

Abstract
Absorption models used in the estimation of pharmacokinetic drug characteristics from plasma concentration data are generally empirical and simple, utilizing no prior information on gastro-intestinal (GI) transit patterns. Our aim was to develop and evaluate an estimation strategy based on a mechanism-based model for drug absorption, which takes into account the tablet movement through the GI transit. This work is an extension of a previous model utilizing tablet movement characteristics derived from magnetic marker monitoring (MMM) and pharmacokinetic data. The new approach, which replaces MMM data with a GI transit model, was evaluated in data sets where MMM data were available (felodipine) or not available (diclofenac). Pharmacokinetic profiles in both datasets were well described by the model according to goodness-of-fit plots. Visual predictive checks showed the model to give superior simulation properties compared with a standard empirical approach (first-order absorption rate + lag-time). This model represents a step towards an integrated mechanism-based NLME model, where the use of physiological knowledge and in vitro–in vivo correlation helps fully characterize PK and generate hypotheses for new formulations or specific populations.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-012-9324-y) contains supplementary material, which is available to authorized users.
Key words: absorption, model, non-linear mixed effect, semi-mechanistic
INTRODUCTION
Oral drug absorption from solid dosage forms depends on a combination of complex processes including disintegration, dissolution, degradation, gastric emptying, intestinal transit, intestinal permeation and transport, intestinal metabolism, and hepatic metabolism (1). The absorption mechanism is governed by physicochemical properties of the drug, and the solubility at the different pH values and other conditions encountered in the gastro-intestinal (GI) tract. Of the physiological aspects, gastric emptying and intestinal transit are known to be critical factors in drug absorption (2–4).
Mechanistic models have been proposed to characterize drug absorption, taking into account these different processes. Examples of advanced absorption models currently in use are the CAT, and later the Advanced Compartmental Absorption and Transit model (5,6) implemented in Gastroplus® (Simulations Plus, Lancaster, CA), and the Advanced Dissolution, Absorption and Metabolism model implemented in SimCyp® (SimCyp Limited, Sheffield, UK) (7), which include physicochemical factors, physiological factors and dosage factors. These physiologically based pharmacokinetic (PBPK) models were designed for predicting drug absorption using physicochemical and physiological principles, but tend to be less well adapted for estimating absorption and disposition model parameters from observed pharmacokinetic data. Also, the GI movements of drug amount in these models are usually taken to be continuous, an assumption more suited to dissolved or dispersed drug rather than single unit modified released formulations.
When analyzing clinical pharmacokinetic observations (typically drug plasma concentrations) it is common to use non-linear mixed-effect modeling, the so-called population approach. Drug absorption is often assumed to follow a zero- or a first-order process. If a delay is observed between oral administration and drug absorption, a lag-time may be introduced either as an estimated parameter or as a chain of 1 or more transit compartments (8). To model erratic absorption profiles such as multiple peak behavior, several empirical models have been proposed. Plusquellec et al. proposed a discontinuous oral absorption model, following the absorption window concept to describe pharmacokinetic (PK) profiles with double peaks (9), then extended this to multiple peaks (10). Recently, Godfrey et al. applied a parallel input model and a stepwise input rate function to describe multiple peak PK data (11). Lindberg-Freijs and Karlsson used a flexible input function with different absorption rates for different time windows (12). In their approach, the number of windows and their duration had to be determined based on the sampling design of the study.
Whilst PBPK models rely on prior knowledge from physiological/physicochemical drug properties to predict absorption, and empirical models use mathematical functions to describe absorption, novel imaging techniques can now provide data on in vivo GI transit and dissolution, pointing to a new approach for the modeling of drug absorption. Tablet movement through the GI tract can be studied with magnetic marker monitoring (MMM), a novel imaging technique for the investigation of the behavior of solid oral dosage forms within the GI tract (13). When these data are combined with simultaneous pharmacokinetic sampling, information on the effect of absorption on observed pharmacokinetic profiles is generated. These types of data have been modeled previously by Bergstrand et al. (14) where models for tablet position measured by MMM have been linked with in vivo drug release (also measured by MMM) and disposition with pharmacokinetic sampling (Fig. 1).
Fig. 1.

Schema of models proposed by Bergstrand et al. (14). Left panel Tablet position modeled as a Markov model for fundus (F), antrum (A), proximal small intestine (P. SI: P1–P4), distal small intestine (D. SI: D1–D3), and colon (C). The tablet can move backward and forward between the upper and lower parts of the stomach (fundus and antrum). The probability of transiting through the proximal small intestine is described by four transit compartments (P 1–P 4). Transit through the distal part is described by three transit compartments (D 1–D 3). Concomitant food intake prolongs the residence of the tablet in the stomach. Right panel Drug release, absorption and disposition model of felodipine extended-release formulation. Compartment 1 represents the amount of drug present in the remaining part of the tablet. Zero-order release rates (D1–D5) are activated one at a time depending on the tablet location in the GI tract. The first-order rate constants K 23 and K 34 describe the distribution of disintegrated drug substance downstream the GI channel. Rate of absorption across the GI wall is governed by the first-order rate constants K 47, K 57, and K 67. The amount of drug absorbed across the GI wall is limited by bioavailability factor FA. Compartment 7 is a semiphysiological representation of a liver with a fixed volume (0.0143 L/kg). Hepatic elimination is governed by allometrically scaled liver blood flow (Q H) and the estimated hepatic extraction ratio (E H). The systemic distribution of felodipine is described by a central observation compartment (8) and two peripheral compartments (9,10)
In this paper, we propose a new mechanism-based approach to model absorption kinetics, taking into account tablet movement in the GI tract that we will refer to as the Gastro-Intestinal Transit Time (GITT) model. This non-linear mixed-effect model relies on some a priori knowledge on tablet transit times derived from MMM data. In the first part, we develop the new approach using a data set where MMM data are available as reference (14). In a second part, we applied this approach to pharmacokinetic data from which no MMM measurements were available.
PATIENTS AND METHODS
MMM Technique, Studies and Models
MMM is a technique for visualizing the transit of a solid oral dosage form through the GI tract, and is based on a determination of the magnetic dipole field generated by a magnetically labeled dosage form. Furthermore, the disintegration properties of the solid dosage form can be monitored during its passage through the GI tract by means of the decrease in magnetic moment (15).
In a cross-over study, six healthy volunteers were administered magnetically labeled extended-release tablets containing felodipine under fasting and fed conditions (16). This study was approved by the ethics committee of Charité, Berlin. Three types of observations were dynamically collected: tablet GI positions and drug release were monitored using MMM technique, and simultaneous plasma concentrations of felodipine were measured. Tablet GI position was categorized into five regions: fundus, antrum, proximal small intestine, distal small intestine, and colon.
Two separate models were previously developed to describe both GI tablet transit and distribution of released drug, and absorption and disposition for an extended-release formulation of felodipine (Fig. 1). Table I and II present the parameter estimates reported by Bergstrand et al. Further details of the model development are presented elsewhere (14).
Table I.
Typical Parameter Estimates and Associated Interindividual Variability for the Application of the GITT Model to the Felodipine Data
| Parameter | Typical value (RSE%) | IIV (%) (RSE%) |
|---|---|---|
| Drug release (mg/h) | ||
| D1 | 0.68 (5.1) | 9.2 (30.8) |
| D2 and D3 | 1.91 (5.0) | |
| D4 and D5 | 1.16 (3.3) | |
| Released substance Absorption (h−1) | ||
| K23 a | 0.43 (9.3) | – |
| K34 fasted/fed a | 3.48 (7.4)/0.81 (5.9) | 46.4 (32.1)/– |
| K47 and K57 | 2.87 (8.4) | – |
| K67 | 1.15 (18.8) | – |
| Disposition | ||
| FA fasted/fed | 0.23 (12.9)/0.39 (13.8) | 17.4 (15.7)/12.3 (14.2) |
| EH b | 0.50 (3.5) | 10.8 (17.5) |
| QH b (L/h/kg0.75) | 3.5 (FIXED) | – |
| Vliver (L/kg) | 0.0143 (FIXED) | – |
| Vcentral (L) | 20.4 (5.0) | 47.9 (30.8) |
| Qperiph1 (L/h) | 174 (3.1) | 23.4 (22.7) |
| Vperiph1 (L) | 88 (3.0) | 25.0 (23.6) |
| Qperiph2 (L/h) | 21.9 (7.3) | – |
| Vperiph2 (L) | 166 (4.1) | 18.4 (21.5) |
| Residual error (%) | 23 (8.9) | – |
In the GITT approach, all the population parameter distributions were fixed, and individual parameters estimated
IIV, interindividual variability; RSE, relative standard error; V central, V periph1, V periph2 are the volumes of the central and peripheral compartments, respectively; Q periph1 and Q periph2 are the inter-compartmental clearances between the central compartment and each peripheral compartment
aRate increased by 5 h−1 after tablet movement to small intestine
bHepatic extraction ratio (E H) and liver blood flow (Q H)
Table II.
Mean and Variance Residence Times in Each GI Region to Describe the Tablet Movement in the GITT Model
| Parameter | MRT (h) | VRT (h2) | CV (%) |
|---|---|---|---|
| Fundusa | 0.4/1.04 | 0.46/1.09 | 100 |
| Antruma | 0.32/1.58 | 0.15/2.50 | 100 |
| Proximal SI | 1.17 | 1.37 | 50 |
| Distal SI | 1.22 | 1.48 | 58 |
| Colon | – | – | – |
aParameters reported under fasted/fed conditions, computed from Bergstrand et al. (14)
MRT mean residence time; VRT variance residence time; CV coefficient of variation
The GI tablet transit model consisted of a Markov chain where each GI region was characterized by one or several states, and by a corresponding instant and time-varying probability of being at each position. In total, ten Markovian states were defined, including a chain of four states for the proximal small intestine, and a chain of three for the distal small intestine.
In the second model, tablet GI position was included as a covariate affecting drug release, GI distribution of released drug substance and absorption rate. A three-compartment model was used to model felodipine disposition, fitted separately to the intravenous data. An allometric relationship to bodyweight was assumed for disposition and elimination parameters. The elimination of felodipine was assumed to be 100% hepatic. Liver compartment volume, blood flow and blood/plasma concentration ratio were fixed to literature values, i.e. 1 L, 90 L/h, and 1.45, respectively (14).
Concomitant food intake was found to significantly prolong the mean tablet transit time from antrum to proximal small intestine, slow the rate of distribution of released drug in antrum and PSI, and to increase of the absorbed fraction over the gut-wall.
Gastro-Intestinal Transit Time Model
To further develop the approach described above, the absorption and disposition models were coupled and estimated simultaneously, using prior population parameter estimates for tablet GI transit, but not the position data measured by MMM. The model for the GI position was transformed to a GI step function (details below). The reason for this modification was that the use of the original model for tablet movement when MMM measurements were not available would require Hidden Markov estimation techniques, which are not readily available in common non-linear mixed-effects modeling software. The discrete movement of tablet was therefore translated into step functions in time, where each position (fundus, antrum, proximal small intestine, distal small intestine, and colon) corresponds to specific absorption characteristics. The following function was used for each position:
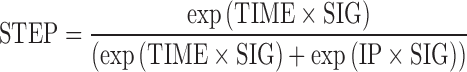 |
1 |
With STEP, a value between 0 and 1; TIME, the dependent variable; IP, the inflection time point corresponding to a STEP value of 0.5; SIG, the sigmoidicity factor (the higher the SIG, the steeper the step function). The choice of the step function is discussed later in the manuscript.
Prior information based on the separate modeling of the GI tablet transit data (14) was included to govern the typical value and interindividual variability for tablet residence time in each GI region. The complete GI transit was thus described by the sum of the step functions, one for each type of movement (as shown on Fig. 2). Besides downstream transit, the model also enables return from antrum to fundus. This movement was taken into account by a mixture model defining three possible patterns: no return, one return, or two returns to fundus. Since profiles with more returns to fundus were not observed in the MMM data, the model was limited to two returns to fundus. Table II reports the mean residence time values and the associated variability, derived from transition probabilities K according to the following equations (17) :
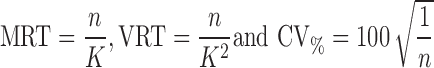 |
2 |
Fig. 2.

Example of GI tablet movement patterns, with or without return from antrum to fundus under fasting (solid line) and fed (dashed line) conditions. The times for each inflection point are governed by MRTs and VRTs, as reported in Table II
With MRT, the mean residence time; VRT, the variance residence time; CV, the associated theoretic coefficient of variation; K, rate constant governing change in probability from one position to the next one; n, the number of serial compartments in the tablet movement Markov model (1 for Fundus, Antrum and Colon, 3 for Distal SI, 4 for Proximal SI).
The individual values for the inflection points IPi in each STEP function, for the tablet movement from position A to B, were defined as: 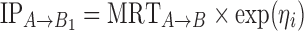 and
and 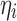 following a normal distribution of mean 0, and variance
following a normal distribution of mean 0, and variance 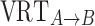 . The population parameter distributions of the inflection points were fixed to the values reported in Table II.
. The population parameter distributions of the inflection points were fixed to the values reported in Table II.
The individual likelihood in mixture modeling results from the sum of individual likelihoods from each mixture, weighted by their probabilities (18). In order to allow the same weight to each mixture, the mixture probabilities were set to the same value (1/3 each). The interindividual variability structure on the mean residence times in fundus and antrum was also simplified. A single IIV parameter was estimated for the mean residence time in stomach (fundus + antrum). This was required to obtain normally distributed and 0-centered Empirical Bayes Estimates for GI transit variability parameters.
Comparison of Models
The mechanism-based approach was compared to a more empirical model where the absorption phase was characterized by a lag-time and an absorption rate constant and corresponding interindividual variability; this approach is referred to as the lag-time model. Disposition parameters were fixed to remain similar across models.
Predictions from three different absorption models were thus compared: a simple lag-time model, a model based on MMM measurements using GI tablet position as a covariate, and the GITT model. The visual adequacy between observed and predicted plasma concentrations was compared between models. The predicted tablet movement, driven only by the observed plasma concentrations was compared between those predicted by the GITT model and those observed with MMM. Visual predictive checks were also performed to compare the performance of the 3 approaches.
Application of the GITT Model to Diclofenac
The next stage was to apply a relatively complex model structure accounting for a priori knowledge on tablet transit through GI to an example where no observations of GI transit were available. Diclofenac is an anti-inflammatory drug, available in numerous formulations, for example: infusion, suspension, immediate-, or extended-release tablets (19,20).
Data from a bioequivalence study including 30 healthy adult volunteers receiving 50 mg diclofenac were used. These data were from a study which aimed to compare the pharmacokinetic properties of three formulations, under fasting conditions: enteric-coated tablet, soluble tablet and suspension (unpublished, Rosemont Pharmaceuticals Ltd, UK) (21). This study was ethically approved by the Irish Medicines Board.
Samples were taken at 0.25, 0.5, 0.75, 1, 1.33, 1.67, 2, 2.5, 3, 3.5, 4, 6, 9, and 12 h after administration. The semi-physiologic GITT approach was applied to the diclofenac enteric-coated data only.
Diclofenac disposition was separately modeled using intravenous pediatric data (22). This study was approved by the Children’s Hospital of Helsinki. Ten patients aged between 4 and 6 years were administered 0.5 mg/kg over 5 or 15 min, and blood samples were taken 2, 5, 15, 60 min, and 2, 4, 6 h after administration. A non-linear mixed-effect model was fitted to the intravenous pediatric data, and an allometric relationship to weight was assumed a priori for all disposition parameters in order to scale to the adult bioequivalence subjects (23).
The absorption model structure applied to the felodipine data had to be modified before application to the diclofenac example. The reason for this was that no information about regional drug release from the tablet was available in the diclofenac data. As a consequence of this, the drug release constant and absorption rate constant could not be separately distinguished. The model was therefore simplified, assuming that released substance was rapidly absorbed as compared to tablet dissolution. As a consequence, a unique absorption parameter for each region, resulting from release and absorption phenomena, was defined as a first-order constant rate. It was also assumed that tablet GI transit times remained unchanged across drugs. Disposition parameter distributions were fixed to population estimates, and total bioavailability and absorption rates for each GI region were estimated using NONMEM 7, SAEM algorithm followed by Importance Sampling evaluation step (24) and PsN suite ([http://psn.sourceforge.net/], version 3.2.14) (25). Data handling and graphical representations were performed in R (26), using the Xpose package ([http://xpose.sourceforge.net/], version 4.2.1) (27). The residual error model consisted of combined additive and proportional errors. An example of NONMEM control file is supplied as electronic supplement file.
In the bioequivalence study, 58% of the observations following the administration of enteric-coated tablet were below the limit of quantification (LOQ = 10 ng mL−1 = 33.767 nmol L−1). The censored observations were handled with the so-called M3 method, which considers the likelihood of observations being below the quantification limit (28–30). VPCs were performed to compare the GITT approach to the lag-time model with respect to both observations above and below LOQ (28).
RESULTS
Application of GITT Model to Felodipine Data
Empirical Bayes Estimates for each parameter were obtained in NONMEM VI or 7. Using the GITT model individual tablet movement profiles were compared to those observed (Fig. 3 and Electronic Supplementary Material (ESM) Fig. 3).
Fig. 3.

Tablet movement through GI tract predicted in two individuals (see Electronic Supplement File for all individuals). Predicted positions (solid black line) are driven by plasma concentrations, given the population model parameters. Predictions are compared to the measured positions with MMM technique (green circles) GI positions are categorized as 1 fundus, 2 antrum, 3 proximal small intestine, 4 distal small intestine, 5 colon
Using only plasma concentrations and the pharmacokinetic model, tablet GI positions were adequately predicted in 9 out of 12 profiles compared to positions measured by MMM. In the case of three individuals (shown in ESM Fig. 3), the predicted tablet movement profiles were quite different from those observed, but were estimated to be the most likely to explain the corresponding PK profile, given the model (ESM Fig. 4). On seven occasions, subjects were estimated to most likely to belong to the first subpopulation (no return to fundus) whereas five were estimated as having one or two returns to fundus, which is in agreement with observed profiles.
Figure 4 compares model predictions for felodipine plasma concentration in two individuals, issued from different models: an empirical model considering a first-order absorption process and a lag-time (“Lag-time model”), the model considering tablet position measurements as a covariate (“MMM model”), and the GITT model. The GITT approach performed as well as the MMM model using GI position as a covariate (similar figure for each individual presented in ESM Fig. 4). Figure 5 presents the visual predictive checks of the median profile, obtained from 1,000 simulated replicates for the lag-time, MMM and GITT models.
Fig. 4.

Individual felodipine plasma concentration profiles in two individuals (see Electronic Supplement File for all individuals). Predicted concentrations were issued from the GITT approach (solid black line), the MMM model using tablet position measurements as covariate (blue dotted line) and a lag-time model (gray dashed line). Observed concentrations are symbolized by red circles
Fig. 5.

Visual Predictive Checks of felodipine plasma concentrations, for the lag-time model (on the left), the MMM model using GI position as a covariate (in the middle) and the GITT approach (on the right), on a log-scale. The observations are symbolized by the blue circles, and the median of observations are represented by red lines. Blue areas represent the 95% prediction interval for the median, computed from 1,000 simulated replicates
Application of GITT Model to Diclofenac Data
Diclofenac disposition was well characterized by a three-compartment model, with parameters scaled to bodyweight (Table III). Goodness-of-fit plots and visual predictive checks are provided as electronic supplement files.
Table III.
Typical Parameter Estimates and Associated Interindividual Variability for the Application of GITT Model to the Diclofenac Data
| Parameter | Typical value (RSE%) | IIV CV% (RSE%) |
|---|---|---|
| Disposition parametersa | ||
| CL (L/h/70 kg0.75) | 16.5 (5) | 16.9 (9) |
| V1 (L/70 kg) | 3.68 (17) | 20.1 (18) |
| Q2 (L/h/70 kg0.75) | 1.75 (21) | 37.5 (32) |
| V2 (L/70 kg) | 7.48 (16) | 32.7 (75) |
| Q3 (L/h/70 kg0.75) | 7.21 (3) | 53.1 (37) |
| V3 (L/70 kg) | 3.79 (7) | 35.4 (24) |
| Absorption parametersb | ||
| KAPSI (nmol/h) | 1.06 (21) | 109 (370) |
| KADSI (nmol/h) | 8.64 (258) | 53 (227) |
| KACol (nmol/h) | 1 (FIXED) | – |
| FA | 0.61 (15) | 8 (13) |
| Residual error | ||
| additive | 10 (0.3) | – |
| proportional | 12.6 (21) | – |
aParameter estimates obtained on the basis of intravenous data; parameters fixed to population distributions on oral data; Disposition parameters were allometrically scaled to weight
bParameter estimates obtained on the basis of oral data in the GITT model
IIV Inter-individual variability, CV coefficient of variation, RSE Relative Standard Error (obtained from Nonmem) CL clearance, V1, V2, V3 volumes of central and peripheral compartments, Q2 and Q3 intercompartmental clearances, KA PSI, KA DSI, KA Col are the absorption first-order rate constants in, respectively, proximal small intestine, distal small intestine and colon. FA denotes the overall absorbed fraction
After transiting intact through the stomach (fundus + antrum), the enteric-coated tablet moves to the proximal small intestine, distal small intestine, and colon. Transit through the stomach was estimated to take 2 h on average (ranging from 1.5 to 3 h across the studied individuals).
Compared with the lag-time model, the GITT model was better able to characterize the variability in lag-time before diclofenac systemic uptake. The goodness-of-fit and individual plots were drawn for both approaches (not shown), and showed a better agreement with the data with the GITT approach. A transit compartment approach (8) has also been applied to those data, giving similarly poor goodness-of-fit and inadequate visual predictive checks as the lag-time model. Figure 6 presents visual predictive checks from the three models, and highlights a better predictive ability of the GITT approach for the diclofenac PK.
Fig. 6.

Visual predictive checks of diclofenac plasma concentrations for the lag-time model (on the left), the transit model (in the middle) and the GITT model (on the right). Upper panel The observations are symbolized by the blue circles, and the median and 95th percentile of observations are represented by red lines. Blue areas represent the 95% confidence intervals for the median and the 95th percentile, computed from 1,000 simulated replicates. Lower panel The blue circles represent the proportion of below the quantification limit (BQL) observations within a given time bin. The blue area represents the corresponding 95% confidence interval
Absorption was estimated to occur mainly in the distal small intestine, and to a smaller extent in most individuals in the proximal small intestine, as shown in Fig. 7 and ESM Fig. 7. Most of the dose was predicted to be absorbed before the remaining tablet reached the colon.
Fig. 7.

Evolution of absorbed amount with time, for three individuals, until 8 h after dose. Predicted amount absorbed in proximal small intestine (blue dashed line), distal small intestine (gray dotted line) and the remaining amount to be absorbed in the tablet (black solid line). Amount absorbed in colon was close to zero in all individuals and is not represented
Table III shows model parameters estimated for diclofenac absorption. Total bioavailability was estimated to be 61% which is in accordance with values reported in the literature (31). Empirical Bayes Estimates for the GI transit parameters were normally distributed around 0, with reasonable shrinkage (respectively, 19%, 11% and 24% for residence times in stomach (antrum + fundus), proximal small intestine, and distal small intestine).
DISCUSSION
In non-linear mixed-effect modeling, most of the proposed PK models consider a relatively simple pattern for absorption, defined by a limited number of parameters. Simplistic approaches to absorption modeling may not be suitable under certain circumstances, especially when the absorption rate is neither a zero- nor first-order process or when multiple peaks are observed. In such cases, more complex approaches are often implemented, and these should ideally be supported by some knowledge on formulation behavior, physiology and absorption mechanism. By including mechanistic parameters that relate to formulation and physiology, models can be used for extrapolation to special populations or for predicting the PK profile of new formulations. We have successfully developed a mechanism-based model for absorption kinetics, which takes into account the tablet movement through the GI tract. As can be seen in Figs. 5 and 6, the GITT approach is clearly superior to the lag-time model in simulating modified release felodipine and enteric-coated diclofenac data, respectively.
To be able to use tablet transit characteristics in absorption modeling when MMM measurements are not available, the Markov model for GI transit proposed by Bergstrand et al. (14) was transformed into a GI step function. Felodipine tablet movement through the GI tract could be adequately predicted using only plasma concentration values and the GI step function (Fig. 3 and ESM Fig. 3). Comparing both approaches on the same felodipine dataset, one can see in Figs. 4 and 5 that the GI step function approach for tablet movement is suitable for the GITT model, and clearly superior to the empirical lag-time model.
Numerous functions exist to reproduce instantaneous on/off switch and non-continuous functions lead to estimation difficulties around the discontinuity point, which is often the parameter of interest. Continuous functions avoid this problem and most authors use a sigmoid function with a high sigmoidicity factor:
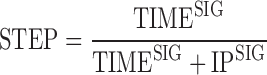 |
3 |
With STEP, a value between 0 and 1; TIME, the dependent variable; IP, the inflection time point corresponding to a STEP value of 0.5; SIG, the sigmoidicity factor (the higher the SIG, the steeper the step function).
The main drawback to the function shown in Eq. 3 is that the slope of the step function depends on the inflection point value: the step flattens with increasing time. Another function, written as Eq. 1 (see “Patients and Methods” section), was thus used in order to guarantee a steep slope for the step independent from the inflection point value.
When the GITT model was applied to diclofenac, simplifications were possible compared with the felodipine example: since the drug release and absorption processes were indistinguishable, a single parameter resulting from both processes was estimated for each GI region. In order to estimate both drug release and absorption parameters, one would need to add information to the model, either in vivo drug release data, either prediction of the drug release from in vitro–in vivo correlation studies. In addition, as an enteric-coated formulation of the diclofenac was studied, the data do not contain information on transit within the stomach, between fundus and antrum due to the fact that no drug would be released in this region.
In the diclofenac example, the “lag-time” model performed poorly (Fig. 6), especially in characterizing the lag-time and its associated inter-individual variability. In this model, a log-normal distribution of lag-time values was assumed, whereas the GITT approach allows a multimodal distribution of absorption kinetics thanks to the mixture model. The multimodal distribution comes about because the GITT approach allows the estimation of several absorption rates across regions of the GI tract. This is similar to the method employed by Lindberg-Freijs and Karlsson, who used a flexible input function with different absorption rates for different time windows, and pre-determined number and lengths of windows (12). In our approach, the transitions from one absorption rate to another are determined by the estimated tablet localization in the GI tract. As opposed to the model by Lindberg-Freijs and Karlsson, the present model is built on a mechanistic basis and utilizes prior information on tablet transit times.
A possible criticism of this work is that tablet movement characteristics were derived from a MMM cross-over study performed in only six individuals. With so few subjects we really cannot be sure that these data are truly representative of tablet movement behavior, but having said this the improvements gained with this approach compared with the lag-time model are encouraging. As more MMM investigations are performed, meta-analysis across MMM studies will yield improved estimates of the tablet transit times and their variability, and therefore predictions, of tablet residence time in each region of the GI tract (32). In addition, the tablet movement pattern was assumed to be similar for felodipine (11 mm diameter tablet) and diclofenac (7.3 mm). Such meta-analysis will intend to explore the role of formulation size in tablet movement patterns.
The main focus of this model was on tablet movement through the GI tract in a mechanism-based manner. Using data from diclofenac enteric-coated tablet pharmacokinetics, and known diclofenac disposition, we were able to show predictions of the fraction absorbed in different parts of the GI tract in individual patients (Fig. 7 and ESM Fig. 7). Whilst care must be exercised when making direct mechanistic conclusions from the results of applying the GITT model to data where no prior information about regional absorption is available, these results could be used to guide formulation development derived from in vitro characteristics, and in physiologically based predictions of absorption and disposition in special populations. In the GITT model, absorption parameters are estimated to best fit the data which raises hypotheses about relative absorption rates in different GI regions. The GITT model is a very flexible model as it allows the estimation of several absorption rates. However, the mechanistic basis allows additional assumptions to be introduced, such as relative rates of absorption (for example absorption in small intestine assumed to be greater than in colon) or using knowledge derived from regional absorption studies.
CONCLUSION
This work represents a first step in the use of prior information on GI tablet movement in absorption kinetics with non-linear mixed-effect modeling. The GITT model promises to enhance both the top-down modeling of observed pharmacokinetic data of drugs with variable absorption, and in the bottom-up predictions of drug absorption made by physiologically based systems.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(DOC 45 kb)
{kind=link}
(JPEG 390 kb)
{kind=link}
(JPEG 453 kb)
{kind=link}
(JPEG 549 kb)
(DOC 38 kb)
Acknowledgments
The authors would like to thank Jeff Rothwell, Rosemont Pharmaceuticals Ltd, UK, and Professor Klaus Olkkola, University of Helsinki, Finland, who provided the diclofenac data. Dr Emilie Henin was supported by a grant to Uppsala University from Novartis, and by the Lavoisier Program from the French Ministry of European Affairs.
References
- 1.Huang W, Lee SL, Yu LX. Mechanistic approaches to predicting oral drug absorption. AAPS J. 2009;11(2):217–224. doi: 10.1208/s12248-009-9098-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirtz J. The gastrointestinal absorption of drugs in man: a review of current concepts and methods of investigation. Br J Clin Pharmacol. 1985;19(Suppl 2):77S–83S. doi: 10.1111/j.1365-2125.1985.tb02746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez N, Garcia JJ, Diez MJ, Sahagun AM, Gonzalez A, Diez R, et al. Effects of slowed gastrointestinal motility on levodopa pharmacokinetics. Auton Neurosci. 2010;156(1–2):67–72. doi: 10.1016/j.autneu.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Hardoff R, Sula M, Tamir A, Soil A, Front A, Badarna S, et al. Gastric emptying time and gastric motility in patients with Parkinson’s disease. Mov Disord. 2001;16(6):1041–1047. doi: 10.1002/mds.1203. [DOI] [PubMed] [Google Scholar]
- 5.Yu LX, Amidon GL. A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm. 1999;186(2):119–125. doi: 10.1016/S0378-5173(99)00147-7. [DOI] [PubMed] [Google Scholar]
- 6.Agoram B, Woltosz WS, Bolger MB. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev. 2001;50(Suppl 1):S41–S67. doi: 10.1016/S0169-409X(01)00179-X. [DOI] [PubMed] [Google Scholar]
- 7.Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami-Hodjegan A. The Simcyp((R)) Population-based ADME Simulator. Expert Opin Drug Metab Toxicol. 2009. doi: 10.1517/17425250802691074 [DOI] [PubMed]
- 8.Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711–726. doi: 10.1007/s10928-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 9.Plusquellec Y, Campistron G, Staveris S, Barre J, Jung L, Tillement JP, et al. A double-peak phenomenon in the pharmacokinetics of veralipride after oral administration: a double-site model for drug absorption. J Pharmacokinet Biopharm. 1987;15(3):225–239. doi: 10.1007/BF01066319. [DOI] [PubMed] [Google Scholar]
- 10.Plusquellec Y, Efthymiopoulos C, Duthil P, Houin G. A pharmacokinetic model for multiple sites discontinuous gastrointestinal absorption. Med Eng Phys. 1999;21(8):525–532. doi: 10.1016/S1350-4533(99)00060-0. [DOI] [PubMed] [Google Scholar]
- 11.Godfrey KR, Arundel PA, Dong Z, Bryant R. Modelling the Double Peak Phenomenon in pharmacokinetics. Comput Methods Programs Biomed. 2010. doi: 10.1016/j.cmpb.2010.03.007 [DOI] [PubMed]
- 12.Lindberg-Freijs A, Karlsson MO. Dose dependent absorption and linear disposition of cyclosporin A in rat. Biopharm Drug Dispos. 1994;15(1):75–86. doi: 10.1002/bdd.2510150107. [DOI] [PubMed] [Google Scholar]
- 13.Weitschies W, Blume H, Monnikes H. Magnetic marker monitoring: high resolution real-time tracking of oral solid dosage forms in the gastrointestinal tract. Eur J Pharm Biopharm. 2010;74(1):93–101. doi: 10.1016/j.ejpb.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Bergstrand M, Soderlind E, Weitschies W, Karlsson MO. Mechanistic modeling of a magnetic marker monitoring study linking gastrointestinal tablet transit, in vivo drug release, and pharmacokinetics. Clin Pharmacol Ther. 2009;86(1):77–83. doi: 10.1038/clpt.2009.43. [DOI] [PubMed] [Google Scholar]
- 15.Weitschies W, Kosch O, Monnikes H, Trahms L. Magnetic marker monitoring: an application of biomagnetic measurement instrumentation and principles for the determination of the gastrointestinal behavior of magnetically marked solid dosage forms. Adv Drug Deliv Rev. 2005;57(8):1210–1222. doi: 10.1016/j.addr.2005.01.025. [DOI] [PubMed] [Google Scholar]
- 16.Weitschies W, Wedemeyer RS, Kosch O, Fach K, Nagel S, Soderlind E, et al. Impact of the intragastric location of extended release tablets on food interactions. J Control Release. 2005;108(2–3):375–385. doi: 10.1016/j.jconrel.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Chan KK, Wnuck K, Bell CL. Microcomputer-based programs for calculating mean and variance residence time by the method of prospective areas. Comput Methods Programs Biomed. 1987;25(1):23–30. doi: 10.1016/0169-2607(87)90074-5. [DOI] [PubMed] [Google Scholar]
- 18.Carlsson KC, Savic RM, Hooker AC, Karlsson MO. Modeling subpopulations with the $MIXTURE subroutine in NONMEM: finding the individual probability of belonging to a subpopulation for the use in model analysis and improved decision making. AAPS J. 2009;11(1):148–154. doi: 10.1208/s12248-009-9093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies NM, Anderson KE. Clinical pharmacokinetics of diclofenac. Therapeutic insights and pitfalls. Clin Pharmacokinet. 1997;33(3):184–213. doi: 10.2165/00003088-199733030-00003. [DOI] [PubMed] [Google Scholar]
- 20.Brogden RN, Heel RC, Pakes GE, Speight TM, Avery GS. Diclofenac sodium: a review of its pharmacological properties and therapeutic use in rheumatic diseases and pain of varying origin. Drugs. 1980;20(1):24–48. doi: 10.2165/00003495-198020010-00002. [DOI] [PubMed] [Google Scholar]
- 21.Standing JF, Howard RF, Johnson A, Savage I, Wong IC. Population pharmacokinetics of oral diclofenac for acute pain in children. Br J Clin Pharmacol. 2008;66(6):846–853. doi: 10.1111/j.1365-2125.2008.03289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korpela R, Olkkola KT. Pharmacokinetics of intravenous diclofenac sodium in children. Eur J Clin Pharmacol. 1990;38(3):293–295. doi: 10.1007/BF00315034. [DOI] [PubMed] [Google Scholar]
- 23.Holford NH. A size standard for pharmacokinetics. Clin Pharmacokinet. 1996;30(5):329–332. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 24.Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User’s Guides (1989–2009) Ellicott City, MD, USA: Icon; 2009. [Google Scholar]
- 25.Lindbom L, Ribbing J, Jonsson EN. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 26.R DCT. R: A Language and Environment for Statistical Computing. R.2.12.0 ed. Vienna, Austria: R Foundation for Statistical Computing; 2010.
- 27.Jonsson EN, Karlsson MO. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64. doi: 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- 28.Bergstrand M, Karlsson MO. Handling data below the limit of quantification in mixed effect models. AAPS J. 2009;11(2):371–380. doi: 10.1208/s12248-009-9112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35(4):401–421. doi: 10.1007/s10928-008-9094-4. [DOI] [PubMed] [Google Scholar]
- 30.Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504. doi: 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- 31.Willis JV, Kendall MJ, Flinn RM, Thornhill DP, Welling PG. The pharmacokinetics of diclofenac sodium following intravenous and oral administration. Eur J Clin Pharmacol. 1979;16(6):405–410. doi: 10.1007/BF00568201. [DOI] [PubMed] [Google Scholar]
- 32.Hénin E, Bergstrand B, Karlsson MO. Meta-analysis of Magnetic Marker Monitoring data to characterize tablet movement through the gastrointestinal tract. PAGE Abstracts of the Annual Meeting of the Population Approach Group in Europe. 2011;PAGE 20 (2011) Abstr 2249 [www.page-meeting.org/?abstract=2249].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 45 kb)
(JPEG 390 kb)
(JPEG 453 kb)
(JPEG 549 kb)
(DOC 38 kb)