by other amines which differed in size and basicity. Such considerations led to the syntheses and cytotoxic evaluation of series 16-19 as indicated in Scheme 3.
Scheme 3.

Formation of the N-acyl products 15-19 from series 14.
The compounds in series 16-19 displayed potent cytotoxicity towards human Molt 4/C8 and CEM T-lymphocytes whereby three quarters of the IC50 values are less than the figures for melphalan (~ 2.9 μM). In particular, 16c, 17c, 18c and 19c where R1 is a nitro group have submicromolar IC50 figures. The murine L1210 cells are less sensitive to these compounds but in 63% of the assays, the IC50 values are less than 10 μM. In general, the greatest cytotoxic potencies are found when the R1 substituent is either nitro or methyl and the dimethylamino, diethylamino and 1-piperidyl groups are present in the N-acyl side chain. Comparisons were made between the IC50 values of the piperidines 14 and 16-19 when the same substituents are present in rings A and B. In 48% of the comparisons, the N-acylated products were more potent while in 35% of the cases, equal potencies were observed. The conclusion drawn is that the N-[4-(2-aminoethoxy) phenylcarbonyl] side chain likely interacts at auxiliary binding sites thereby increasing potencies.
A number of the compounds in series 16-19 were evaluated against approximately 49 human tumour cell lines. The average IC50 values ranged from <0.52 (19b) to >11.5 (18b) μM. Compound 19b was particularly cytotoxic towards the colon cancer HCC-2998 having an IC50 figure of less than 5 nM. A huge differential between the IC50 values were noted for most of these compounds, i.e., there was a selective toxicity towards certain tumour cell lines which may translate into a preferential lethality for neoplastic cells rather than the corresponding normal tissues. A TUNEL assay using human HepG2 liver cancer cells revealed that 19b caused apoptosis. A subsequent study revealed that two representative molecules 16a and 19d stimulated respiration in rat liver mitochondria vide infra and none of these compounds caused mortalities when 300 mg/kg was administered to mice [31].
A major problem in treating cancers with drugs is that resistance often develops. Thus compounds are urgently required which will enable anticancer drugs to be retained in the malignant cells rather than be extruded rapidly by a drug efflux pump such as P-glycoprotein (P-gp). Recent studies have revealed that the mounting of the diaryldienone moiety on a N-acyl piperidinyl scaffold led to potent revertants of P-gp associated multidrug resistance. The biological assay employed in these studies used murine L-5178 lymphoma cells transfected with the human MDR1 gene [32]. The concentration of rhodamine 123 in treated and untreated transfected and parental cells are measured and the fluorescent activity ration (FAR) values calculated. A FAR value of greater than 1 indicates that reversal of MDR has occurred. This assay is referred to subsequently as the L-5178 P-gp reversal screen. Using concentrations of 4 μg/mL, the FAR figures of 16-19 ranged from 46–179 which are substantially greater than was noted for a reference drug verapamil which has a FAR value of 4.2 at this concentration [33]. The optimal basic group is the 1-piperidinyl ring present in series 18 while higher FAR values are associated with the compounds containing 4-methyl and 4-chloro substituents in the arylidene aryl rings. One may note that the precursor amines 14 are virtually bereft of MDR revertant properties using concentrations of both 4 and 40 μg/mL.
A brief summary is now presented of four series of compounds 20-23 which were principally designed to explore the tolerance of auxiliary binding sites to the acyl group attached to the nitrogen atom of various 3,5-bis(benzylidene)-4-piperidones. In general, evaluation of the cytotoxic properties of these compounds involved the use of human Molt 4/C8 and CEM T-lymphocytes and murine L1210 leukemic cells. Many of the compounds possessed submicromolar IC50 values. Furthermore in short-term toxicity studies, doses up to and including 300 mg/kg of these dienones did not produce mortalities.
The first study involved the preparation of series 20 [34] in which the chalcone motif is present in the N-acyl group. It is conceivable that the compounds per se interact strongly at receptors or possibly they may undergo hydrolysis liberating the cytotoxic piperidinone and chalcones, many of which have cytotoxic properties [35,36]. In general, the compounds in series 20 (IC50 values of 1.7–94 μM) were less potent than 14a (IC50 range is 1.7–8 μM) which may have been due to the bulk of ring A impeding alignment at a binding site. However a second series of compounds 21 in which ring A had been excised has similar potencies as series 20.
Series 22 was designed in which the olefinic double bond in the N-acyl group not only provided an additional site for thiol alkylation but also conferred rigidity to the molecules thereby controlling the location of terminal aryl ring B relative to the 3,5-bis(benzylidene)-4-oxopiperidinyl moiety [37]. In general, these compounds were more potent than the diaryldienones 13 (R1=R2=H) and 14a. The IC50 values are submicromolar in over half of the compounds and less than 5 μM in all cases. All of these compounds are more potent than melphalan towards the T-lymphocytes. Changing the stereochemistry of the olefinic group in the N-acyl side chain from Z to E led to series 23 which, in general, was accompanied by a reduction in potency [38].
Attempts have been made to determine the modes of action of representative compounds in series 22 and 23. First, the enzyme myristoyl-CoA:protein N-myristoyl transferase (HMT) catalyzes the attachment of the myristoyl group onto the N-terminal glycine residues of certain polypeptides [39]. In particular the cysteine-169 residue of human NMT (hNMT) is believed to be involved in the myristoylation process [40] and the activity of this enzyme is higher in some colorectal tumours than in the corresponding normal tissues [41]. hNMT may therefore be considered a molecular target in cancer chemotherapy. All of the compounds in series 22 were examined for their effect on human NMT (hNMT) using concentrations up to and including 40 mM. No inhibition or stimulation of the enzyme was observed [37]. On the other hand, two representative compounds in series 23 (R1=3-Cl, R2=4-Cl and R1=4-CH3, R2=H) both inhibited the action of hNMT by 52% using concentrations of 100 and 50 μM, respectively [38]. A COMPARE program [42] of a number of compounds in series 23 revealed that the correlation coefficients were greatest in regard to tyrosine kinases which indicates a possible way that these diaryldienones exert their bioactivity.
Previously the impressive cytotoxic potencies of both various 3,5-bis(benzylidene)-4-piperidones 12 and the corresponding N-acryloyl derivatives 13 have been described. These bioevaluations utilized neoplastic and transformed cells. An important question to be resolved in considering the potential of these molecules for further development is whether selective toxicity to malignant cells is exhibited by these compounds. Furthermore in order to consider the possibility of increasing the selective toxicity of candidate cytotoxins for neoplasms, the decision was made to form adducts between series 13 and 2-mercaptoethanesulphonic acid leading to the formation of the acids 24. This suggestion was made on the basis that coadministration of sodium mercaptoethanesulfonate (mesna) and various anticancer drugs led to reduction of the side effects in normal cells but did not alter the antineoplastic properties of the anticancer drugs [43–45]. Thus the compounds 12a,b,e,g,h, 13a,b,e,g,h and 24a,b,e,g,h were prepared and evaluated against both malignant and tumourous cell lines [46].
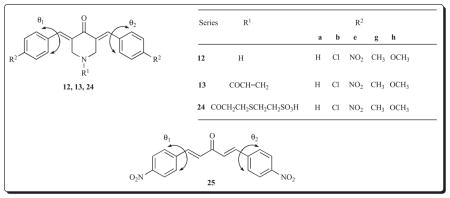
The compounds in series 12, 13 and 24 were evaluated against the following human neoplastic cell lines, namely HSC-2 and HSC-4 squamous cell carcinomas as well as HL-60 promyelocytic leukemia cells. Three normal human cells were used viz HGF gingival fibroblasts, HPC pulp cells and HPLF peridontal ligament fibroblasts. In regard to the malignant cells, the average CC50 values (the concentrations of the compounds to reduce the number of viable cells by 50%) of 12a,b,e,g,h, 13a,b,e,g,h and 24a,b,e,g,h are 29.4, 1.27 and 81.3 μM, respectively, revealing the marked potencies of the compounds in series 13. In fact, the CC50 figures of the compounds in series 13 are submicromolar in 73% of the bioassays. In addition, since the average CC50 value of melphalan is 40.7 μM, the far greater potencies of the compounds in series 13 than this established drug are revealed.
Selectivity index (SI) figures were calculated which are the quotients of the average CC50 values of the normal and neoplastic cells. A SI figure of 10 was arbitrarily chosen as demonstrating noteworthy selectivity and was met by 12a,e,g and 13a,b,e,g,h. The SI figure for melphalan is >4.9. Thus the compounds in series 13 are clearly important lead molecules in terms of cytotoxic potencies and high SI values. The disappointing results of the compounds in series 24 was attributed to two phenomena. First, a stability study of a solution of a representative compound in series 24, namely 24e, revealed that no decomposition occurred after incubation at 37° C for 24h (the temperature and time of the cytotoxicity assays). In other words, mesna and 13e were not liberated from 24e. Second, the intact molecules 24 contain a polar sulfonic acid group which may impede the penetration of cell membranes and hence in future the corresponding ethyl esters should be made.
An experiment was undertaken to ascertain whether the compression of the 1,5-diaryl-3-oxo-1,4-pentadienyl group into rigid cyclic scaffolds influences cytotoxic potencies and SI values. Consequently the acyclic analog of 12e, 13e and 24e, namely 25, was prepared. The average CC50 values towards the three malignant cell lines of 12e, 13e, 24e and 25 are 3.5, 0.26, 4.7 and 94 μM, respectively, and the SI figures are 13, 15, 7.7 and >4.3. These results indicate that in general the incorporation of part of the 1,5-bis-(4-nitrophenyl)-3-oxo-1,4-pentadienyl group into a piperidine ring makes an important contribution to the desired bioactivities in terms of potency and selectivity. In order to determine the reason for this phenomenon, models were built of these compounds and their shapes compared. While various interatomic distances are similar in 12e, 13e, 24e and 25, the torsion angles θ1 and θ2 as indicated in structures 12, 13, 24 and 25 are substantial in the case of 12e, 13e and 24e (42–61°) but very small in 25 (0.1–0.3°) which may have contributed to the divergence in biological properties. The possibility of correlations between θ values and IC50 as well as SI figures should be explored in the future.
Substitutions on the C2 and C6 Piperidyl Carbon Atoms
A further structural modification of 3,5-bis(benzylidene)-4-piperidones involved the placement of a dimethylene bridge between the two ring carbon atoms attached to the piperidyl nitrogen atom. The dimethylene bridge could increase binding at receptors by van der Waals bonding; alternatively, it could cause steric impedance to alignment at a binding site thereby lowering cytotoxic potencies. As mentioned previously, there is a lack of coplanarity between the aryl rings and the adjacent olefinic groups in the 3,5-bis(benzylidene)-4-piperidones which is caused by non-bonded interactions between the equatorial proton attached at the C2 and C6 atoms and one of the ortho protons of the aryl ring [22]. The decision was made to replace the axial protons attached to the C2 and C6 carbon atoms in order that the torsion angles θ1 and θ2 present in compounds with and without a dimethylene chain will be similar and interpretation of the biodata will not be confused by more variables than necessary. Thus series 26 was prepared and also the corresponding analogs 27 which lack the dimethylene bridge [47].
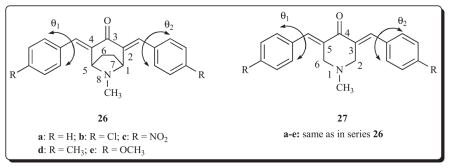
Apart from the anomolous behaviour of 26b and 26c, the torsion angles θ1 and θ2 are in the range of 43–47° for all of the compounds in series 26 and 27. Thus substitution on the axial C2 and C6 atoms of 27a–e leading to the tropinones 26 does not appear to affect the magnitude of the torsion angles in general. The biodata generated for 26 and 27 using the Molt 4/C8, CEM and L1210 bioassays reveal clearly that the insertion of a dimethylene bridge between the C2 and C6 atoms of series 27 leading to 26a–e causes a reduction in cytotoxic potencies. This conclusion was drawn from the fact that the compounds in series 27 are more potent than the tropinones 26 which have the same R1 values in the aryl rings except that 26e is more cytotoxic than 27e in the L1210 assay. The most potent compound in each series is the un-substituted analog, namely 26a and 27a, which have average IC50 values of 9.77 and 4.69 μM, respectively. These results may indicate that an E4 operating parameter is in effect [48], i.e., that para substitution exerts an unfavourable steric effect at binding sites. The compounds were examined against HSC-2, HSC-3, HSC-4 and HL-60 neoplasms as well as HGF, HPC and HPLF normal cells. SI figures of over 10 were obtained for 26a (>15), 27b (>19) and 27c (>11) which serve as lead molecules for future development.
An attempt was made to determine some of the ways that representative compounds exert their bioactivity and to provide an explanation for the differences in potencies between various compounds. A number of the acyclic Mannich bases derived from conjugated arylidene ketones affect respiration in isolated mitochondria [49, 50]. Since 4-piperidone can be considered to be a cyclic Mannich base, two representative compounds with widely differing potencies were evaluated for their effects on respiration in rat liver mitochondria. The average IC50 values of 26a and 26d in the Molt 4/C8, CEM and L1210 bioassays are 9.77 and > 500 μM, respectively. Both compounds stimulate respiration in mitochondria but the latent period is much shorter with 26a. Furthermore, 26a produced rapid swelling of the mitochondria whereas 26d does so but more slowly. Thus interference with mitochondrial function appears to be one way in which cytotoxicity is caused and influences the magnitude of the cytotoxic potencies.
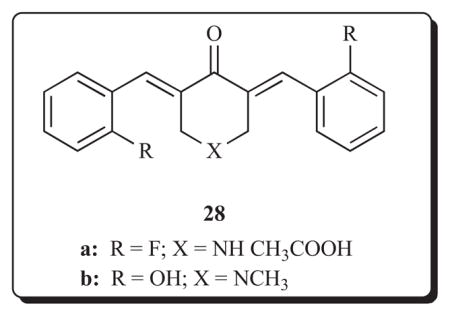
Some further aspects of the potential of 3,5-bis(benzylidene)-4-piperidones for utilization in cancer chemotherapy may be illustrated with compound 28a. This cytotoxic molecule may be linked to other structural units in order to increase selective toxicity to neoplasms, is effective in vivo towards certain tumours and has anti-angiogenic properties. A complex of compound 28a-linker-tripeptide-methylketone-factor VIIa has been prepared [51, 52]. Factor VIIa interacts with tissue factor which is expressed on the surface of tumours and also on the endothelial cells of the tumour vasculature. Hence the complex should hone in preferentially on malignant cells, undergo endocytosis and then release the cytotoxin. This complex inhibits the growth of both human MDA-MB-231 breast cancer cells and human RPMI-7951 melanoma cells to a greater extent than when 28a was used alone. This compound 28a, which demonstrated promising activity in the NCI in vitro screen, was administered subcutaneously to nude mice inoculated with human MDA-MB-231 breast cancer cells [53]. The average tumour weight was reduced by 30% and 55%, respectively, using doses of 20 and 100 mg/kg, respectively. The 100 mg/kg dose did not cause any liver, kidney or spleen toxicity and the animals had normal weight gain. Anti-angiogenic compounds have a suppressive effect on proliferating endothelial cells and selectively inhibit the blood supply to tumours. Compounds 28a and 28b inhibit endothelial cell growth in vitro and further evaluation revealed that 28a reduced cord formation and cell migration in human umbilical vein endothelial cells (HUVEC) at a concentration of approximately 1 μM [53]. Thus such compounds, even if bereft of antineoplastic properties, could be administered with anti-cancer drugs. In addition, development of enones possessing both cytotoxic and anti-angiogenic properties may well prove to be useful agents to combat carcinogenesis as is the case, for example, with certain combretastatins [54,55].
A number of diaryldienones are antioxidants and the importance of this property is deemed to be as follows. The body has a number of natural defences to harmful chemically reactive species such as various enzymes, e.g., superoxide dismutase, glutathione peroxidase and catalyse. In addition, reduced glutathione plays a part in the strategies to eliminate various chemically reactive species. However when these defences are overcome, then a variety of toxic reactions can occur such as the initiation of carcinogenesis. In order to counteract these problems, antioxidants may be used to scavenge harmful reactive oxygen and nitrogen species [56], and hence may be referred to as chemoprotectant agents. In the case of phenolic compounds, the free radical scavenging capacity is associated with their ability to donate hydrogen atoms [57].
A variety of diaryldienones which contain a 4-hydroxy substituent in the aryl rings have demonstrated antioxidant properties. The series of compounds 29 and 30 were prepared as antioxidants and their relative ability to scavenge free radicals was determined towards diphenylpicrylhydrazyl [58]. The pseudo first order rate constants (K min−1 × 10−2) were determined from which the following correlations between structure and antioxidant properties were noted. First, in series 29, the placement of one or two methoxy, ethoxy or methyl substituents ortho to the 4-hydroxy group increased the radical scavenging properties compared to the parent compound (R1 groups are hydrogen atoms). Increasing the size of the N-alkyl group in series 29 from methyl to ethyl to propyl led to compounds displaying a corresponding increased reaction rate to the diphenylpicrylhydrazyl free radical. This observation is surprising as the diaryldienone moiety is separated from the N-alkyl group by three sigma bonds. Second, replacement of the 4-piperidone ring by a cycloheptyl scaffold produced series 30 with similar K min−1 × 10−2 values. In addition, the ability to scavenge the oxygen free radical produced by the addition of phorbol-12-myristate-13-acetate to peripheral multinuclear neutrophil cells revealed that all of the compounds inhibited free radical formation with the highest activity being present in series 29.
2. bis(Benzylidene)cycloalkanones
The previous section outlined in some detail the cytotoxicity of a number of 3,5-bis(benzylidene)-4-piperidones and indicated some of the ways by which bioactivity is mediated. The 1,5-diaryl-3-oxo-1,4-pentadienyl group has also been mounted on alicyclic scaffolds which are now presented. In general, the compounds 31-38 were evaluated against human Molt 4/C8 and CEM T-lymphocytes and the IC50 values are generally in the range of 0.1–10 μM, while somewhat higher figures of 0.5–15 μM are found in the murine L1210 screen. In addition, doses up to and including 300 mg/kg could be administered to mice without causing lethal effects. In this section emphasis is placed on important SAR correlations, probing for the mechanisms of action of representative compounds, the identification of lead molecules and guidelines for future development.
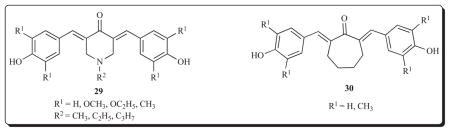
A series of 12 bis(benzylidene)cyclohexanones 31 were assessed against murine L1210 cells and the IC50 values ranged from 0.2 to >50 μM [59]. A Topliss analysis [48] based on the biodata of five compounds revealed a +σ dependency. This result indicates that extension of this project should include the placement of strongly electron-withdrawing groups in the aryl rings. The most potent compound [31, R1 = O(CH2)3N(CH3)2, R2 = H] possesses an IC50 value of 0.2 μM revealing its potency to be twice that of the reference drug melphalan. In addition, the average IC50 figure of this compound towards a number of human tumour cell lines from different neoplastic conditions (the NCI screen) is 1.55 μM which is 17 times lower than the average IC50 value of melphalan. This compound is clearly a lead molecule.
In order to obtain an appreciation of the importance of the relative locations of the olefinic carbon atoms, two analogs of 31 (R1 = R2 = H), namely 32 and 33 were prepared. An overlap of the three compounds as indicated in Fig. (3) reveals that the nature of the alicyclic scaffold controls the relative positions of the olefinic protons where interactions with thiols is considered to occur. The IC50 figures towards L1210 cells of 31 (R1 = R2 = H), 32 and 33 are 4.4, 7.5 and 16 μM, respectively, suggesting that retention of the six-membered ring is preferred.
Fig. 3.
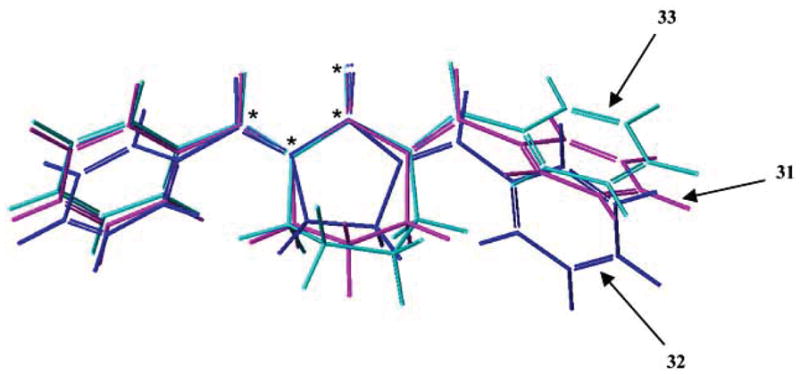
Overlap of models of 31 (R1 = R2 = H), 32 and 33. The four atoms which are superimposed are indicated with an asterisk.
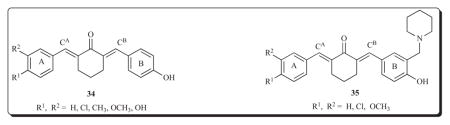
Two further series of 2,6-bis(benzylidene)cyclohexanones 34 and 35 were prepared in which the group in rings A and B were different in most cases [60]. This strategy led to variations in the atomic charges on the olefinic carbon atoms CA and CB which should facilitate the concept of sequential cytotoxicity taking place. A number of tumours are more acidic than the corresponding normal cells [61]. The placement of a 1-piperidinylmethyl group in ring B in series 35 should lead to a higher percentage of charged species in malignant cells than the corresponding normal tissues which would deplete the electron density on the CB atom and could make it more reactive towards thiols. In both series of compounds, linear plots between the atomic charges on the CA atoms and the Hammett σ values were observed (p < 0.05). The IC50 values of the majority of the compounds are in the low micromolar range (3–6 μM) and have comparable potencies to melphalan.
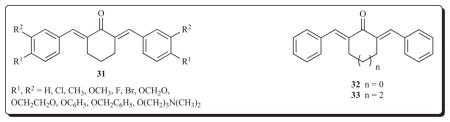
A continuation of the concept of exacerbating the differential in the atomic charges on the olefinic carbon atoms has been made recently [62]. In these compounds, the strongly electron-withdrawing nitro group was placed in the ortho, meta or para locations in ring A while a variety of substituents having different σ values was placed in ring B. Such considerations give rise to series 36-38. The differences in the charge densities on the olefinic carbon atoms CA and CB are approximately 0.02–0.07 esu. Interestingly the atomic charges are lower on the CB atom, indicating that the polarization of the π electrons in the 1,5-diaryl-3-oxo-1,4-pentadienyl group is towards ring A. The compounds were evaluated in the Molt 4/C8, CEM and L1210 bioassays and the IC50 values are generally in the 1–10 μM range.
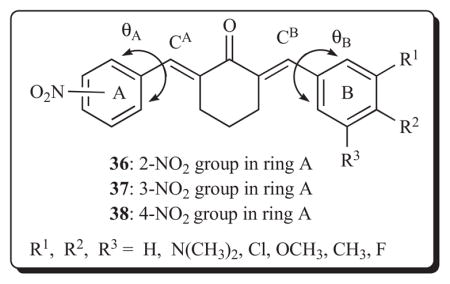
The following correlations between structures and cytotoxic potencies were noted. First, positive correlations (p < 0.05) were found when linear plots were constructed between the electron densities at carbon atom CB and the IC50 values in all three screens for series 36 and 38 and in the L1210 assay in series 37. Thus potency increases (IC50 values diminish) as the charge densities are reduced at the site of the initial alkylation at CB. Hence in the future, substituents with large σ values should be placed in ring B. Second, the same groups are present in ring B in series 36-38. A comparison of the IC50 values in the Molt 4/C8, CEM and L1210 bioassays revealed that the order of potencies is 36 > 37 > 38. The rate and extent of thiolation at the CA and CB atoms may be influenced by the magnitude of the torsion angles θA and θB. The θB angles in all three series of compounds are virtually constant viz 51.4±0.5°, whereas the θA figures in series 36, 37 and 38 are 76.8°, 51.3° and 51.1°, respectively. Hence the creation of new series of analogs in which larger θA values are manifested should be prepared such as the formation of congeners with large Taft ES constants. Third, the average log P figures of series 36, 37 and 38 are 4.86, 5.06 and 5.08, respectively, and hence the lower lipophilicity in series 36 may have contributed to its superior potencies.
Since a number of thiol alkylators react with mercapto groups in mitochondria [63], three representative compounds 36d, 37d and 38d were chosen, each of which has the 3,4,5-trimethoxy substitution pattern in ring B. The average IC50 figures of 36d, 37d and 38d in the three bioassays are 0.88, 43.8 and 6.00 μM, respectively. Using a concentration of 10μM, the two most potent compounds, viz 36d and 38d, stimulated the rate of respiration in rat liver mitochondria while 37d had no effect at 10 μM nor when the concentration of this compound was raised to 100 μM. While the variation in cytotoxicity is likely multifactorial, the different effects on mitochondrial function may contribute to the differences in the observed potencies.
A further aspect of the contribution that some of these compounds may make to cancer chemotherapy is the ability to reverse multidrug resistance. For example, using a concentration of 4 μg/mL, 38d and a related analog 38 (R1 = R2 = OCH3; R3 = H) have FAR values of 16 and 20, respectively, in the L-5178 P-gp reversal screen [64].
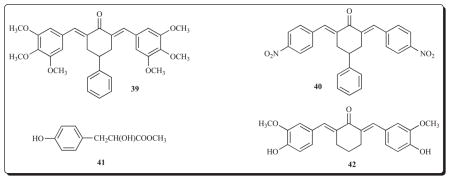
Various 2,6-bis(benzylidene)-4-phenylcyclohexanones have IC50 values in the low micromolar range when evaluated against murine B16 melanoma and L1210 leukemia cell lines and in the case of 39 and 40, the IC50 figures in the B16 and L1210 assays are 0.51 and 0.35 μM, respectively, and are clearly lead molecules [65]. There is a structural similarity of some of these compounds, particularly those possessing several methoxy aryl substituents, to agents which inhibit tubulin polymerization. Two representative compounds, namely 39 and 40, were examined for this property. However using concentrations at least 25 times greater than the IC50 values did not disrupt microtubules. Hence the way in which these compounds exert their cytotoxic properties is by mechanisms other than inhibition of tubulin polymerization.
Another important example of a 2,6-bis(benzylidene) cyclohexanone is RSV-101. Compound 41, known as methyl p-hydroxyphenyllactate, is a cell growth regulating agent which occurs naturally [66]. It binds to nuclear type II sites in non-malignant cells thereby ensuring a relatively low level of cell proliferation. However 41 is metabolized in tumour cells which leads to high levels of unoccupied nuclear type II sites and hence a loss of regulatory control. The enone RSV-101 (42) binds with high affinity to nuclear type II sites in neoplastic cells which inhibits malignant cell proliferation [67, 68].
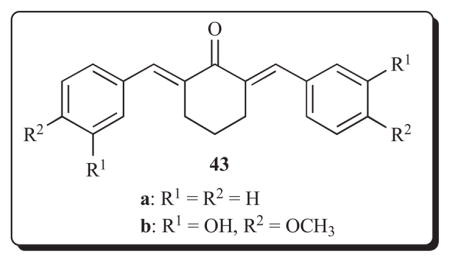
The 2,6-bis(benzylidene)cyclohexanones 43 have anti-angiogenesis properties causing a 90–94% inhibition of SVR endothelial cells using a concentration of 3 mg/mL [69].
3. 3,4-Dihydro-1H-naphthalen-2-ones
A further investigation designed to create molecules which permit successive reactions with cellular thiols to occur involved the preparation of a series of 1,3-bis(benzylidene)-3,4-dihyro-1H-naphthalen-2-ones 44 [70]. In these compounds the olefinic carbon atoms CA and CB are in different topographical environments. Thus when both rings A and B are unsubstituted, the Mulligen charges on CA and CB are −0.105 and −0.095, respectively. All of the diaryldienones 44 were examined in the Molt 4/C8, CEM, L1210 and P388 bioassays and selected compounds in the NCI screen. The IC50 figures are generally in the low micro-molar range. Three representative compounds, namely the unsubstituted, 2-nitro and 4-nitro congeners, inhibited DNA, RNA and protein biosynthesis while the unsubstituted and 2-nitro analogs did not bind to calf thymus DNA. Thus, in part at least, the cytotoxicity of these compounds is by interference with macromolecular biosynthesis.
A subsequent investigation with three representative compounds was undertaken in an attempt to find additional ways by which cytotoxicity is mediated [71]. The three compounds chosen, namely 45-47 have IC50 values in the 1.0–1.6 μM range towards Molt 4/C8 and CEM cells. First, a TUNEL assay revealed that 45 caused apoptosis in human HepG2 liver cancer cells at a concentration of 1 μM, suggesting that apoptosis is an important way whereby cytotoxicity is mediated. Second, there is higher activity and expression of hNMT in some colon cancers than the surrounding tissues vide supra. At the maximum concentration utilized, namely 1 mM, 46 inhibited hNMT by 23%. Third, a number of arylidene compounds which have several methoxy groups on the aryl ring inhibit tubulin polymerization [72]. However 47 inhibited this biochemical process by 16% only using a concentration of 50 μM. In summary, an important mechanism of action of these novel cytotoxins is by induction of apoptosis while the inhibition of hNMT and tubulin polymerization appear at most to be marginal contributors to the bioactivity observed.
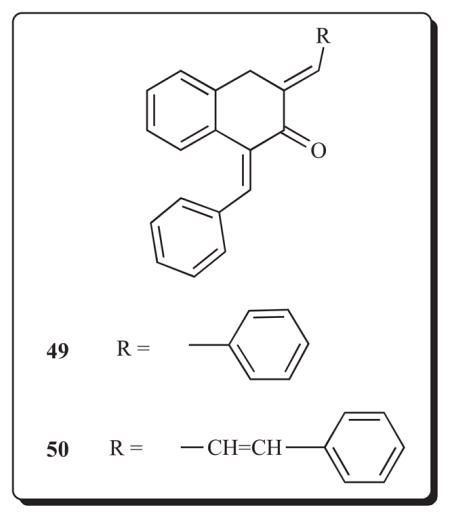
A further consideration is whether this cluster of diaryldienones possesses selective toxicity to malignant cells. Accordingly 45-47 and several analogs were evaluated against HSC-2, HSC-3, HSG and HL-60 neoplastic cell lines as well as non-malignant HGF, HPC and HPLF cells [64]. These compounds display high potencies towards the tumorous cells, e.g., the average CC50 value of 46 is 1.75 μM. On the other hand, the CC50 figures for several of these compounds towards the non-malignant cells is in excess of 400 μM. The selective index (SI) figures range from 12 to 229 (in the case of 46) which affords further evidence of the importance of developing these compounds. Of interest is the observation that the nor analog of 46, namely 48, was far less potent towards the malignant cells (average CC50 value of >335 μM) and had a SI figure of 0.6. This observation confirms the importance of the relative locations of different functional groups in these compounds with the bioactivity observed. This conclusion was also reached by comparing the cytotoxic potencies of 49 with its vinylog 50 [73]. In the Molt 4/C8, CEM and L1210 assays, 50 is on average nine times less potent than 49 (average IC50 value is 4.68 μM) and further experimentation is required in order to determine whether the optimal spacer between the aryl rings is an 3-oxo-1,4-pentadienyl group.
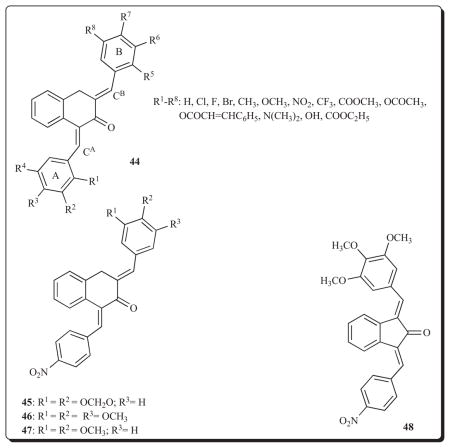
The importance of developing 3,4-dihydro-1H-naphthalen-2-ones is that a number of these molecules demonstrate significant potencies in the L-5178 P-gp reversal screen. In particular 46 and 47 have FAR values of 134 and 44, respectively, using a concentration of 4 μg/mL [64].
4. Acyclic 1,5-diaryl-3-oxo-1,4-pentadienes
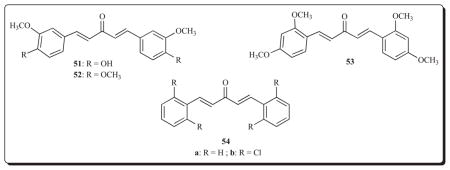
Recently the acyclic derivatives 51-53 were evaluated using two human prostate cancer cell lines, namely PC-3 cells which are androgen-independent and LNCaP cells which are androgen-dependent [74]. Both 51 and 52 display excellent growth-inhibiting properties with IC50 values in the 1.4–3.8 μM range towards both cell lines. On the other hand, when the two methoxy groups are located in the 2 and 4 positions of the aryl rings as in 53, the potencies towards PC-3 and LNCaP cells compared to 52 are lowered by approximately sixfold. In this study involving a number of structurally related compounds with larger unsaturated conjugated spacer groups between the aryl rings than the five carbon chain in 51-53, the importance of 3,4-disubstitution on the aryl rings in conferring marked potencies was noted.
Another feature of certain acyclic diaryldienones is their anti-angiogenic properties. Thus the unsaturated ketones 54a and 54b inhibited the growth of SVR endothelial cells by 97% and 87%, respectively, using a concentration of 3 mg/mL [69].
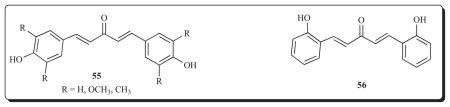
Series 55 were prepared as candidate antioxidants and shown to possess radical-scavenging properties as determined towards diphenylpicrylhydrazyl [58]. Other studies with compounds related to 55 displayed good antioxidant properties and maximum potencies are displayed by compounds possessing two small substituents ortho to the aryl hydroxy group [75]. However another investigation revealed that the presence of an aryl hydroxy group does not automatically confer antioxidant properties [76]. It is of interest to note that 56 not only inhibited superoxide formation and lipid peroxidation but displayed marked cytotoxicity towards Dalton’s lymphoma ascites cells and Ehrlich ascites cells [77].
5. Concluding Remarks on Cytotoxicity
The account so far has revealed that marked potencies are displayed by a variety of compounds in which the 1,5-diaryl-3-oxo-1,4-pentadienyl groups is present in a number of piperidines, cycloalkanes, arylalicyclics and acyclic molecules. In the future, the diaryldienone moiety should be mounted on further scaffolds with the goal of optimizing the biological responses. In this review is described the marked potencies of some diaryldienones which is accompanied by such favourable properties as greater toxicity to neoplasms than non-malignant cells. Various physicochemical parameters of some of the molecules described herein such as atomic charges and torsion angles influence the magnitude of the biological responses. The modes of action include the induction of apoptosis, stimulation of respiration in mitochondria and possibly a minimal effect on human myristoyl-CoA:protein N-myristoyltransferase (hNMT). In virtually all cases the series of compounds are well tolerated by mice.
Mention may be made of three further studies which aim to unify some of these observations. First, the 3D QSAR methodologies of comparative molecular field analysis (CoMFA) [78,79] and comparative molecular similarity indices analyses (CoMSIA) [80] were applied to the cytotoxic potencies of various 1,3-bis(benzylidene)-3,4-dihydro-1H-naphthalen-2-ones, 2,6-bis(benzylidene)cyclohexanones and 3,5-bis(benzylidene)-4-piperidones in the Molt 4/C8, CEM and L1210 screens [81]. From these determinations, the compounds interact at binding sites which have the following characteristics, namely two electropositive pockets to accommodate the electron-withdrawing group in the para position of the aryl rings, the aryl rings align at an electro-negative pocket and no groups should be present in the ortho and meta locations, a sterically forbidden site at which one of the olefinic groups interacts (i.e., no substitution on one of the methine carbon atoms) and a bulky hydrophobic pocket into which the group beta to the carbonyl function fits (e.g., the N-acyl group in the case of the 4-piperidones).
Second, a recent review indicated that many bioactive compounds contain an allylic group with an oxygen atom at one end of this substituent [82]. The oxygen atom can be located in a hydroxy, ester, ether, ketal or carbonyl substituents. Furthermore various compounds which lack this functionality may be converted into one of these groups by metabolism and in particular by the cytochrome P-450 enzymes. The allylic bioactive molecules may exert their influence in a variety of ways including controlling the phosphorylation of proteins, reducing the electron flow in mitochondria and lowering the concentration of glutathione in cells.
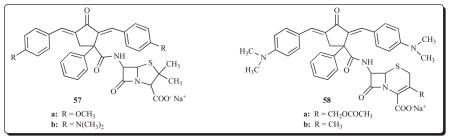
Third, as mentioned in the introductory remarks of this article, compounds interacting with multiple targets may exert more than one type of bioactivity. An example of the success of this strategy is as follows. An investigation was launched with a view to producing novel compounds with both antitumour and antimicrobial properties. In the molecules 57 and 58, the penicillin and cephalosporin side chains contain the antineoplastic diaryldienone group [83]. The percentage total growth inhibition in the murine S-180 screen for 57a, 57b, 58a and 58b are 36, 68, 41 and 58, respectively, and 38, 69, 0 and 54, respectively, in the murine S-37 bioassay. The percentage increases in the survival times of mice inoculated with Ehrlich’s ascitic carcinoma are 25, 100, 0 and 0, respectively. Clearly 57b in particular is a promising lead molecule. These compounds are well tolerated in mice insofar as the LD50 values ranged from 1250–3000 mg/kg compared to 2.6 mg/kg for the reference compound carminomycin. In addition, these molecules suppressed the growth of Staphylococcus aureus and Staphylococcus albus in the concentration range of 0.39–1.56 μg/mL. Two further positive attributes of these compounds are their greater resistance to both β-lactamase and acidic conditions than benzylpenicillin. This investigation illustrates the importance of the 1,5-diaryl-3-oxo-1,4-pentadienyl group in producing compounds capable of combating multiple pathological conditions.
MODES OF ACTION
Some of the ways in which certain diaryldienones exert their antineoplastic properties have been mentioned previously such as inducing apoptosis, inhibiting macromolecular biosyntheses and stimulating respiration in mitochondria. Some further comments revealing the array of different molecular targets of these compounds will now be presented.
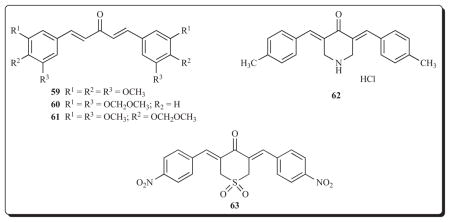
The diaryldienones 59-61 possess an IC50 value of 0.25 μM towards human HCT116 colon cancer cells and the way whereby cytotoxicity is mediated has been investigated [84]. Cell cycle analysis revealed that treatment of HCT116 cells by 59-61 impaired the synthesis of DNA. In addition, both 60 and 61 caused an increase in the sub-G1 population of the cells which indicates that apoptosis has occurred. Apoptosis can proceed via two major pathways. First, Fas (CD95/APO-1)-associated death domain (FADD) binds to procaspase 8 leading to the formation of caspase-8. Second, cytochrome c is released from the mitochondria leading to a complex with Apaf-1 which binds to procaspase-9 and activates various caspases including caspase-9. When 60 and 61 were added to HCT116 cells treated with an inhibitor of caspase-3 and caspase-8, namely Z-DEVD-fmk, the sub-G1 population returned to normal values. Thus two modes of action of these compounds are by inhibiting DNA synthesis and inducing apoptosis. Furthermore 60 and 61 induced down regulation of a number of oncoproteins, including ErbB-2, c-Myc, cy-clin D1 and Ki-ras.
Both 62 and 63 cause apoptosis by a Bcl-2-dependent but apoptosome-independent pathway of caspase activation in E1A/C9 cells [85]. This pathway is not followed by many anticancer agents [86]. Both 62 and 63 inhibit ubiquitin isopeptidases as do other cross-conjugated α,β;-unsaturated dienones with two sterically accessible electrophilic β-carbon atoms [87]. Hence ubiquitin-dependent protein degradation is impeded.
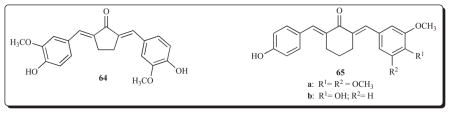
Another site of action of a diaryldienone 64 is topoisomerase 1. This unsaturated ketone, which inhibits the growth of a number of human tumour cell lines in the low micromolar range, interferes with two functions of topoisomerase 1, namely DNA relaxation and DNA single strand breaks [88]. It does not intercalate with DNA but binds to AT-rich sites in the minor groove of this nucleic acid. Cell cycle analysis revealed that treatment of KB cells with 64 caused accumulation in the G2/M phase. Camptothecin is an anticancer drug which interferes with the function of topoisomerase-1 but no cross-resistance was noted with two camptothecin-resistant cell lines. This compound also inhibits tubulin polymerization and causes apoptosis in KB cells whereby activation of caspase-3 was observed. A further study with 64 indicated that this molecule activated p53 but not Bcl-2 in KB cells [89]. Furthermore it did not cause perturbed mitochondrial membrane potential in contrast to 69, vide infra, which emphasized how minor structural changes affect the modes of action of the 1,5-diaryl-3-oxo-1,4-pentadienes.
Thioredoxin reductase catalyzes NADPH-dependent reduction of the disulfide group in thioredoxin. Hence this enzyme is important in controlling the redox potential and proliferation in cells. A number of tumours have elevated concentrations of thioredoxin reductase [90] and thus molecules which inhibit this enzyme may lead to products which are useful in cancer chemotherapy.
Following the observation that curcumin inhibits rat thioredoxin reductase [91], a number of analogs of this naturally occurring compound have been evaluated, including 65a and 65b [92]. The IC50 values of 61a and 61b towards thioredoxin reductase are 24.9 and 5.1 μM, respectively, and this mechanism of action may contribute to the antineoplastic properties of some or all conjugated dienones.
Recently a quest was initiated with the goal of finding compounds which stimulate an enzyme associated with cellular proliferation for the following reasons. The theory of sequential cytotoxicity is based on the differences in the vulnerability between neoplastic and non-malignant cells to chemical insults vide supra. The possibility exists that this differential in sensitivity could be exacerbated by the rate of proliferation in cancer cells being increased to a greater extent than occurs with normal tissue. Hence the first stage to explore this hypothesis is to find a cluster of compounds which stimulates an enzyme which controls cell proliferation, such as a protein kinase (PK). Fyn kinase is a PK and is overexpressed in certain tumours [93]. The 4-piperidone 5 stimulates fyn kinase, albeit weakly, namely approximately 15% using the concentration range of 25–100 μM [94]. However conversion of 5 to various N-acyl analogues led to compounds with increased enzyme-stimulating properties, e.g., 15 caused a 56% increase in fyn kinase activity at a concentration of 0.1 nM [94]. In addition, this compound is a cytotoxin having an average IC50 value of 5.11 μM in the Molt 4/C8, CEM and L1210 screens [30]. Hence the structural features of 15 could be included in the design of more complex molecules which initially release 15 or related compounds. Hence a greater increase in sensitivity to a subsequent liberation of a cytotoxin in malignant cells rather than the corresponding normal cells may result.
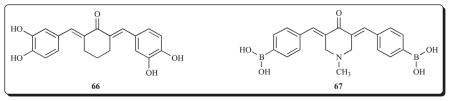
Another way in which diaryldienones exert their antineoplastic properties is by interactions with nuclear type II sites. Ligands which react with this receptor inhibit cellular growth and proliferation since the type II sites located in the nuclear matrix are believed to assist DNA synthesis and replication [95,96]. Compounds 42 and 66 bind to type II sites in rat uterine cells with very high affinity [68]. A correlation was noted between the extent of binding and the percentage inhibition of the growth of human MCF-7 breast cancer cells. In addition, 66 reduced the growth of T-364 L mammary tumours in mice revealing it possessing anticancer properties in vivo.
The proteasome is an enzyme complex involved in the degradation of proteins which is accomplished by ubiquitin conjugation. The diaryldienone 67 inhibits the activity of the 20S proteasome in vitro with an IC50 value of approximately 1 μM [97]. Thus the cytotoxicity of 67 towards cancer cells may be via inhibition of the proteasome leading to the accumulation of p53 and ubiquitinated proteins.
Another site of action of certain cyclic and acyclic diaryldienones is NFkappaB (NFκB) expression. This transcription factor is activated in a number of tumours and is important in metastasis [98]. NFkB protects certain malignant cells from apoptosis and hence compounds which inhibit its expression may be valuable in cancer chemotherapy. An acyclic and cyclic series of compounds have been prepared as exemplified by 42, 56 and 68 [99]. These diaryldienones were evaluated for their capacity to inhibit TNFα-induced activation of NFκB in cells stably infected with NFkB-dependent reporter construct using the Panomics Reporter Stable Cell Line. Compounds 42, 56 and 68 have IC50 values of approximately 4.5 μM.
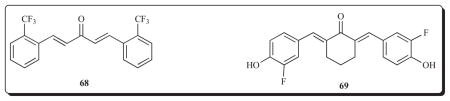
An important cellular target of compounds containing the 1,5-diaryl-3-oxo-1,4-pentadienyl group is the mitochondria. In particular these compounds have the capacity to increase the rates of respiration in these organelles. Compound 69 increases respiration rates in mitochondria and causes a collapse of the membrane potential [100]. This effect was reversed by 6-ketocholestanol which is a recoupling agent suggesting that 69 is an uncoupler. This compound inhibits the production of superoxide and oxidizes certain thiol groups which lead to swelling of mitochondria and the release of cytochrome c.
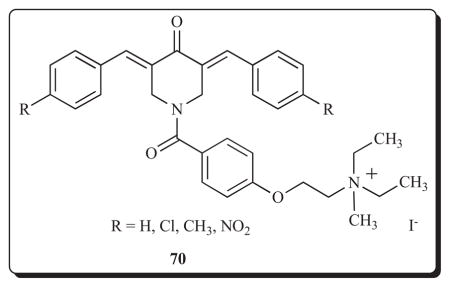
The possibility that different effects on mitochondria contribute to variation in cytotoxic potencies and murine toxicity was addressed recently [101]. Reference has already been made to the excellent cytotoxicity to series 17 [30] and that these compounds are well tolerated in rodents [31]. Quaternization of the diaryldienones 17 led to 70 which have reduced potencies [30] and most of the compounds are lethal when 300 mg/kg is administered to mice [31]. Using a concentration range of 10–100 μM, all of the compounds in series 17 and 70 stimulate respiration in rat liver mitochondria revealing one of the ways in which these compounds exert their bioactivity. However in virtually all cases when the same substituent was present in the arylidene aryl rings, respiration was greater in the quaternary ammonium salts. Furthermore using a concentration of 50 μM, all of the members of 70 caused swelling of mitochondria in contrast to a representative member of series 17 (R=H). While the greater effect on mitochondria by 70 does not explain their diminished cytotoxic properties compared to 17, the pronounced murine toxicity noted in the quaternary ammonium salts may be due to a greater biochemical effect on mitochondria.
CONCLUSIONS
A number of diaryldienones have IC50 values towards neoplastic and transformed cells in the submicromolar range (0.1–0.9 μM) while single and double digit nanomolar values are observed in some cases. The diaryldienones have the capacity to alkylate cellular thiols sequentially which may permit greater toxicity to malignant cells rather than the corresponding normal cells. In this regard, the clear demonstration of the preferential toxicity to neoplastic cells has been observed. Arguments have been presented in favour of compounds which have multiple sites of action and this review has revealed a number of different ways in which diaryldienones exert their modes of action. The observation that antiangiogenic, MDR-revertant and antioxidant properties coexist with antineoplastic activity affords further support for the development of these compounds as candidate anticancer agents.
Scheme 4.

Synthesis of 20-23 from 14a.
Acknowledgments
The authors thank the Canadian Institutes of Health Research for financial support of some of the research which is described in this review. Appreciation is extended to Beryl McCullough who typed various drafts of this article and Erin Watson who helped with some of the literature retrieval.
References
- 1.Dimmock JR, Wong MLC. Bioactivities and potential uses in drug design of acyclic α, β-unsaturated ketones. Can J Pharm Sci. 1976;11:35–53. [Google Scholar]
- 2.Dimmock JR, Raghavan SK, Logan BM, Bigam GE. Antileukemic evaluation of some Mannich bases derived from 2-arylidene-1,3-diketones. Eur J Med Chem. 1983;18:248–254. [Google Scholar]
- 3.Mutus B, Wagner JD, Talpas CJ, Dimmock JR, Phillips OA, Reid RS. 1-p-Chlorophenyl-4.4-dimethyl-5-diethylamino-1-penten-3-one hydrobromide, a sulfhydryl-specific compound which reacts irreversibly with protein thiols but reversibly with small molecular weight thiols. Anal Biochem. 1989;177:237–243. doi: 10.1016/0003-2697(89)90045-6. [DOI] [PubMed] [Google Scholar]
- 4.Benvenuto JA, Connor TH, Monteith DK, Laidlaw JA, Adams SC, Matney TS, Theiss JC. Degradation and inactivation of antitumor drugs. J Pharm Sci. 1993;82:988–991. [PubMed] [Google Scholar]
- 5.Okey AB, Harper PA. In: Principles of Medical Pharmacology. 7. Kalant H, Grant DM, Mitchell J, editors. Elsevier; Toronto, Canada: 2007. p. 902. [Google Scholar]
- 6.Dimmock JR, Elias DW, Beazely MA, Kandepu NM. Bio-activities of chalcones. Curr Med Chem. 1999;6:1125–1149. [PubMed] [Google Scholar]
- 7.Dimmock JR, Kumar P. Anticancer and cytotoxic properties of Mannich bases. Curr Med Chem. 1997;4:1–22. [Google Scholar]
- 8.Dimmock JR, Sidhu KK, Chen M, Reid RS, Allen TM, Kao GY, Truitt GA. Evaluation of some Mannich bases of cycloalkanones and related compounds for cytotoxic activity. Eur J Med Chem. 1993;28:313–322. [Google Scholar]
- 9.Chen G, Waxman DJ. Role of cellular glutathione and glutathione S-transferase in the expression of alkylating agent cytotoxicity in human breast cancer cells. Biochem Pharmacol. 1994;47:1079–1087. doi: 10.1016/0006-2952(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell JB, Russo A. The role of glutathione in radiation and drug induced cytotoxicity. Br J Cancer. 1987;55:96–104. [PMC free article] [PubMed] [Google Scholar]
- 11.Tsutsui K, Komuro C, Ono K, Nishidia T, Shibamoto Y, Takahashi M, Abe M. Chemosensitization by buthionine sulfoximine in vivo. Int J Radiat Oncol Biol Phys. 1986;12:1183–1186. doi: 10.1016/0360-3016(86)90254-3. [DOI] [PubMed] [Google Scholar]
- 12.Workman P, Collins I. In: Cancer Drug Design and Discovery. Neidle S, editor. Academic Press; London: 2008. p. 6. [Google Scholar]
- 13.Reidy D, Saltz L. Targeted strategies in the treatment of metastatic colon cancer. J Natl Compr Cancer Netw. 2007;5:983–990. doi: 10.6004/jnccn.2007.0082. [DOI] [PubMed] [Google Scholar]
- 14.Espinoza-Fonseca LM. The benefits of the multi-target approach in drug design and discovery. Bioorg Med Chem. 2006;14:896–897. doi: 10.1016/j.bmc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 15.Frantz S. Playing dirty. Nature. 2005;437:942–943. doi: 10.1038/437942a. [DOI] [PubMed] [Google Scholar]
- 16.Galanski M, Keppler BK. Searching for the magic bullet: anti-cancer platinum drugs which can be accumulated or activated in the tumor tissue. Anti-Cancer Agents Med Chem. 2007;7:55–73. doi: 10.2174/187152007779314017. [DOI] [PubMed] [Google Scholar]
- 17.Quinn FR, Milne GWA. Toxicities derived from anti-tumor screening data. Fundam Appl Toxicol. 1986;6:270–277. doi: 10.1016/0272-0590(86)90240-x. [DOI] [PubMed] [Google Scholar]
- 18.Jha A, Duffield KM. 3,5-Bis(arylmethylene)-4-piperidone derivatives as novel anticancer agents. Indian J Chem. 2006;45B:2313–2320. [Google Scholar]
- 19.Dimmock JR, Smith LM, Smith PJ. The reaction of some nuclear substituted acyclic conjugated styryl ketones and related Mannich bases with ethanethiol. Can J Chem. 1980;58:984–991. [Google Scholar]
- 20.Dimmock JR, Taylor WG. Evaluation of nuclear-substituted styryl ketones and related compounds for antitumor and cytotoxic properties. J Pharm Sci. 1975;64:241–249. doi: 10.1002/jps.2600640210. [DOI] [PubMed] [Google Scholar]
- 21.Dimmock JR, Patil SA, Shyam K. Evaluation of some Mannich bases of 1-aryl-ethanones and related ketones for anticonvulsant activities. Pharmazie. 1991;46:538–539. [PubMed] [Google Scholar]
- 22.Dimmock JR, Arora VK, Wonko SL, Hamon NW, Quail JW, Jia Z, Warrington RC, Fang WD, Lee JS. 3,5-bis-Benzylidene-4-piperidones and related compounds with high activity towards P388 leukemia cells. Drug Des Deliv. 1990;6:183–194. [PubMed] [Google Scholar]
- 23.Dimmock JR, Arora VK, Semple HA, Lee JS, Allen TM, Kao GY. 3,5-Bis-arylidene-1-methyl-4-piperidone methohalides and related compounds with activity against L1210 cells and DNA binding properties. Pharmazie. 1992;47:246–248. [PubMed] [Google Scholar]
- 24.Albert A. Selective Toxicity. 7. Chapman and Hall; London: 1985. pp. 642–643. [Google Scholar]
- 25.Dimmock JR, Arora VK, Duffy MJ, Reid RS, Allen TM, Kao GY. Evaluation of some N-acyl analogues of 3,5-bis(arylidene)-4-piperidones for cytotoxic activity. Drug Des Discov. 1992;8:291–299. [PubMed] [Google Scholar]
- 26.Dimmock JR, Arora VK, Quail JW, Pugazhenthi U, Allen TM, Kao GY, De Clercq E. Cytotoxic evaluation of some 3,5-diarylidene-4-piperidones and various related quaternary ammonium compounds and analogs. J Pharm Sci. 1994;83:1124–1130. doi: 10.1002/jps.2600830811. [DOI] [PubMed] [Google Scholar]
- 27.Dimmock JR, Padmanilayam MP, Puthucode RN, Nazarali AJ, Motaganahalli NL, Zello GA, Quail JW, Oloo EO, Kraatz HB, Prisciak JS, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK. A conformational and structure-activity relationship study of cytotoxic 3,5-bis(arylidene)-4-piperidones and related N-acryloyl analogues. J Med Chem. 2001;44:586–593. doi: 10.1021/jm0002580. [DOI] [PubMed] [Google Scholar]
- 28.Pandeya SN, Dimmock JR. An Introduction to Drug Design. New Age International (P) Limited, Publishers; New Delhi: 1997. p. 136.p. 137. [Google Scholar]
- 29.Dimmock JR, Kandepu NM, Hetherington M, Quail JW, Pugazhenthi U, Sudom AM, Chamankhah M, Rose P, Pass E, Allen TM, Halleran S, Szydlowski J, Mutus B, Tannous M, Manavathu EK, Myers TG, De Clercq E, Balzarini J. Cytotoxic activities of Mannich bases of chalcones and related compounds. J Med Chem. 1998;41:1014–1026. doi: 10.1021/jm970432t. [DOI] [PubMed] [Google Scholar]
- 30.Das U, Alcorn J, Shrivastav A, Sharma RK, De Clercq E, Balzarini J, Dimmock JR. Design, synthesis and cytotoxic properties of novel 1-[4-alkylaminoethoxy)phenylcarbonyl]-3,5-bis(arylidene)-4-piperidones and related compounds. Eur J Med Chem. 2007;42:71–80. doi: 10.1016/j.ejmech.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Das U, Das S, Bandy B, Stables JP, Dimmock JR. N-Aroyl-3,5-bis(benzylidene)-4-piperidones: a novel class of antimycobacterial agents. Bioorg Med Chem. 2008;16:3602–3607. doi: 10.1016/j.bmc.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawase M, Sakagami H, Motohashi N, Hauer H, Chatterjee SS, Spengler G, Vigyikanne AV, Molnár A, Molnár J. Coumarin derivatives with tumor-specific cytotoxicity and multidrug resistance reversal study. In Vivo. 2005;19:705–712. [PubMed] [Google Scholar]
- 33.Das U, Molnár J, Baráth Z, Bata Z, Dimmock JR. 1-[4-(2-Aminoethoxy)phenylcarbonyl]-3,5-bis-(benzylidene)-4-oxopiperidines: a novel series of highly potent revertants of P-glycoprotein associated multidrug resistance. Bioorg Med Chem Lett. 2008;18:3484–3487. doi: 10.1016/j.bmcl.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dimmock JR, Jha AJ, Zello GA, Quail JW, Oloo EO, Nienaber KH, Kowalczyk ES, Allen TM, Santos CL, De Clercq E, Balzarini J, Manavathu EK, Stables JP. Cytotoxic N-[4-(3-aryl-3-oxo-1-propenyl)phenylcarbonyl]-3,5-bis(phenylmethylene)-4-piperidones and related compounds. Eur J Med Chem. 2002;37:961–972. doi: 10.1016/s0223-5234(02)01414-9. [DOI] [PubMed] [Google Scholar]
- 35.Ducki S. The development of chalcones as promising anticancer agents. IDrugs. 2007;10:42–46. [PubMed] [Google Scholar]
- 36.Go ML, Wu X, Liu XL. Chalcones: an update on cytotoxic and chemopreventive properties. Curr Med Chem. 2005;12:483–499. doi: 10.2174/0929867053363153. [DOI] [PubMed] [Google Scholar]
- 37.Dimmock JR, Jha A, Zello GA, Sharma RK, Shrivastav A, Selvakumar P, Allen TM, Santos CL, Balzarini J, De Clercq E, Manavathu EK, Stables JP. 3,5-Bis(phenylmethylene)-1-(N-arylmaleomyl)-4-piperidones: a novel group of cytotoxic agents. J Enzyme Inhib Med Chem. 2003;18:325–332. doi: 10.1080/1475636031000121938. [DOI] [PubMed] [Google Scholar]
- 38.Jha A, Mukherjee C, Prasad AK, Parmar VS, De Clercq E, Balzarini J, Stables JP, Manavathu EK, Shrivastav A, Sharma RK, Nienaber KH, Zello GA, Dimmock JR. E,E,E-1-(4-Arylamino-4-oxo-2-butenoyl)-3,5-bis(arylidene)-4-piperidones: a topographical study of some novel potent cytotoxins. Bioorg Med Chem. 2007;15:5854–5865. doi: 10.1016/j.bmc.2007.05.065. [DOI] [PubMed] [Google Scholar]
- 39.Boutin JA. Myristoylation. Cell Signal. 1997;9:15–35. doi: 10.1016/s0898-6568(96)00100-3. [DOI] [PubMed] [Google Scholar]
- 40.Peseckis SM, Resh MD. Fatty acyl transfer by human N-myristoyltransferase is dependent upon conserved cysteine and histidine residues. J Biol Chem. 1994;269:30888–30892. [PubMed] [Google Scholar]
- 41.Raju RVS, Moyana TN, Sharma RK. N-myristoyltransferase overexpression in human colorectal adenocarcinomas. Exp Cell Res. 1997;235:145–154. doi: 10.1006/excr.1997.3679. [DOI] [PubMed] [Google Scholar]
- 42.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J Natl Cancer Inst. 1989;21:1088–1092. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 43.Ypsilantis P, Tentes I, Assimakopoulos SF, Kortsaris A, Scopa CD, Simopoulos C. Mesna ameliorates intestinal mucosa damage after ifosfamide administration in the rabbit at a dose-related manner. J Surg Res. 2004;121:84–91. doi: 10.1016/j.jss.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Haselberger MB, Schwinghammer TL. Efficacy of mesna for prevention of hemorragic cystitis after high-dose cyclophosphamide therapy. Ann Pharmacother. 1995;29:918–921. doi: 10.1177/106002809502900914. [DOI] [PubMed] [Google Scholar]
- 45.Allan SG, Smyth JF, Hay FG, Leonard RC, Wolf CR. Protective effect of sodium-2-mercaptoethanesulfonate on the gastrointestinal toxicity and lethality of cis-diammine dichloroplatinum. Cancer Res. 1986;46:3569–3573. [PubMed] [Google Scholar]
- 46.Pati HN, Das U, Quail JW, Kawase M, Sakagami H, Dimmock JR. Cytotoxic 3,5-bis(benzylidene)piperidin-4-ones and N-acyl analogs displaying selective toxicity for malignant cells. Eur J Med Chem. 2008;43:1–7. doi: 10.1016/j.ejmech.2007.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pati HN, Das U, Das S, Bandy B, De Clercq E, Balzarini J, Kawase M, Sakagami H, Quail JW, Stables JP, Dimmock JR. The cytotoxic properties and preferential toxicity to tumour cells displayed by some 2,4-bis(benzylidene)-8-methyl-8-azabicyclo[3.2.1]octan-3-ones and 3,5-bis(benzylidene)-1-methyl-4-piperidones. Eur J Med Chem. 2009;44:54–62. doi: 10.1016/j.ejmech.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Topliss JG. A manual method for applying the Hansch approach to drug design. J Med Chem. 1977;20:463–469. doi: 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- 49.Hamon NW, Bassendowski DL, Wright DE, Dimmock JR, Noble LM. Effect of antineoplastic and cytotoxic Mannich bases derived from conjugated styryl ketones on mitochondrial respiration in rat liver cells. J Pharm Sci. 1978;67:1539–1542. doi: 10.1002/jps.2600671112. [DOI] [PubMed] [Google Scholar]
- 50.Dimmock JR, Hamon NW, Noble LM, Wright DE. Nuclear-substituted styryl ketone analogs: effects on neoplasms, microorganisms, and mitochondrial respiration of tumorous and normal cells. J Pharm Sci. 1979;68:1033–1039. doi: 10.1002/jps.2600680830. [DOI] [PubMed] [Google Scholar]
- 51.Sun A, Shoji M, Lu YJ, Liotta DC, Snyder JP. Synthes of EF-24-tripeptide chloromethyl ketone: a novel curcumin-related anticancer drug delivery system. J Med Chem. 2006;49:3153–3158. doi: 10.1021/jm051141k. [DOI] [PubMed] [Google Scholar]
- 52.Shoji M, Snyder JP, Liotta DD, Sun A. Cytotoxic compound-protein conjugates as suppressors of tumor growth and angiogenesis. 2005/0069551 A1. US. 2005 Mar 31;
- 53.Adams BK, Ferstl EM, Davis MC, Herold M, Kurtkaya S, Camalier RF, Hollingshead MG, Kaur G, Sausville EA, Rickles FR, Snyder JP, Liotta DC, Shoji M. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg Med Chem. 2004;12:3871–3883. doi: 10.1016/j.bmc.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Nam NH. Combretastatin A-4 analogues as antimitotic anti-tumor agents. Curr Med Chem. 2003;10:1697–1722. doi: 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- 55.Tozer GM, Prise VE, Wilson J, Locke RJ, Vojnovic B, Stratford MR, Dennis MF, Chaplin DJ. Combretastatin A-4 phosphate as a tumor vascular-targeting agent: early effects in tumors and normal tissues. Cancer Res. 1999;59:1626–1634. [PubMed] [Google Scholar]
- 56.Gomes A, Fernandes E, Silva AMS, Santos CMM, Pinto DCGA, Cavaleiro JAS, Lima JLFC. 2-Styrylchromones: novel strong scavengers of reactive oxygen and nitrogen species. Bioorg Med Chem. 2007;15:6027–6036. doi: 10.1016/j.bmc.2007.06.046. [DOI] [PubMed] [Google Scholar]
- 57.Gomes A, Fernandes E, Garcia MBQ, Silva AMS, Pinto DCGA, Santos CMM, Caveleiro JAS, Lima JLFC. Cyclic voltammetric analysis of 2-styrylchromanones: relationship with the antioxidant activity. Bioorg Med Chem. 2008;16:7939–7943. doi: 10.1016/j.bmc.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 58.Youssef KM, El-Sherbeny MA, El-Shafie FS, Faraq HA, Al-Deeb OA, Anadalla SAS. Synthesis of curcumin analogues as potential antioxidant, cancer chemopreventive agents. Arch Pharm Pharm Med Chem. 2004;337:42–54. doi: 10.1002/ardp.200300763. [DOI] [PubMed] [Google Scholar]
- 59.Dimmock JR, Sidhu KK, Chen M, Li J, Quail JW, Allen TM, Kao GY. Synthesis and cytotoxic evaluation of some cyclic arylidene ketones and related oximes, oxime esters and analogs. J Pharm Sci. 1994;83:852–858. doi: 10.1002/jps.2600830619. [DOI] [PubMed] [Google Scholar]
- 60.Dimmock JR, Kumar P, Nazarali AJ, Motaganahalli NL, Kowalchuk TP, Beazely MA, Quail JW, Oloo EO, Allen TM, Szydlowski J, De Clercq E, Balzarini J. Cytotoxic 2,6-bis(arylidene)cyclohexanones and related compounds. Eur J Med Chem. 2000;35:967–977. doi: 10.1016/s0223-5234(00)01173-9. [DOI] [PubMed] [Google Scholar]
- 61.Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2:343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 62.Das U, Doroudi A, Das S, Bandy B, Balzarini J, De Clercq E, Dimmock JR. E,E-2-Benzylidene-6-(nitrobenzylidene)cyclohexanones: syntheses, cytotoxicity and an examination of their electronic, steric and hydrophobic properties. Bioorg Med Chem. 2008;16:6261–6268. doi: 10.1016/j.bmc.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bryla J. In: Inhibition of Mitochondrial Function. Erecińska M, Wilson DF, editors. Pergamon Press; Oxford: 1981. pp. 244–246. [Google Scholar]
- 64.Dimmock JR, Das U, Gul HI, Kawase M, Sakagami H, Baráth Z, Ocsovsky I, Molnár J. 3-Arylidene-1-(4-nitrophenylmethylene)-3,4-dihydro-1H-naphthalen-2-ones and related compounds displaying selective toxicity and reversal of multidrug resistance in neoplastic cells. Bioorg Med Chem Lett. 2005;15:1633–1636. doi: 10.1016/j.bmcl.2005.01.054. [DOI] [PubMed] [Google Scholar]
- 65.Davis R, Das U, Mackay H, Brown T, Mooberry SL, Dimmock JR, Lee M, Pati H. Syntheses and cytotoxic properties of the curcumin analogs 2,6-bis(benzylidene)-4-phenylcyclohexanones. Arch Pharm Chem Life Sci. 2008;341:440–445. doi: 10.1002/ardp.200800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Markaverich BM, Gregory RR, Alejandro MA, Clark JH, Johnson GA, Middleditch BS. Methyl p-hydroxyphenyllactate. J Biol Chem. 1988;263:7203–7210. [PubMed] [Google Scholar]
- 67.Markaverich BM, Varma RS. Combination therapy using bioflavonoid compounds with anti-cancer drugs. 93/01824. PCTWO. 1993 Feb 4;
- 68.Markaverich BM, Schauweker TH, Gregory RR, Varma M, Kittrell FS, Medina D, Varma RS. Nuclear type II sites and malignant cell proliferation: inhibition by 2,6-bis-benzylidenecyclohexanones. Cancer Res. 1992;52:2482–2488. [PubMed] [Google Scholar]
- 69.Robinson TP, Ehlers T, Hubbard RB, IV, Bai X, Arbiser JL, Goldsmith DJ, Bowen JP. Design, synthesis, and biological evaluation of angiogenesis inhibitors: aromatic enone and dienone analogues of curcumin. Bioorg Med Chem Lett. 2003;13:115–117. doi: 10.1016/s0960-894x(02)00832-6. [DOI] [PubMed] [Google Scholar]
- 70.Dimmock JR, Pamanilayam MP, Zello GA, Quail JW, Oloo EO, Prisciak JS, Kraatz HB, Cherkasov A, Lee JS, Allen TM, Santos CL, Manavathu EK, De Clercq E, Balzarini J, Stables JP. Cytotoxic 1,3-diarylidene-2-tetralones and related compounds. Eur J Med Chem. 2002;37:813–824. doi: 10.1016/s0223-5234(02)01402-2. [DOI] [PubMed] [Google Scholar]
- 71.Das U, Gul HI, Alcorn J, Shrivastav A, George T, Sharma RK, Nienaber KH, De Clercq E, Balzarini J, Kawase M, Kan N, Tanaka T, Tari S, Werbovetz KA, Yakovich A, Manavathu EK, Stables JP, Dimmock JR. Cytotoxic 5-aryl-1-(4-nitrophenyl)-3-oxo-1,4-pentadienes mounted on alicyclic scaffolds. Eur J Med Chem. 2006;41:577–585. doi: 10.1016/j.ejmech.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 72.Petit GR, Singh SB, Boyd MR, Hamel E, Petit RK, Schmidt JM, Hogan F. Antineoplastic agents. 291. Isolation and synthesis of combretastatins A-4, A-5, and A-6. J Med Chem. 1995;38:1666–1672. doi: 10.1021/jm00010a011. [DOI] [PubMed] [Google Scholar]
- 73.Jha A, Zhao J, Cameron TS, De Clercq E, Balzarini J, Manavathu EK, Stables JP. Design, synthesis and biological evaluation of novel curcumin analogues as anti-neoplastic agents. Lett Drug Des Discov. 2006;3:304–310. [Google Scholar]
- 74.Lin L, Nyarko AK, Baston KF, Wu CC, Su CY, Shih CCY, Lee KH. Antitumor agents. 250. Design and synthesis of new curcumin analogues as potential anti-prostate cancer agents. J Med Chem. 2006;49:3963–3972. doi: 10.1021/jm051043z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sardjiman SS, Reksohadiprodjo MS, Hakim L, van der Goot H, Timmerman H. 1,5-Diphenyl-1,4-pentadiene-3-ones and cyclic analogues as antioxidative agents. Synthesis and structure-activity relationship. Eur J Med Chem. 1997;32:625–630. [Google Scholar]
- 76.Weber WM, Hunsaker LA, Abcouwer SF, Deck LM, Vander Jagt DL. Anti-oxidant activities of curcumin and related enones. Bioorg Med Chem. 2005;13:3811–3820. doi: 10.1016/j.bmc.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 77.Anto RJ, Sukumaran K, Kuttan G, Rao MNA, Subbaraju V, Kuttan R. Anticancer and antioxidant activity of synthetic chalcones and related compounds. Cancer Lett. 1995;97:33–37. doi: 10.1016/0304-3835(95)03945-s. [DOI] [PubMed] [Google Scholar]
- 78.Cramer RD, III, Patterson DE, Bunce JD. Comparative Molecular Field Analysis (COMFA). 1. Effect of shape on binding of steroids to carrier proteins. J Am Chem Soc. 1988;110:5959–5967. doi: 10.1021/ja00226a005. [DOI] [PubMed] [Google Scholar]
- 79.Cramer RD, III, Bunce JD, Patterson DE. Crossvalidation, bootstrapping, and partial least squares compared with multiple regression in conventional QSAR studies. Quant Struct-Act Relat. 1988;7:18–25. [Google Scholar]
- 80.Klebe G, Abraham U, Mietzner T. Molecular similarity indices in a comparative analysis (COMSIA) of drug molecules to correlate and predict their biological activity. J Med Chem. 1994;37:4130–4146. doi: 10.1021/jm00050a010. [DOI] [PubMed] [Google Scholar]
- 81.Patel MR, Dimmock JR, Talele TT. CoMFA and CoMSIA studies on 1,3-bis(benzylidene)-3,4-dihydro-1H-naphthalen-2-one, 2,6-bis(benzylidene)cyclohexanone, and 3,5-bis(benzylidene)-4-piperidone series of cytotoxic compounds. J Chem Inf Model. 2007;47:2110–2123. doi: 10.1021/ci700210z. [DOI] [PubMed] [Google Scholar]
- 82.Radin NS. Drug design: hiding in full view. Drug Dev Res. 2008;69:15–25. [Google Scholar]
- 83.Martirosyan AO, Gasparyan SP, Oganesyan VE, Mantirosyan VV, Chachoyan AA, Kazaryan ÉV, Garibdzhanyan BT. Synthesis, antibacterial and antitumor activity of semisynthetic penicillins and cephalosporins based on new sarcomycin analogs. Pharm Chem J. 2005;39:67–69. [Google Scholar]
- 84.Ohori H, Yamakoshi H, Tomizawa M, Shibuya M, Kakudo Y, Takahashi A, Takahashi S, Kato S, Suzuki T, Ishioka C, Iwabuchi Y, Shibata H. Synthesis and biological analysis of new curcumin analogues bearing an enhanced potential for the medicinal treatment of cancer. Mol Cancer Ther. 2006;5:2563–2571. doi: 10.1158/1535-7163.MCT-06-0174. [DOI] [PubMed] [Google Scholar]
- 85.Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006;66:9235–9244. doi: 10.1158/0008-5472.CAN-06-0702. [DOI] [PubMed] [Google Scholar]
- 86.Henderson CJ, Aleo E, Fontanini A, Maestro R, Paroni G, Brancolini C. Caspase activation and apoptosis in response to proteasome inhibitors. Cell Death Differ. 2005;12:1240–1254. doi: 10.1038/sj.cdd.4401729. [DOI] [PubMed] [Google Scholar]
- 87.Mullally JE, Fitzpatrick FA. Pharmacophore model for novel inhibitors of ubiquitin isopeptidases that induce p53-independent cell death. Mol Pharmacol. 2002;62:351–358. doi: 10.1124/mol.62.2.351. [DOI] [PubMed] [Google Scholar]
- 88.Chang JY, Hsieh HP, Pan WY, Liou JP, Bey SJ, Chen LT, Song JS. Dual inhibition of topoisomerase I and tubulin polymerization by BPROY007, a novel cytotoxic agent. Biochem Pharmacol. 2003;65:2009–2019. doi: 10.1016/s0006-2952(03)00197-7. [DOI] [PubMed] [Google Scholar]
- 89.Juang SH, Pan WY, Kuo CC, Liou JP, Hung YM, Chen LT, Hsieh HP, Chang JY. A novel bis-benzylidenecyclopentanone derivative, BPROY007, inducing a rapid caspase activation involving upregulation of Fas (CD95/APO-1) and wild-type p53 in human oral epidermoid carcinoma cells. Biochem Pharmacol. 2004;68:293–303. doi: 10.1016/j.bcp.2004.03.036. [DOI] [PubMed] [Google Scholar]
- 90.Yoo MH, Xu XM, Carlson BA, Gladyshev VN, Hatfield DL. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J Biol Chem. 2006;281:13005–13008. doi: 10.1074/jbc.C600012200. [DOI] [PubMed] [Google Scholar]
- 91.Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anti-cancer activity. J Biol Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 92.Qiu X, Liu Z, Shao WY, Liu X, Jing DP, Yu YJ, An LK, Huang SL, Bu XZ, Huang ZS, Gu LQ. Synthesis and evaluation of curcumin analogues as potential thioredoxin reductase inhibitors. Bioorg Med Chem. 2008;16:8035–8041. doi: 10.1016/j.bmc.2008.07.054. [DOI] [PubMed] [Google Scholar]
- 93.Jardines L, Weiss M, Fowble B, Green M. Neu(c-erbB-2/HER2) and the epidermal growth factor receptor (EGFR) in breast cancer. Pathobiology. 1993;61:268–282. doi: 10.1159/000163805. [DOI] [PubMed] [Google Scholar]
- 94.Das U, Selvakumar P, Sharma RK, Haas TH, Dimmock JR. N-Acyl-3,5-bis(arylidene)-4-piperidones and related compounds which stimulate fyn kinase. J Enzyme Inhib Med Chem. 2007;22:451–455. doi: 10.1080/14756360701192515. [DOI] [PubMed] [Google Scholar]
- 95.Markaverich BM, Clark JH. Two binding sites for estradiol in rat uterine nuclei: relationship to uterotropic response. Endocrinology. 1979;105:1458–1462. doi: 10.1210/endo-105-6-1458. [DOI] [PubMed] [Google Scholar]
- 96.Markaverich BM, Upchurch S, Clark JH. Progesterone and dexamethasone antagonism of uterine growth: a role for a second nuclear binding site for estradiol in estrogen action. J Steroid Biochem. 1981;14:125–132. doi: 10.1016/0022-4731(81)90164-3. [DOI] [PubMed] [Google Scholar]
- 97.Achanta G, Modzelewska A, Feng L, Khan SR, Huang P. A boronic-chalcone derivative exhibits potent anticancer activity through inhibition of the proteasome. Mol Pharmacol. 2006;70:426–433. doi: 10.1124/mol.105.021311. [DOI] [PubMed] [Google Scholar]
- 98.Andela VB, Gordon AH, Zotalis G, Rosier RN, Goater JJ, Lewis GD, Schwartz EM, Puzas JE, O’Keefe RJ. NFkappa B: a pivotal transcription factor in prostate cancer metastasis to bone. Clin Orthop Relat Res. 2003;415(Suppl):S75–S85. doi: 10.1097/01.blo.0000093048.96273.aa. [DOI] [PubMed] [Google Scholar]
- 99.Weber WM, Hunsaker LA, Roybal CN, Bobrovnikova-Marjon EV, Abcouwer SF, Royer RE, Deck LM, Vander Jagt DL. Activation of NFκB is inhibited by curcumin and related enones. Bioorg Med Chem. 2006;14:2450–2461. doi: 10.1016/j.bmc.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 100.Ligeret H, Barthélémy S, Doulakas GB, Carrupt PA, Tillement JP, Labidalle S, Morin D. Fluoride curcumin derivatives: new mitochondrial uncoupling agents. FEBS Lett. 2004;569:37–42. doi: 10.1016/j.febslet.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 101.Das S, Das U, Bandy B, Gorecki DKJ, Dimmock JR. The effect of some 1-[4-(2-diethylaminoethoxy)phenylcarbonyl]-3,5-bis(benzylidene)-4-piperidone methoiodides and related compounds on respiration and swelling of rat liver mitochondria. Pharmazie. 2008;63:827–829. [PMC free article] [PubMed] [Google Scholar]