

Abstract
Critical reagents are essential components of ligand binding assays (LBAs) and are utilized throughout the process of drug discovery, development, and post-marketing monitoring. Successful lifecycle management of LBA critical reagents minimizes assay performance problems caused by declining reagent activity and can mitigate the risk of delays during preclinical and clinical studies. Proactive reagent management assures adequate supply. It also assures that the quality of critical reagents is appropriate and consistent for the intended LBA use throughout all stages of the drug development process. This manuscript summarizes the key considerations for the generation, production, characterization, qualification, documentation, and management of critical reagents in LBAs, with recommendations for antibodies (monoclonal and polyclonal), engineered proteins, peptides, and their conjugates. Recommendations are given for each reagent type on basic and optional characterization profiles, expiration dates and storage temperatures, and investment in a knowledge database system. These recommendations represent a consensus among the authors and should be used to assist bioanalytical laboratories in the implementation of a best practices program for critical reagent life cycle management.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9334-9) contains supplementary material, which is available to authorized users.
Key words: characterization, critical reagents, data management, inventory, knowledge database, ligand binding assays
INTRODUCTION
Ligand binding assays (LBAs) are used at all stages of the drug development process, from discovery and candidate screening/selection to post-marketing monitoring. These bioanalytical assays are designed to support in vitro screening, product development/release, pharmacokinetic (PK), biomarker, and immunogenicity analyses. Robust and reproducible assay performance is primarily dependent on reagent quality and consistency. Critical reagents are those essential components of LBAs whose unique characteristics are crucial to assay performance and therefore require thorough characterization and documentation. Unlike other analytical technologies, such as liquid chromatography-mass spectrometry (LC-MS), LBAs require specific and unique biomolecular interactions between the critical reagents and the analyte. This feature places great importance on the structural integrity and stability of LBA reagents.
Common protein-based, chemically derived, and complex biological critical reagents can be classified into multiple categories (Table I). This paper focuses on antibodies (monoclonal antibodies (MAbs) and polyclonal antibodies (PAbs)), engineered proteins and peptides, and their conjugates. Chemically synthesized reagents, complex biologics, solid supports, and assay biological matrices will not be addressed in this paper. Calibrators and quality controls (QCs) made from reference drug material, although key components of LBAs (1) are also not addressed in this paper. Additionally, best practices are described elsewhere for maintaining and using cell lines (2) and aptamers (3,4) as reagents.
Table I.
Common Critical Reagents Utilized in LBA
| Critical reagent category | Examples |
|---|---|
| Antibodies | Monoclonal antibodies (MAbs) |
| Polyclonal antibodies (PAbs) | |
| Engineered proteins | Soluble receptors |
| Cytokines and growth factors | |
| Non-cytokine drug ligands | |
| Fusion proteins | |
| Enzymes | |
| Engineered proteins | |
| Conjugates of antibodies and engineered proteins and peptides | Small organic molecules ( e.g. , biotin) |
| Enzymes ( e.g. , HRP, alkaline phosphatase) | |
| Detection molecules ( e.g. , fluorescent dyes; chemiluminescent or luminescent dyes/labels) | |
| Chemically synthesized molecules | Aptamers (peptides or nucleic acids) |
| Non-protein hormones | |
| Carbohydrates | |
| Toxins | |
| Small organic molecules | |
| Radiolabels (e.g., 125I) | |
| Complex biologics | Live or fixed cells |
| Tissues and cell lysates | |
| Cell membranes | |
| Organelles | |
| Viruses | |
| Bacteria | |
| Solid supports and matrices | Plates |
| Beads | |
| Chips | |
| Biological matrices (e.g., serum, plasma, synovial fluid, milk) |
Categories of critical reagents in bold font are discussed in this paper
Critical reagents are typically produced via biological processes and thus may be inherently prone to variability as different lots are prepared. This can impact the performance of the LBA considerably. For example, cell line production changes of MAbs or other engineered protein critical reagent lots could lead to changes in impurity profiles, post-translational modifications, and altered protein primary or tertiary structure. In addition, variability in the production of PAb reagents occurs as animal immune responses mature. To address these common production issues, a risk mitigation strategy is a helpful tool to employ during all stages of a critical reagent life cycle (Table II).
Table II.
Risk Mitigation Activities at Various Stages of Critical Reagents’ Life Cycle
| Critical reagent stage | Risk-mitigating activities |
|---|---|
| Initial and subsequent generation | Allow sufficient lead time to generate appropriate reagents |
| Assess quality of available reagents relative to the complexity and stage of the LBA (e.g., for non-regulated versus regulated LBA) | |
| Document and standardize generation of reagents, including back up reagents | |
| Characterization | Generate a panel of biophysical and biochemical characterization parameters |
| Determine reagent assay qualification and performance assessment criteria for initial and subsequent lots | |
| Assess stability | |
| Inventory management | Assure sufficient supply for intended use including stage of program, multiple laboratory usage, estimated usage, and forecast users consumption |
| Allow sufficient time for reagent resupply | |
| Archive critical reagent cell line(s) | |
| Knowledge database | Document appropriate use and performance in assays |
Depending on the business structure of each organization, assay methods and associated reagents may be transferred to other laboratories within the organization or externally to a CRO. Thus, the characterization, maintenance, long-term storage, inventory, and knowledge database of associated LBA critical reagents, even at early stages of drug development, are crucial (Fig. 1). The most important benefit of a critical reagent management system is to assure successful support of analytical methods and longitudinal studies, enabling the smooth transition of assays between laboratories and timely data collection. Clearly defined responsibilities pertaining to the management of critical reagents need to be assigned in order to assure reagents are well characterized and that the supply chain, knowledge database, and inventory are appropriately maintained. Critical reagent life cycle management by the organization is crucial, whether the reagents are prepared internally or outsourced to vendors, and whether they are utilized as individual reagents of a custom assay or as part of commercial kits.
Fig. 1.
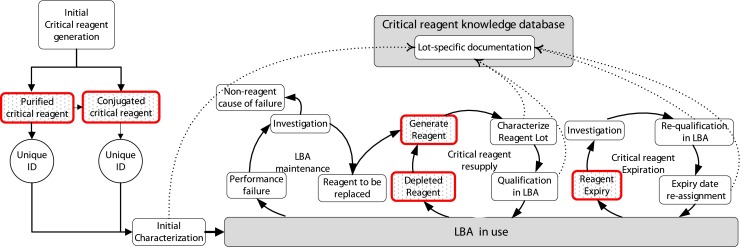
Critical reagent life cycle management. The schematic shows critical reagent life cycle management, from initial reagent generation through repeat use and resupply cycles. Inventory information is identified by red outlined boxes. Transfer of critical reagent information to the knowledge database is depicted by dotted lines
Several publications have described recommendations for LBA method validation, including methods for biomarker assays (5,6), PK assays (7), anti-drug antibody (ADA)/neutralizing antibody (NAb) assays (8–10), and flow cytometric assays (11). However, minimal guidance is available on how to manage reagents used in these LBAs. The FDA has included reagents and solutions in their regulations on good laboratory practice (GLP) for nonclinical laboratory studies (12), indicating that reagent and solution information should include identity, titer or concentration, storage requirements, and expiration date. The purpose of this paper is to describe best practices and provide recommendations for the life cycle management, characterization, and supply of critical reagents used in these LBA. Recommendations are made for each type of critical reagent on basic and optional characterization profiles, a range of expiration dates and storage temperatures, and investment in a knowledge database system with appropriately assigned responsibilities. The initiative to develop and publish a consensus among the authors originated from a 2009 American Association of Pharmaceutical Scientists workshop on the Twenty-First Century Bioanalytical Laboratory, sponsored by the Ligand Binding Assay Bioanalytical Focus Group. This publication is one of several articles written to build alignment across the industry on the expectations and operations of a bioanalytical laboratory.
The data and examples described herein are for illustrational purposes only and may represent hypothetical, blinded, and/or modified data. Although the opinions and recommendations expressed in this paper have been broadly discussed, the authors note that the principles expressed herein may not be applicable to all critical reagents and/or LBAs. Appropriate LBA development, qualification, validation, and implementation will depend upon the specific critical reagent or LBA and the context of their use. The paper has been organized into six main areas that describe the initial generation and selection of reagents, the characterization of these reagents, the maintenance and long-term supply of critical reagents, the maintenance of LBA, the considerations for use of commercial LBA kits, and, finally, the best practices for inventory and knowledge database management.
INITIAL GENERATION AND SELECTION OF REAGENTS
To ensure that critical reagents for LBAs meet their intended purposes, early strategic discussions should address specific reagent requirements for a given therapeutic development program. The sensitivity and specificity of the LBA is influenced by the quality of specific lots of critical reagents used in the assay (13,14). An attempt to considerably improve LBA sensitivity may require the incorporation of alternative reagents or efforts to develop new reagents.
LBA reagents generally include capture and detector reagents, positive controls for immunogenicity assays, and calibrator reagents that are needed for biomarker assay. When several types of assay formats and reagents are considered for the same application, it is advisable to initiate the generation of the different types of reagents in parallel, increasing the likelihood of obtaining optimal reagents with the appropriate and pre-specified characteristics. Conducting an appropriate risk assessment provides an opportunity to adequately balance resources and timelines.
A cost-effective and efficient strategy is to consider reagents already available for assay development, whether internally produced or from a commercial source, prior to initiating the generation of new reagents. The generation and selection of LBA reagents are determined by the objectives of the study supported by the LBA, the stage of the drug development program, potential assay formats, species of origin and type of matrices of samples to be tested, required assay sensitivity and specificity, as well as the potential need for reagent reactivity to multiple analytes, e.g., host cell protein (HCP) LBAs. Therefore, prior to initiating the generation of new reagents, key information should be gathered to understand the characteristics of the therapeutic and the requirements of the LBA. Examples of useful information to be collected include: (a) the requirements for sensitivity as well as specific and selective interactions between a reagent and the target analyte; (b) the need to reduce potential non-specific interactions with other assay components, such as heterophilic antibodies (15–17), and their interactions in PK assays (18,19), biomarker assays (20,21), as well as in clinical diagnostic assays (22); (c) the need to reduce potential cross-reactivity in the target sample population [e.g., rheumatoid factor, (23–25)]; (d) the need for discrimination between analyte isoforms and its proteolytic byproducts (13); (e) the requirement for detecting free analytes versus those in larger complexes (26); and (f) detecting anti-drug antibodies bound to the therapeutic with or without treatment of the sample with acid (27).
PAbs are typically generated in rabbits, goats, sheep, and chickens (Fig. 2). At a minimum, two animals should be initially immunized. However, a larger number of animals may be needed to accommodate for heavier reagent use or a higher than usual variation in animal immune response. MAbs are typically generated from immunized and fused spleen cells from mice (28–31) rats, hamsters, guinea pigs, and, more recently, rabbits (32), though MAbs may also be selected from genomic libraries (33–35). If purified protein antigens are unavailable or difficult to produce, genetic immunizations using cDNA have proven very useful in generating antibodies to natively expressed molecules (36–39). Non-Ab biological reagents are generated by initial cloning, followed by expression and purification (40).
Fig. 2.
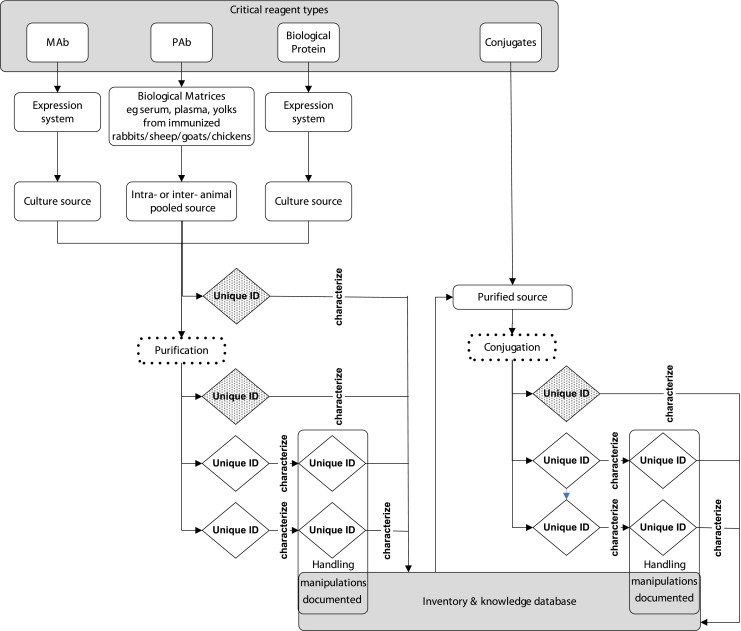
Generation and characterization of critical reagents. Purification and conjugation (dotted boxes) processes generate lot specific reagents with unique identifiers (diamond). Initial characterization of generated reagents (hatched diamonds) includes the full characterization profile, subsequent reagents (open diamonds) may undergo characterization according to the organizations practices and procedures. Bulk reagents may be manipulated further, including thawing from frozen or diluting then re-aliquoting and re-freezing. Handling manipulations are documented, and new designations may be assigned, though the original source lot number is either maintained or cross-referenced in the inventory and knowledge database
Factors that influence the immune response to the therapeutic in immunized animals are as follows: (a) the degree of sequence homology in related proteins from the animals used for immunization; in cases of high degree of homology, avian species should be considered for PAb reagents and knockout animals (if available) for MAb reagents; (b) the immunization route; (c) choice of adjuvant and frequency of injection, for example, the use of Freunds complete and incomplete adjuvants; (d) the molecular size of the immunogen; in case of molecules with <15KDa molecular weight, consider conjugation to haptens such as KLH; and (e) the presence of immunogenic epitopes on the antigens.
Common sequences and structural motifs on biotherapeutics often enable the use of common reagents across multiple LBAs, which may be advantageous when project timing is particularly critical. For instance, during early MAb therapeutic discovery programs, generic anti-IgG reagents may be used, which may later become less desirable as target-specific reagents are generated. Additionally, reagents specific to covalent modifications on a therapeutic protein may be developed, for example, a linked protein, a linker sequence, a molecular tag, a small chemical moiety, or a polyethylene glycol chain (41,42) of varying size and complexity.
Once protein reagents are generated and purified, reagent conjugates are frequently generated by standardized methods that employ known ratios of protein to solid supports (such as beads) or molecular label reactants (such as biotin, fluorophores, or enzymes). For a newly conjugated reagent, multiple protein/label ratios should be employed and evaluated, allowing for the selection of optimal conditions to be used, one eventually being selected for larger scale production. To assure reproducibility among lots, excess reactants (e.g., free molecular label) should be removed from the product and ratios of labeled components to protein reagent should be determined.
Critical Reagent Screening Strategies
Prior to reagent generation, designing an assay format similar to the intended LBA can be an effective screening tool to rank reagents for required attributes. Various assay formats and platforms can be used to screen for appropriate reagents. At a minimum, screening methods must be used to demonstrate that core requirements of the reagents are met. Several examples of screening methods include: (a) an assessment of binding of the reagent to the analyte in the absence and presence of its ligand (26); (b) for HCP impurity assays, screening polyclonal anti-HCP antibodies may include Western blot analyses to ensure that antibody reagents demonstrate a good breadth of coverage of the spectrum of proteins present in the calibration standard; (c) for neutralizing activity assays, a simplified screening assay includes the therapeutic and target and would detect this activity and distinguish these reagents from non-neutralizing antibodies. Consider platforms that provide multiplexing capabilities or label-free measurements to leverage the ability to identify appropriately compatible reagents in a LBA format.
Small amounts (1–5 mg) of each of the reagents selected during the screening process are sufficient for the BA laboratory to assess reagent pairing, neutralizing activity, different LBA formats, and/or possibly different LBA platforms.
Critical Reagent Quantities
Once selection has been made for the reagent to be used in a LBA, a larger scale lot is produced. The amount to be produced depends on the anticipated usage, the duration of a program, the program decision points, and the number of studies to be supported. While it would be preferable to have sufficient material generated to reduce the need for multiple reagent lots, this should be balanced with the required timing of the assay development activities and the knowledge of reagent stability and reproducibility. This bulk critical reagent should be made in sufficient quantities for use in the LBA and for subsequent comparability studies with future lots.
Considerations for Outsourcing Critical Reagent Generation and Supply
Outsourcing reagent generation activities to a vendor is often necessary and may include some or all of the steps described above. Successful partnerships require clear communication, conflict resolution, and critical thinking skills. It is important to understand the supplier practices and capabilities prior to vendor selection (43,44). Use of questionnaires, audits, or site visits provide valuable information. With a new supplier, it may be prudent to consider undertaking a pilot reagent preparation project prior to entrusting a pivotal or large scale project to the laboratory.
Critical Reagent Cell Lines
The sources of the majority of critical reagents are cell lines generated from cloned hybridoma cells or transfected cell lines expressing recombinant engineered proteins. To ensure the continued supply and quality of critical reagents, it is vital to carefully manage the generation, storage, and documentation of master and/or working stocks of cell lines (1).
CRITICAL REAGENT CHARACTERIZATION
In this section, characterization refers to the process of identifying physico-chemical attributes of reagents. Critical reagents must possess the appropriate characteristics to meet LBA parameters of sensitivity, specificity, and reproducibility. Whether the assay is quantitative (e.g., PK or biomarker assays) or quasi-quantitative (e.g., immunogenicity assays), these assay parameters are essential to assure optimal performance over time. Examples of critical reagent characteristics and their relevance to assay parameters are presented in Table III, with recommended basic and optional characterization profiles indicated for each reagent type.
Table III.
Recommended Characterization Profile of Critical Ligand Binding Reagents Using Orthogonal Methods
| Attributes affecting LBA | Antibody | Engineered protein | Conjugate |
|---|---|---|---|
| Sensitivity | |||
| Concentration | ✓ | ✓ | ✓ |
| Binding activity | ✓ | ✓ | ✓ |
| Potency | O | O | O |
| Binding kinetics and affinity determination (SPR) | O | O | O |
| Neutralization activity | O | O | O |
| Aggregation level (e.g., SEC, etc.) | ✓ | ✓ | ✓ |
| Specificity | |||
| Cross-reactivity | O | O | O |
| Reproducibility | |||
| Molecular weight (e.g., SEC, SDS-PAGE, or LC-MS, etc.) | ✓ | ✓ | ✓ |
| Purity (SEC, SDS-PAGE, or LC-MS, etc.) | ✓ | ✓ | ✓ |
| Isotype (for MAbs) | ✓ | N | N |
| Formulation buffer selection | O | O | O |
| Isoelectric point | O | O | O |
| Conjugate incorporation ratio (absorbance, LC-MS, etc.) | N | N | ✓ |
| Endotoxin level (for bioassays) | O | O | O |
| Protein A level | O | O | O |
| Host cell protein level | O | O | O |
| Stability | O | O | O |
| Bovine IgG level (from fetal calf/bovine serum) | O | O | O |
| Functional assays | O | O | O |
Check mark recommended, O optional as needed for intended use in LBA, N generally not applicable
Based on the characterization data generated across multiple lots of reagents, along with the LBA performance data collected over time, a core set of requisite physico-chemical attributes should be assigned to a particular critical reagent. These important characteristics provide the means to: (a) guide the generation of reagents and screen different lots that may be qualified for use in an assay; (b) identify root causes of assay performance problems; and (c) determine when a reagent is deteriorating and must be replaced.
Sensitivity of a quantitative assay refers to the lowest amount of analyte in a sample which can be reliably quantified with acceptable precision and accuracy. Reagent attributes that play a role in assay sensitivity include Ab titer (corresponds to reagent molecular interactions or neutralizing activity), potency (corresponds to reagent function), and binding activity (corresponds to affinity and/or avidity). Specificity is characterized by the ability of reagents (and the assay) to differentiate the analyte of interest from endogenous components in the matrix or other components in the sample. Reagent attributes that impact assay specificity include cross-reactivity, which may be caused by structural and functional homology of the analyte to biological components, and non-specific binding (e.g., due to reagent and matrix incompatibilities). Studies undertaken with critical reagents that lack appropriate specificity in LBAs may lead to inappropriate or erroneous conclusions (45) and demonstrates the importance of characterization early in a program. Reproducibility (precision) describes the closeness of repeated individual measures of analyte. Reagent attributes that impact assay reproducibility include physico-chemical parameters of the protein such as molecular weight (glycoslyation levels and isoforms), pI, isotype, structure, aggregation, stability, and source (generation, purification, and modification processes). These physico-chemical parameters also may be impacted by formulation and storage conditions.
Recommended Characterization Profile of Critical Reagents
During the early discovery and lead optimization phase of a therapeutic program, it is generally acceptable to apply a fit-for-purpose approach to assay sensitivity and specificity. When assays are validated for IND-enabling studies, the focus should be to maximize reagent quality and the reproducibility of method performance among different lots of critical reagents. As assays are transitioned into human matrices, a strategy should be defined to ensure the assay remains fit-for-purpose across the often lengthy clinical program and subsequent post-marketing phase. Once assay reagents are used in a clinical (and post-marketing) setting, further improvement in assay reproducibility may be needed to meet greater demands.
A basic characterization profile is recommended for each reagent type (Table III) and includes the assessment of concentration, binding activity, aggregation, purity level, and molecular weight. While a high level of purity of a protein reagent may not be needed at the early stages of drug development, some level of purification may be applied to reduce the impact of impurities, which can vary from lot-to-lot and may affect reagent stability during long-term storage. The purity of protein reagents is critical when they are used to coat a solid support or if they are to be conjugated to molecular labels (e.g., biotin, ruthenium chelate, or HRP). Therefore, once a protein reagent is purified, it is recommended that methods such as SDS-PAGE or SEC be employed to verify reagent purity and monodispersity prior to use in the assay. Aggregation levels are frequently measured by SEC though other methods such as DLS can also be used. IgG aggregates have been characterized further by recently developed methods using extrinsic fluorescent dyes (46,47). Orthogonal methods, such as SPR or bioassays, can also be very helpful to assure that the reagent is functioning as expected. However, it is important to note that reagent physico-chemical characterization results are not always predictive of functional properties, such as sensitivity and specificity, in the chosen assay format. In these cases, the functional assessment should be used as the final lot-release criteria for the critical reagent.
As drug development progresses, it may be helpful to express MAb reagents in a well-characterized stable mammalian cell line to assure long-term genetic stability and greater consistency across reagent lots. Optional characterization parameters to assure consistent, high-performing lots of critical reagents include: determining levels of protein A in a reagent that has been purified using a protein A column; bovine IgG levels from tissue culture expansion; and levels of residual HCP, in cases when this may contribute to interferences in LBAs.
Vendor laboratories may provide a baseline level of reagent characterization followed by further physico-chemical characterization and qualification performed at the requesting laboratory. Beyond the basic level of physico-chemical characterization of the reagent performed at the point of initial generation and first use in the LBA, more extensive characterization may not need to be repeated, particularly if the reagent is performing appropriately in the assay.
Five different examples of critical reagent characterization profiles are given in Electronic supplementary material S1, with the corresponding effect on LBA performance.
CRITICAL REAGENT MAINTENANCE
Formulation of Critical Reagents
Critical reagents are produced and stored as bulk lots in their most concentrated liquid form. Proteins may degrade via chemical (e.g., oxidation or deamidation) (48–50) or physical (e.g., aggregation or misfolding) means (51). High pH formulations can accelerate deamidation processes, and thus, neutral pH formulation buffers are recommended, unless data suggest otherwise. Aggregation is the most common form of protein alteration reported for reagents. Aggregate levels should be monitored, for example, by SECs described in Table III.
Formulation buffers used for storage of critical reagents may impact reagent stability. For example, during freezing and thawing, the local pH of PBS drops, which could impact the stability and function of the stored reagent in the LBA (29,52). Addition of preservatives (e.g., azide, KATHON™, Proclin® or thimersol) can slow microbial growth when reagents are stored under non-sterile conditions. However, the use of specific preservatives may not be allowed due to environmental hazards (e.g., mercurials). Stabilizers can be added (e.g., BSA, gelatin) to act as carrier proteins and minimize non-specific adsorption of reagents. Cryoprotectant additives (e.g., glycerol or sucrose) can be incorporated into formulation buffers to minimize effects of freeze/thaw. While all of these additives often have predefined shelf-lives, these dates cannot be used to estimate reagent expiration date. Furthermore, additives should be chosen with caution because some additives are incompatible with reagents and other assay components (e.g., sodium azide is not compatible with the use of HRP conjugates).
Assignment of Critical Reagent Expiration
The initial expiration or re-evaluation date of the critical reagent is given from the date of production, purification, or conjugation. Recommended examples of expiration dates for bulk and concentrated preparations of critical reagent types are shown in Table IV. These are recommended ranges of expiration dates and storage temperatures that are based on experience and organizations use in their best practices. Certainly, exceptions for each reagent type will occur that can be identified by monitoring the LBA performance and potentially re-evaluating the expiration date and/or storage temperature of each reagent. When bulk critical reagent lots are diluted to generate working stocks for the LBA, storage temperatures may remain the same whereas the expiration date may be set for an earlier date than that of the bulk critical reagent, following the organizational best practices. Expiration date extension for a critical reagent is possible by confirming the reagent still meets assay performance criteria with no observable trends predicting reagent failure and is monitored during the extension. There are various strategies employed to extend expiration dates; some laboratories use archived reagents that meet the established assay acceptance criteria as comparators; others use trending information of assay performance to justify continued use of a critical reagent; others prefer to apply statistical tools to experimental data in order to determine the appropriate expiration date. Regardless of the approach taken, expiration date re-assignment should be well documented and justified. Unexpected performance of an assay, independent of reagent expiration date, should prompt an investigation and possible re-evaluation of reagents.
Table IV.
Recommended Storage Temperatures and Expiration Dates for Each Reagent Type
| Reagent type | Storage temperature | Expiration of bulk critical reagent |
|---|---|---|
| Mabs | Purified, at −60°C to −100°C | 2–10 years from purification |
| PAbs | Serum, at −15°C to −30°C or −60°C to −100°C | 2−10 years from preparation |
| Purified, at −60°C to −100°C | 2–10 years from purification | |
| Engineered proteins | Purified, at −60°C to −100°C | 1−5 years from purification |
| Conjugates of Abs and biological proteins | Conjugates, at −60°C to −100°C | 2−5 years from preparation |
Critical Reagent Storage Considerations
Recommended storage temperatures for each reagent type are described in Table IV. Other storage considerations include: (a) preparation of single-use aliquots that can reduce the impact of freeze/thaw damage and potential for contamination and (b) the use of temperature alarm systems and an uninterrupted power supply with generator back-up for refrigerators and freezers, both invaluable for ensuring proper storage conditions of the reagents at all times.
Critical Reagent Stability
In most BA laboratories, acceptable performance of a LBA over time is indicative of a critical reagent’s functional stability. However, orthogonal basic or extended physico-chemical characterization(s) methods are essential to associate physical reagent parameter(s) with functional performance in the LBA, and the data can assist in the determination of causal relationships of reagent instability (53).
Qualification of LBA Critical Reagents Lots
Requirements for critical reagent qualification (and re-qualification) are dependent on the purpose of the LBA, the platform and technology employed in the assay, the molecular nature of the reagent, and the function of that reagent in the assay. As lots of critical reagents are depleted, or as they approach a pre-determined re-qualification date, it is essential to demonstrate that critical reagents continue to perform appropriately in the assay. The performance of the LBA, once well-established, serves as the primary baseline for critical reagent qualification. The new lot must meet a clear set of assay performance criteria that are established a priori, well documented, and scientifically driven. A noteworthy risk to point out is of “assay drift”, where a consistent bias could result in a very different assay mean being established over time (54). It is therefore important to adhere to the original assay acceptance criteria, which may mitigate this risk.
The level of evaluation of new reagent lots is also dependent on the extent of the change to generate the new lot. Examples of minor and major changes to critical reagents are given in Table V with suggested approaches for evaluation that are related to the magnitude of change.
Table V.
Examples of Minor and Major Changes to Critical Reagents Affecting LBA Performance and Suggested Evaluations
| Changes affecting LBA performance |
|---|
| Major change |
| New cell line and/or new reagent generation |
| Re-characterize the reagent |
| For calibrators, check alignment to reference standard |
| Reassign value if necessary to maintain calibration |
| Evaluate LBA performance |
| Re-optimize in LBA if function is not comparable to prior lot in LBA; revalidation for a GLP study is necessary |
| New reagent conjugation |
| Check molar incorporation ratio |
| Measure relative affinity for ligand compared with unlabeled molecule |
| Assess LBA performance |
| Re-optimize in LBA if needed |
| Minor change |
| New reagent purification |
| Measure concentration |
| Check purity |
| Assess LBA performance |
| New working lot of QCs from reagent stock |
| Measure concentration |
| Reassign nominal concentration value if necessary to maintain calibration |
| Assess LBA performance |
Long-Term Supply of Critical Reagents
The life span of an LBA is generally dependent on the stage of the program, ranging typically from 6 weeks to 6 months in the preclinical stage, 6 months to 5 years during the clinical development stage, and possibly as long as 20+ years to support post-marketing commitments and drug manufacturing. During the time span of drug development and study support, it is possible that the LBA may be transferred to different laboratories within the same organization, externally, domestic, or internationally. Thus, planning for and ensuring a continuous supply of critical reagents for multiple users and over the lifespan of the assay is a challenging endeavor. Critical reagents are often prepared, inventoried, and managed in large lots. Inventory management for long-term supply is greatly aided by the use of forecasting tools, including end-user surveys, processes for monitoring usage, or inventory queue systems where re-order is triggered when a pre-set threshold quantity is reached. All of these reagent management tools should factor in lead time for process steps, such as re-immunizations (for PAbs), expression, purification, and conjugation as well as reagent qualifications.
An additional challenge to maintaining a long-term reagent supply is the increased use of portfolio-specific processes for method development, which results in the same reagent used between several LBA. Examples are: the use of PAb critical reagents for HCP LBAs supporting multiple drug development programs; the common capture or detector reagent used in multiple LBAs for PK or Biomarker support; and the positive control antibody used in ADA and NAb immunogenicity assays. In these situations, a best practice is to identify users and continuously assess their usage needs, adjusting reagents production accordingly.
LIGAND BINDING ASSAY MAINTENANCE
A variety of changes in critical reagent sources and LBA controls are likely to occur throughout the life span of a LBA. Over time, unexpected challenges may be encountered that could necessitate LBA re-validation with new reagent(s), such as a commercially available reagent being discontinued, accelerated reagent usage, unforeseen catastrophic loss of a reagent, or LBA performance shifts due to an unpredicted reagent stability failure. The types of assay maintenance challenges should be anticipated so that risks can be mitigated through appropriate planning and resource management. Common practices used to track assay performance and how this information may be used to monitor the performance of reagents in the method are described here.
Ligand Binding Assay Performance Tracking and Trending
Tracking and trending LBA performance in an analytical laboratory is essential for ensuring that data are comparable between runs over time. Regulatory guidance (55) and industry white papers (7,10) recommend inclusion of LBA QC samples in analytical runs to ensure the reliability of data generated.
The use of an appropriately designed and routinely monitored quality control program accelerates the detection of trends in the data that may include, among other factors, a systematic bias between different reagent lot preparations, evidence of reagent degradation, and equipment failures, which will lead to the need for an investigation. LBA trending data may also be used to support the extension of critical reagent expiration date as long as enough reagent stability-indicating information exists to draw a relevant conclusion.
The most basic method to monitor assay performance is to plot QCs’ and the assay’s background signal data over time. If employed, this approach may provide additional data and information to that derived from the use of the QC samples accuracy data alone. This approach can be applied to quantitative methods such as those designed for PK, biomarker, or HCP measurements, for quasi-quantitative assays, such as those used for immunogenicity assessments, as well as for qualitative assays (9,10). Monitoring reagent performance can be based on the use of a binary outcome of the positive and negative controls (qualitative assays only) for run acceptance (56).
There are several software packages that can be utilized for tracking and trending analyses. These include Microsoft Excel, SAS-JMP, Westgard QC Validator, and various LIMS applications. Limits are predefined using the QC rules that are appropriate for the assay type (7,10) and should be identified on the graphical charts.
Ideally, a QC chart (or another form of a bioanalytical quality summary) may be included in a final data package for completed analyses supporting a given study. Whether sample testing is conducted by a laboratory internal or external to the organization, specifications for the number and types of failures during sample testing that trigger notification by the testing laboratory should be set early on (e.g., QC chart parameters, number of consecutive run failures, etc.).
CONSIDERATIONS FOR USING COMMERCIAL LIGAND BINDING ASSAY KITS
Commercial LBA kits provide the convenience of purchasing a set of reagents that have been paired in a LBA format, tested to produce reproducible results in specified matrices, and contain consistent and readily available supply of critical reagents. The use of bioanalytical kits from commercial sources requires selection, planning, and anticipation of study timelines. Commercial LBA kits may range from research grade to in vitro diagnostic kits (57,58); the latter are provided with quality assurance, though for a limited context which may not be applicable to other testing conditions. The level of qualification of the LBA reagent and kit performed by the sponsor may vary. Furthermore, the proprietary nature of some kit reagents makes assessment and management of assay performance parameters an empirical exercise.
Commercially produced kits may be susceptible to vendor supply problems or kit discontinuation, which are magnified if the supplier is the sole producer (59). Common considerations to mitigate the risks associated with use of commercial LBA kits are: (a) assessing in advance the ability of manufacturers to ensure a consistent supply of the desired reagents; (b) ideally, identifying a comparable kit (if available) from a secondary manufacturer; (c) assessing multiple lots of kits from vendor(s) to ensure consistent results over a long period of time; (d) if possible, securing a large purchase of a single kit lot to support the length of a study prior to its start; (e) negotiating customization of individual reagents, e.g., dilution buffer, detection reagents for purchase; and (f) developing a plan with the vendor to allow early testing of new kit lots.
Changes in kit lots may not include a corresponding change in reagent lots. Although measures are usually taken to assure consistent kit quality and performance by the vendor, the nature of the changes from one kit lot to another is not always transparent to the consumer. Vendors should provide documentation of changes in reagent lot specifications or processes on the kit inserts. Vendors should have a quality notification process to rapidly notify customers of defects (e.g., instability of components, failure to meet specifications) in kits or additional information generated regarding kit performance over time. Ideally, this information should be available electronically and/or a recall notification should be sent electronically.
CRITCAL REAGENT INVENTORY AND KNOWLEDGE DATABASE
A well-managed inventory and knowledge database can greatly facilitate the supply and monitoring of critical reagents, assuring assay consistency throughout the life cycle of a therapeutic program (60). While a reagent inventory system is used to capture critical reagent availability as well as tracking usage (or “burn rates”), a knowledge database is used to provide a reference tool to guide its use and application. It is not crucial that they be integrated since they are related but are used for different purposes. The critical reagent inventory describes the source of the critical reagent, the quantity, and the physical location of the reagent. It may also capture the shipment and receipt of reagents which can be used to gauge reagent supply and demand, usage trends, predictions, and the identity of high use requestors. In contrast, the knowledge database is viewed as a repository of disparate information, somewhat analogous to a catalog where bioanalytical users can identify if a suitable type of critical reagent may be available and useful for a particular assay.
Depending on the business needs, accessibility to the inventory and/or knowledge database systems can be limited to a single group or encompass multiple departments, but does not have to serve as a raw data source, and therefore validation of the management tool is not required.
Critical Reagent Inventory
The variety of inventories used to manage critical reagents range from manual paper processes to sophisticated searchable electronic databases that can be used remotely, can cross-reference information, and send E-mail communications. Examples of commercial inventory systems include: Freezerworks (Lynnwood, MA), Bellhawk® (Millbury, MA), Mosaic from Titian (London, UK), and other laboratory information management system (LIMS). These inventory systems provide the ability to manage long-term storage of critical reagents in aliquots and/or bulk amounts. Key attributes for an inventory system include: ease of querying and retrieving information (and the reagent), as well as automated trigger points to request a resupply of a critical reagent or a re-issue of documentation. Restricted access to critical reagents in the inventory (but not necessarily the knowledge database) may be necessary when reagents are either rare (i.e., difficult to replace), have been dedicated to a study, or are yet untested, before they are catalogued and available for request. Some of the more sophisticated inventory systems incorporate information about the requestor and bioanalytical laboratory, with the advantage of enabling follow-up, if new information becomes available for a particular critical reagent.
The naming convention of critical reagents in the inventory can be challenging because of the large number of gene and protein synonyms, variant spellings, idiosyncratic names, and species differences (61). However, consistency in reagent naming conventions (either by electronic drop down boxes or internal codes) is crucial for the effective utility of any inventory system.
An inventory system may be physically located and managed from a single area (centralized) or located in multiple laboratories and managed locally (dispersed). A centralized inventory has the advantage of clear responsibilities and associated resources, whereby all reagent generation groups and end-users have a single point of contact. Depending on the size and need of the organization, consideration of a dispersed inventory system may be appropriate. The responsibility for marshalling a resupply order can reside with the end-user, laboratories manager, or a specialized critical reagents operations individual or coordinator.
Critical Reagent Knowledge Database
The purpose of the knowledge database is to consolidate, into a unified location, scientific information on a single lot or multiple lots of a critical reagent that is often widely dispersed or known only by a limited number of individuals. Types of knowledge database systems range from laboratory notebooks to multi-functional LIMS systems with EXCEL spreadsheets or Access databases serving as alternate intermediary approaches. The knowledge database should minimally include: a characterization summary document, descriptions of methods of generation of the critical reagent, ideally contain, or have links to reagent qualification data as well as historical assay performance (trending) information, although these data sets may be in different spaces (Table VI).
Table VI.
Desirable Requirements for Critical Reagent Inventory and Knowledge Base Systems
| Critical reagent inventory | Critical reagent knowledge database | |
|---|---|---|
| What it contains | Information on physical location of reagent and resupply | Information on attributes of the reagent |
| Reagent identification | Name and identification of critical reagent, lot number, request contact information | Name and identification of critical reagent, lot number, request contact information |
| Desirable requirements | Operations | Attributes |
| • Storage of bulk and individual materials | Characterization summary data and documentation | |
| • Ability to segregate, quarantine, and restrict physical access to reagent materials | • Source, i.e., polyclonal serum, cell line, purified material | |
| Storage | • Purification method (or cross-referenced) | |
| • Storage temperature | • Modification method (or cross-referenced, e.g., fragmentation, conjugation to other molecules) | |
| • Location | Assay performance | |
| • Quantity | • Screening and back-up reagent information | |
| • Concentration or activity units | • Bioanalytical assay type (report or SOP number) | |
| • Preparation and expiration dates | • Reagent qualification data (reports or certificates) | |
| Commercial source | • Historical performance and trending information | |
| • Receipt information from vendor | • Project and study cross-references | |
| • Equivalent reagent if need to switch vendor source reagent | ||
| Request | ||
| • Requests for material | ||
| • Requestor name and lab, date, and amount | ||
| Resupply | ||
| • Triggering of re-supply requests based on remaining inventory, expiration dates, project-related needs, or lead time to re-provision |
The source lots of the critical reagent should also be catalogued, to ensure traceability. Such reagent sources include the polyclonal antibody serum that the critical reagent is purified from, or a purified protein source that is then conjugated to become the critical reagent.
The reagent knowledge database represents a continuum of data collated over the development and in-study phases of the reagent’s lifespan. It begins with formal documentation that captures the unique generation methodology of each critical reagent. For antibody reagents, this information should include details for the immunogen and immunization procedure, as well as purification and formulation conditions and, for engineered proteins, the expression system, cell line, culture, harvest, purification, and formulation conditions. As scientific understanding of the reagent increases, relevant physico-chemical characterizations of each production lot and LBA results from reagent qualification experiments should be documented in the database and serve as a foundation for creation of the lot-specific batch record. The knowledge database should be updated as new production or characterization information becomes available, as such documentation serves as a benchmark for desired attributes of new lots. External suppliers of reagents usually provide documentation of reagent characterization data which should be included in the knowledge database. However, it may be important to request additional data from the supplier or to conduct additional experiments if outstanding questions about reagent attributes exist.
An accessible knowledge database provides improved efficiency in accessing the reagent information, reducing the likelihood of using valuable resources for needless experiments and duplicate reagent generation efforts. Without it, data-driven decision processes could be compromised. While it is recognized that the constraints on resources vary in bioanalytical laboratories and are based on business needs, it is highly recommended to invest resources in a knowledge database system and assign appropriate responsibilities for managing the quality and consistency of entries.
SUMMARY
The immense resources and costs associated with drug development and the integrated use of LBAs to support studies and decision making during all its phases places LBA critical reagents as a high-impact asset of the organization. At the Twenty-First Century Bioanalytical Laboratory Workshop 2009, a parallel analogy was made to the phrase: “to enjoy world class wine—start with world class grapes”, where critical reagents (the grapes) are the source for developing a world class and successful LBA (the wine).
Historically, more attention has been given to these assets when issues related to critical reagent stability, depletion, changes in production processes, or unforeseen catastrophes arise. Even in some organizations that have expertise in protein expression and purification, resources are not allocated for applying this expertise to supply of critical reagents, due to the complexities of critical reagent’s life cycle management. Critical reagents have often been a proverbial “hot potato”, where responsibility for critical reagent generation and management are ill-defined.
It is the bioanalytical laboratory that remains invariably dependent on quality and quantity of critical reagents as tools to develop and maintain LBA. We therefore recommend investing attention and resources into the critical reagent work stream which will benefit the organization by saving time during vital studies, whether early in discovery or conducting post-marketing commitments. Paying attention to the characteristics of a critical reagent by lot secures and ensures reproducibility and performance of the LBA. Therefore, appropriate resources (personnel, equipment, and expertise) should be allocated to the generation and selection of critical ligand binding reagents. Once reagents have been selected and their use in a LBA has been initiated, the importance of the knowledge database systems increases considerably.
In the ultimate Twenty-First Century BA laboratory: critical reagents would be generated in shorter time periods and characterization data files could be located and viewed electronically; an integrated critical reagent inventory system would track use and automate resupply requests and stock refills; integrating the in-house critical reagent knowledge database system with information on performance of the LBA would assist global compliance and harmonization efforts with health authorities and regulatory agencies; the LBA community could be served by a global reagent knowledge database system cataloguing information on the performance of a critical reagent in a LBA and thus minimizing duplicative efforts in selecting commercial reagents; industry consortium would enable access to a common target reagent or platform critical reagents; harmonized critical reagent characterization documents would simplify locating information and identifying when data is missing; and global shipping documents for reagents would allow seamless international shipping activities. While this would be an idyllic scenario, there is still a lot of work to be done. However, these areas should be pursued to integrate and leverage parallel technological advances as they become available, locally, regionally, and globally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
{kind=link}
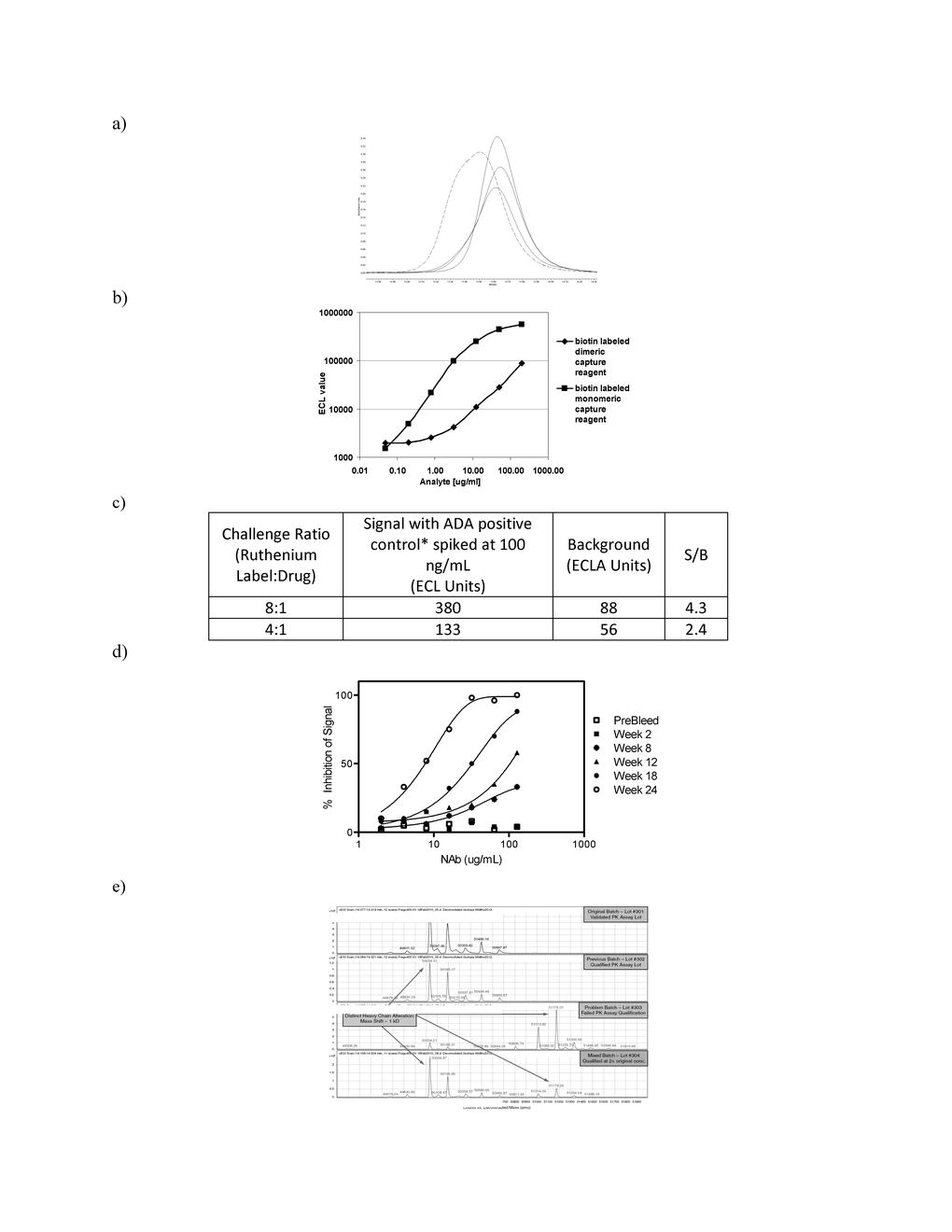
Examples of reagent characterizations and assay performance. a A large quantity of a critical reagent was requested from a vendor who had previously supplied it using a soluble expression system in CHO cells. To supply material for the large request, the vendor changed the expression system to Escherichia coli, harvested inclusion bodies, and refolded the protein reagent. Performance of the E. coli expressed protein in an established LBA showed similar maximum and minimum signal responses compared with the earlier preparation. However, three times the amount of this protein was required to coat the plate and the IC50 of the ELISA changed by twofold relative to the LBA, using the reagent from the mammalian secreted process. Characterization of the reagents, derived from the different expression systems, by SEC showed that the reagent from the E. coli process eluted earlier and had an apparent higher molecular weight than the corresponding reagent from the CHO-derived process. Expressing, harvesting, and refolding protein reagents from bacterial inclusion bodies may have resulted in misfolded and/or aggregated material that was not present in the reagent lot from the soluble mammalian expression system. Qualification of the new reagent lot relative to the soluble mammalian expressed reagent demonstrated differences in performance of the LBA, and the SEC profile of the reagent provided a means to screen new lots of this critical reagent. b The biophysical state of an LBA capture reagent can affect assay performance. The example here is of a recombinant protein that naturally exists in a dimeric state. The protein was purified as monomeric and dimeric proteins and subsequently conjugated with biotin. Under identical conditions (10% mouse plasma) and concentrations, these capture reagents were coated onto streptavidin plates to compare their ability to capture a biotherapeutic recognizing the target protein. While the assay background remained the same, the signal/noise ratio was higher, by at least tenfold with the monomeric capture reagent than the dimeric capture reagent. c Asterisk: sheep polyclonal anti-human IgG antibody spiked into cynomolgus pooled serum. A bridging assay format was used, in which assay signal derived from ADA molecules that bridged two drug molecules, one drug molecule labeled with ruthenium, the other with biotin. ECL units were measured using a commercial chemiluminescence analyzer. A therapeutic MAb was conjugated with a ruthenium-label at different molar challenge ratios. The effect of different molar challenge ratios is evident in the LBA signal, which corresponds to electrochemiluminescence (ECL) units measured in an immunogenicity assay. The ADA specific for the therapeutic MAb was detected using a bridging assay format where the assay signal is obtained from molecules that bridged two labeled drug molecules (one labeled with ruthenium, the other labeled with biotin). The assay background and the positive-sample assay signal increased with increasing challenge ratios. The signal-to-background (S/B) ratio also increased with higher molar-ratio challenges, indicating the potential for improved method sensitivity with greater ratios of ruthenium to protein. d To develop a NAb assay, a positive control (PC) with neutralizing activity is required as a critical reagent. Several animals may be initially immunized initially to generate a PAb PC, and it is essential to gather information on the sample time points (bleeds) for neutralizing activity. In this assay, a dramatic decrease in IC50 in the LBA is observed as the neutralizing activity of PAbs matures with time a dramatic decrease in IC50 in the LBA is observed. This demonstrates that bleeds collected at different times from the same animal are not always equivalent in reactivity. It is important to maximize the amount of each lot of PAb reagent, perhaps by pooling several bleeds with appropriate activity. As part of a strategic plan to maintain the assay and to assure appropriate controls, subsequent lots may require a partial validation to determine new assay acceptance criteria for that new assay reagent. e Four lots of reagent Abs were produced and purified on separate occasions from the same hybridoma cell bank. The purification procedures for all lots were similar, and no differences between lots were observed when the Abs were evaluated by SEC or by reducing and non-reducing SDS-PAGE. The purified reagent Abs were then conjugated with ruthenium for use as a detection Ab in a PK assay. A conjugated MAb from Lot #301 was initially validated at a working concentration of 1.25 μg/mL in the PK assay, and attempts were made to qualify the subsequent lots at the same concentration. A conjugated antibody made from Lot #302 was qualified in the assay at an identical concentration as Lot #301. However, a conjugate made from Lot #303 could not be qualified in the assay and a conjugate made from Lot #304 could only be qualified in the assay at twice the concentration (2.5 μg/mL) of earlier lots. The source lots of these conjugated reagent antibodies were then evaluated for intact and reduced forms by LC-MS (reversed-phase HPLC coupled to time-of-flight mass spectrometry). The LC-MS experiments revealed that Lot # 301 and 302 had nearly identical spectra for their heavy chains, while a set of larger (∼1 kD shift) peaks were in the spectra from the heavy chains of Lots 303 and 304. These experiments indicate that as the relative signal of this higher mass species (when compared with the signal of the original mass) increased, the assay performance also decreased. Therefore, the LC-MS method could be used to prescreen subsequent lots for use in the assay (JPEG 70 kb)
ACKNOWLEDGMENTS
We extend our thanks to all the reviewers of this manuscript, in particular, Jean Lee, Valerie Quarmby, Marian Kelley, all departmental reviewers, and the Ligand Binding Assay Bioanalytical Focus Group (LBABFG) of the American Association of Pharmaceutical Scientists (AAPS) steering committee for their critical review of this manuscript and helpful comments. We also thank Terri Caiazzo (Pfizer), Rosemary Lawrence-Henderson (Pfizer), Brian J. Geist (Janssen R&D, LLC), Michele Frigo (Janssen R&D, LLC), Tong-Yuan Yang (Janssen R&D, LLC), and Yanhong Li (Genentech/Roche) for providing data shown in the Electronic supplementary material S1. We acknowledge the AAPS Twenty-First Century Bioanalytical Laboratory Workshop: planning committee members Jean Lee (co-chair), Valerie Quarmby (co-chair), Ago Ahene, Marian Kelley, Sheldon Leung, Chad Ray, Huifen Faye Wang, Melvin Weinswig; the programming committee (Jean Lee and Valerie Quarmby); and the steering committee (Ago Ahene, Chad Ray and Sheldon Leung). This manuscript was prepared by members of the Twenty-First Century Bioanalytical Laboratory: Reagent Subcommittee of the LBABFG of AAPS.
ABBREVIATIONS
- Ab(s)
Antibody(ies)
- ADA
Anti-drug antibody
- BA
Bioanalytical
- BSA
Bovine serum albumin
- CRO
Contract research organization
- DLS
Dynamic light scattering
- ECL
Electro-chemiluminescence
- GLP
Good laboratory practices (21CFR part 58)
- HCP
Host cell protein
- HPLC
High-performance liquid chromatography
- HRP
Horseradish peroxidase
- IgG
Immunoglobulin G
- IND
Investigational new drug
- KLH
Keyhole limpet hemocyanin
- LBA(s)
Ligand binding assay(s)
- LC
Liquid chromatography
- LIMS
Laboratory information management system
- MAb(s)
Monoclonal antibody(ies)
- MS
Mass spectrometry
- NAb(s)
Neutralizing antibody(ies)
- PAb(s)
Polyclonal antibody(ies)
- PC
Positive control
- PK
Pharmacokinetics
- QC(s)
Quality control(s)
- S/B
Signal-to-background ratio
- SDS-PAGE
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SEC
Size exclusion chromatography
- SOP
Standard operating procedure
- SPR
Surface plasmon resonance
REFERENCES
- 1.Nowatzke W, Woolf E. Best practices during bioanalytical method validation for the characterization of assay reagents and the evaluation of analyte stability in assay standards, quality controls, and study samples. AAPS J. 2007;9(2):E117-E122. [DOI] [PMC free article] [PubMed]
- 2.Cawkill D, Eaglestone SS. Evolution of cell-based reagent provision. Drug Discov Today. 2007;12(19–20):820–825. doi: 10.1016/j.drudis.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 3.Mairal T, Cengiz Özalp V, Lozano Sánchez P, Mir M, Katakis I, O’Sullivan CK. Aptamers: molecular tools for analytical applications. Anal Bioanal Chem. 2008;390(4):989–1007. doi: 10.1007/s00216-007-1346-4. [DOI] [PubMed] [Google Scholar]
- 4.Tremblay GA, Oldfield PR. Bioanalysis of siRNA and oligonucleotide therapeutics in biological fluids and tissues. Bioanalysis. 2009;1(3):595–609. doi: 10.4155/bio.09.66. [DOI] [PubMed] [Google Scholar]
- 5.Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23(2):312–328. doi: 10.1007/s11095-005-9045-3. [DOI] [PubMed] [Google Scholar]
- 6.Lee JW, Weiner RS, Sailstad JM, Bowsher RR, Knuth DW, O’Brien PJ, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm Res. 2005;22(4):499–511. doi: 10.1007/s11095-005-2495-9. [DOI] [PubMed] [Google Scholar]
- 7.Desilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res. 2003;20(11):1885–1900. doi: 10.1023/B:PHAM.0000003390.51761.3d. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S, Indelicato SR, Jethwa V, Kawabata T, Kelley M, Mire-Sluis AR, et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321(1–2):1–18. doi: 10.1016/j.jim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 9.Mire-Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289(1–2):1–16. doi: 10.1016/j.jim.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–1281. doi: 10.1016/j.jpba.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 11.O’Hara DM, Xu Y, Liang Z, Reddy MP, Wu DY, Litwin V. Recommendations for the validation of flow cytometric testing during drug development: II assays. J Immunol Methods. 2011;363(2):120–134. doi: 10.1016/j.jim.2010.09.036. [DOI] [PubMed] [Google Scholar]
- 12.Good Laboratory Practice for Nonclinical Laboratory Studies, Title 21 CFR Part 58.83 (2007).
- 13.Rup B, O’Hara D. Critical ligand binding reagent preparation/selection: when specificity depends on reagents. AAPS J. 2007;9(2):E148-E155. [DOI] [PMC free article] [PubMed]
- 14.Staack RF, Stracke JO, Stubenrauch K, Vogel R, Schleypen J, Papadimitriou A. Quality requirements for critical assay reagents used in bioanalysis of therapeutic proteins: what bioanalysts should know about their reagents. Bioanalysis. 2011;3(5):523–534. doi: 10.4155/bio.11.16. [DOI] [PubMed] [Google Scholar]
- 15.Bjerner J, Børmer OP, Nustad K. The war on heterophilic antibody interference. Clin Chem. 2005;51(1):9–11. doi: 10.1373/clinchem.2004.042994. [DOI] [PubMed] [Google Scholar]
- 16.Ismail AAA. Interference from endogenous antibodies in automated immunoassays: what laboratorians need to know. J Clin Pathol. 2009;62(8):673–678. doi: 10.1136/jcp.2008.055848. [DOI] [PubMed] [Google Scholar]
- 17.Levinson SS, Miller JJ. Towards a better understanding of heterophile (and the like) antibody interference with modern immunoassays. Clinica Chimica Acta. 2002;325(1–2):1–15. doi: 10.1016/S0009-8981(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 18.Altinier S, Varagnolo M, Zaninotto M, Boccagni P, Plebani M. Heterophilic antibody interference in a non-endogenous molecule assay: an apparent elevation in the tacrolimus concentration. Clinica Chimica Acta. 2009;402(1–2):193–195. doi: 10.1016/j.cca.2008.12.021. [DOI] [PubMed] [Google Scholar]
- 19.Kahn MN, Findlay JWA, editors. Ligand binding assays: development, validation and implementation in the drug development arena. Hoboken: John Wiley & Sons; 2010. [Google Scholar]
- 20.DeForge LE, Loyet KM, Delarosa D, Chinn J, Zamanian F, Chuntharapai A, et al. Evaluation of heterophilic antibody blocking agents in reducing false positive interference in immunoassays for IL-17AA, IL-17FF, and IL-17AF. J Immunol Methods. 2010;362(1–2):70–81. doi: 10.1016/j.jim.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 21.Preissner CM, Dodge LA, O’Kane DJ, Singh RJ, Grebe SKG. Prevalence of heterophilic antibody interference in eight automated tumor marker immunoassays. Clin Chem. 2005;51(1):208–210. doi: 10.1373/clinchem.2004.040501. [DOI] [PubMed] [Google Scholar]
- 22.Spencer DV, Nolte FS, Zhu Y. Heterophilic antibody interference causing false-positive rapid human immunodeficiency virus antibody testing. Clinica Chimica Acta. 2009;399(1–2):121–122. doi: 10.1016/j.cca.2008.09.030. [DOI] [PubMed] [Google Scholar]
- 23.Hennig C, Rink L, Fagin U, Jabs WJ, Kirchner H. The influence of naturally occurring heterophilic anti-immunoglobulin antibodies on direct measurement of serum proteins using sandwich ELISAs. J Immunol Methods. 2000;235(1–2):71–80. doi: 10.1016/S0022-1759(99)00206-9. [DOI] [PubMed] [Google Scholar]
- 24.Levinson SS. Antibody multispecificity in immunoassay interference. Clin Biochem. 1992;25(2):77–87. doi: 10.1016/0009-9120(92)80048-L. [DOI] [PubMed] [Google Scholar]
- 25.Tatarewicz S, Miller JM, Swanson SJ, Moxness MS. Rheumatoid factor interference in immunogenicity assays for human monoclonal antibody therapeutics. J Immunol Methods. 2010;357(1–2):10–16. doi: 10.1016/j.jim.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Lee JW, Kelley M, King LE, Yang J, Salimi-Moosavi H, Tang MT, et al. Bioanalytical approaches to quantify “total” and “free” therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 2011;13(1):99–110. doi: 10.1208/s12248-011-9251-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patton A, Mullenix MC, Swanson SJ, Koren E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J Immunol Methods. 2005;304(1–2):189–195. doi: 10.1016/j.jim.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Wild DG, editor. The immunoassay handbook. 3. Kidlington: Elsevier Science; 2005. [Google Scholar]
- 29.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 30.Milstein C. With the benefit of hindsight. Immunol Today. 2000;21(8):359–364. doi: 10.1016/S0167-5699(00)01660-1. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian G, editor. Antibodies: volume 2: novel technologies and therapeutic use. New York, NY: Kluwer Academic/Plenum Publishers; 2004. [Google Scholar]
- 32.Spieker-Polet H, Sethupathi P, Yam PC, Knight KL. Rabbit monoclonal antibodies: generating a fusion partner to produce rabbit–rabbit hybridomas. Proc Natl Acad Sci U S A. 1995;92(20):9348–9352. doi: 10.1073/pnas.92.20.9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradbury ARM, Marks JD. Antibodies from phage antibody libraries. J Immunol Methods. 2004;290(1–2):29–49. doi: 10.1016/j.jim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 34.Lipovsek D, Plückthun A. In-vitro protein evolution by ribosome display and mRNA display. J Immunol Methods. 2004;290(1–2):51–67. doi: 10.1016/j.jim.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 35.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 36.Cohen AD, Boyer JD, Weiner DB. Modulating the immune response to genetic immunization. FASEB J. 1998;12(15):1611–1626. [PubMed] [Google Scholar]
- 37.Sundaram P, Xiao W, Brandsma JL. Particle-mediated delivery of recombinant expression vectors to rabbit skin induces high-titered polyclonal antisera (and circumvents purification of a protein immunogen) Nucleic Acids Res. 1996;24(7):1375–1377. doi: 10.1093/nar/24.7.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang D, DeVit M, Johnston SA. Genetic immunization is a simple method for eliciting an immune response. Nature. 1992;356(6365):152–154. doi: 10.1038/356152a0. [DOI] [PubMed] [Google Scholar]
- 39.Witkowski PT, Bourquain DR, Hohn O, Schade R, Nitsche A. Gene gun-supported DNA immunisation of chicken for straightforward production of poxvirus-specific IgY antibodies. J Immunol Methods. 2009;341(1–2):146–153. doi: 10.1016/j.jim.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Englebienne P. Immune and receptor assays in theory and practice. Boca Raton: CRC Press; 2000. [Google Scholar]
- 41.Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and pitfalls in the PEGylation of therapeutic proteins. Curr Opin Drug Discov Dev. 2008;11(2):242–250. [PubMed] [Google Scholar]
- 42.Su YC, Chen BM, Chuang KH, Cheng TL, Roffler SR. Sensitive quantification of PEGylated compounds by second-generation anti-poly(ethylene glycol) monoclonal antibodies. Bioconjug Chem. 2010;21(7):1264–1270. doi: 10.1021/bc100067t. [DOI] [PubMed] [Google Scholar]
- 43.Babu S. Criteria for CMO selection. Am Pharm Outsourcing. 2009;10(5):10–15. [Google Scholar]
- 44.Dellva P. Managing outsourcing relationships: what to do? What not to do? Am Pharm Outsourcing. 2003;4(4):30–33. [Google Scholar]
- 45.Bock I, Dhayalan A, Kudithipudi S, Brandt O, Rathert P, Jeltsch A. Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics. 2011;6(2):256–263. doi: 10.4161/epi.6.2.13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm Res. 2008;25(7):1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawe A, Friess W, Sutter M, Jiskoot W. Online fluorescent dye detection method for the characterization of immunoglobulin G aggregation by size exclusion chromatography and asymmetrical flow field flow fractionation. Anal Biochem. 2008;378(2):115–122. doi: 10.1016/j.ab.2008.03.050. [DOI] [PubMed] [Google Scholar]
- 48.Cleland JL, Powell MF, Shire SJ. The development of stable protein formulations: a close look at protein aggregation, deamidation, and oxidation. Crit Rev Ther Drug Carrier Syst. 1993;10(4):307–377. [PubMed] [Google Scholar]
- 49.Tsai PK, Bruner MW, Irwin JI, Ip CCY, Oliver CN, Nelson RW, et al. Origin of the isoelectric heterogeneity of monoclonal immunoglobulin h1B4. Pharm Res. 1993;10(11):1580–4586. doi: 10.1023/A:1018912417607. [DOI] [PubMed] [Google Scholar]
- 50.Liu H, Caza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci. 2008;97(7):2426–2447. doi: 10.1002/jps.21180. [DOI] [PubMed] [Google Scholar]
- 51.Manningc M, Patel K, Borchardt RT. Stability of protein pharmaceuticals. Pharm Res. 1989;6(11):903–918. doi: 10.1023/A:1015929109894. [DOI] [PubMed] [Google Scholar]
- 52.Kueltzo LA, Wang W, Randolph TW, Carpenter JF. Effects of solution conditions, processing parameters, and container materials on aggregation of a monoclonal antibody during freeze-thawing. J Pharm Sci. 2008;97(5):1801–1812. doi: 10.1002/jps.21110. [DOI] [PubMed] [Google Scholar]
- 53.Jiskoot W, Beuvery EC, De Koning AAM, Herron JN, Crommelin DJA. Analytical approaches to the study of monoclonal antibody stability. Pharm Res. 1990;7(12):1234–1241. doi: 10.1023/A:1015925519154. [DOI] [PubMed] [Google Scholar]
- 54.Ritter N, Wiebe M. Validating critical reagents used in CGMP analytical testing: Eensuring method integrity and reliable assay performance. BioPharm. 2001;14(5):12–21. [Google Scholar]
- 55.ICH. Q2b. Guidance for industry: validation of analytical procedures methodology 2001.
- 56.Simonet BM. Quality control in qualitative analysis. TrAC-Trends Anal Chem. 2005;24(6):525–531. doi: 10.1016/j.trac.2005.03.011. [DOI] [Google Scholar]
- 57.Bowsher RR, Sailstad JM. Insights in the application of research-grade diagnostic kits for biomarker assessments in support of clinical drug development: bioanalysis of circulating concentrations of soluble receptor activator of nuclear factor κB ligand. J Pharm Biomed Anal. 2008;48(5):1282–1289. doi: 10.1016/j.jpba.2008.09.026. [DOI] [PubMed] [Google Scholar]
- 58.Nowatzke W. Systematic analytical validation of commercial kits for the determination of novel biomarkers for clinical drug development. Bioanalysis. 2010;2(2):237–247. doi: 10.4155/bio.09.191. [DOI] [PubMed] [Google Scholar]
- 59.Crowther JR. The ELISA guidebook. 2. New York: Humana Press; 2010. [Google Scholar]
- 60.Ezzelle J, Rodriguez-Chavez IR, Darden JM, Stirewalt M, Kunwar N, Hitchcock R, et al. Guidelines on good clinical laboratory practice: bridging operations between research and clinical research laboratories. J Pharm Biomed Anal. 2008;46(1):18–29. doi: 10.1016/j.jpba.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruttenberg A, Clark T, Bug W, Samwald M, Bodenreider O, Chen H, et al. Advancing translational research with the Semantic Web. BMC Bioinformatics. 2007;8(SUPPL. 3). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Examples of reagent characterizations and assay performance. a A large quantity of a critical reagent was requested from a vendor who had previously supplied it using a soluble expression system in CHO cells. To supply material for the large request, the vendor changed the expression system to Escherichia coli, harvested inclusion bodies, and refolded the protein reagent. Performance of the E. coli expressed protein in an established LBA showed similar maximum and minimum signal responses compared with the earlier preparation. However, three times the amount of this protein was required to coat the plate and the IC50 of the ELISA changed by twofold relative to the LBA, using the reagent from the mammalian secreted process. Characterization of the reagents, derived from the different expression systems, by SEC showed that the reagent from the E. coli process eluted earlier and had an apparent higher molecular weight than the corresponding reagent from the CHO-derived process. Expressing, harvesting, and refolding protein reagents from bacterial inclusion bodies may have resulted in misfolded and/or aggregated material that was not present in the reagent lot from the soluble mammalian expression system. Qualification of the new reagent lot relative to the soluble mammalian expressed reagent demonstrated differences in performance of the LBA, and the SEC profile of the reagent provided a means to screen new lots of this critical reagent. b The biophysical state of an LBA capture reagent can affect assay performance. The example here is of a recombinant protein that naturally exists in a dimeric state. The protein was purified as monomeric and dimeric proteins and subsequently conjugated with biotin. Under identical conditions (10% mouse plasma) and concentrations, these capture reagents were coated onto streptavidin plates to compare their ability to capture a biotherapeutic recognizing the target protein. While the assay background remained the same, the signal/noise ratio was higher, by at least tenfold with the monomeric capture reagent than the dimeric capture reagent. c Asterisk: sheep polyclonal anti-human IgG antibody spiked into cynomolgus pooled serum. A bridging assay format was used, in which assay signal derived from ADA molecules that bridged two drug molecules, one drug molecule labeled with ruthenium, the other with biotin. ECL units were measured using a commercial chemiluminescence analyzer. A therapeutic MAb was conjugated with a ruthenium-label at different molar challenge ratios. The effect of different molar challenge ratios is evident in the LBA signal, which corresponds to electrochemiluminescence (ECL) units measured in an immunogenicity assay. The ADA specific for the therapeutic MAb was detected using a bridging assay format where the assay signal is obtained from molecules that bridged two labeled drug molecules (one labeled with ruthenium, the other labeled with biotin). The assay background and the positive-sample assay signal increased with increasing challenge ratios. The signal-to-background (S/B) ratio also increased with higher molar-ratio challenges, indicating the potential for improved method sensitivity with greater ratios of ruthenium to protein. d To develop a NAb assay, a positive control (PC) with neutralizing activity is required as a critical reagent. Several animals may be initially immunized initially to generate a PAb PC, and it is essential to gather information on the sample time points (bleeds) for neutralizing activity. In this assay, a dramatic decrease in IC50 in the LBA is observed as the neutralizing activity of PAbs matures with time a dramatic decrease in IC50 in the LBA is observed. This demonstrates that bleeds collected at different times from the same animal are not always equivalent in reactivity. It is important to maximize the amount of each lot of PAb reagent, perhaps by pooling several bleeds with appropriate activity. As part of a strategic plan to maintain the assay and to assure appropriate controls, subsequent lots may require a partial validation to determine new assay acceptance criteria for that new assay reagent. e Four lots of reagent Abs were produced and purified on separate occasions from the same hybridoma cell bank. The purification procedures for all lots were similar, and no differences between lots were observed when the Abs were evaluated by SEC or by reducing and non-reducing SDS-PAGE. The purified reagent Abs were then conjugated with ruthenium for use as a detection Ab in a PK assay. A conjugated MAb from Lot #301 was initially validated at a working concentration of 1.25 μg/mL in the PK assay, and attempts were made to qualify the subsequent lots at the same concentration. A conjugated antibody made from Lot #302 was qualified in the assay at an identical concentration as Lot #301. However, a conjugate made from Lot #303 could not be qualified in the assay and a conjugate made from Lot #304 could only be qualified in the assay at twice the concentration (2.5 μg/mL) of earlier lots. The source lots of these conjugated reagent antibodies were then evaluated for intact and reduced forms by LC-MS (reversed-phase HPLC coupled to time-of-flight mass spectrometry). The LC-MS experiments revealed that Lot # 301 and 302 had nearly identical spectra for their heavy chains, while a set of larger (∼1 kD shift) peaks were in the spectra from the heavy chains of Lots 303 and 304. These experiments indicate that as the relative signal of this higher mass species (when compared with the signal of the original mass) increased, the assay performance also decreased. Therefore, the LC-MS method could be used to prescreen subsequent lots for use in the assay (JPEG 70 kb)