

Abstract
Keratin 16 (KRT16 in human, Krt16 in mouse), a type I intermediate filament protein, is constitutively expressed in epithelial appendages and is induced in the epidermis upon wounding and other stressors. Mutations altering the coding sequence of KRT16 cause Pachyonychia Congenita (PC), a rare autosomal dominant disorder characterized by hypertrophic nail dystrophy, oral leukokeratosis, and palmoplantar keratoderma (PPK). PPK associated with PC are extremely painful and compromise patient mobility, making them the most debilitating PC symptom. In this study, we show that, although inherited in a recessive fashion, the inactivation of Krt16 in mice consistently causes oral lesions as well as PPK-like hyperkeratotic calluses on Krt16−/− front and hind paws, which severely compromise the animals’ ability to walk. Our findings call into question the view that PC-related PPK arise exclusively as a gain-of-function on the account of dominantly acting mutated keratins, and highlight the key role of modifiers in the clinical heterogeneity of PC symptoms.
Introduction
Keratin 16 (KRT16 in human, Krt16 in mouse), a type I intermediate filament protein, is constitutively expressed in a variety of epithelial appendages, including the tongue and the hair follicle, and in glabrous skin (Moll et al, 1982; Bernot et al, 2002; Swensson et al, 1998). Upon stressful epithelial stimuli, such as wounding or chronic inflammation, Krt16 and its binding partner Krt6 are selectively induced in the suprabasal layers of the epidermis (Paladini et al, 1996). Mutations in KRT16 are associated with the development of Pachyonychia Congenita (PC), a rare autosomal dominant disorder characterized by hypertrophic nail dystrophy, palmoplantar keratoderma (PPK), and oral leukokeratosis (McLean et al, 1995). PC symptoms are highly variable in penetrance, age of onset, and severity, even between individuals with the same mutation (Leachman et al, 2005; Liao et al, 2007; Fu et al, 2011). In patients carrying KRT16 mutations, palmoplantar keratoderma are the most prominent symptom and may sometimes appear as focal PPK (FPPK) with little or no nail involvement (Leachman et al, 2005; Liao et al, 2007; Shamsher et al, 1995; Smith et al, 2000; Smith et al, 2005). KRT16 mutation-linked PPK is typically non-epidermolytic (McLean et al, 1995; Liao et al, 2007; Shamsher et al, 1995), although blistering underneath and around PPK calluses has been linked to palmoplantar pain in PC patients (Dahl et al, 1995). Previous mouse models harboring both dominant and recessive mutations in Krt6 and Krt75 recapitulate several PC-like symptoms (Wong et al, 2000, Wojcik et al, 2001; Wong et al, 2005; Chen et al, 2008), but have failed to phenocopy palmoplantar keratoderma, which are considered the most debilitating PC symptom as they are extremely painful and significantly impact patient mobility and quality of life (Leachman et al, 2005; Dahl et al, 1995). In this study, we show that adult mice lacking Krt16 develop hyperkeratotic calluses on their front and hind paws that are strikingly similar to human palmoplantar keratoderma and significantly compromise the animals’ ability to walk.
Results
Failure to thrive and increased postnatal mortality in Krt16−/− mice
Krt16−/− mice were born alive at approximately Mendelian ratios and were initially visually indistinguishable from wild-type (WT) and Krt16+/− littermates (Figure S1A). However, 34% of Krt16−/− mice died within the first 24 h after birth, as opposed to 6% of WT and 11% of Krt16+/− mice (Figure 1A). Toluidine Blue dye exclusion assays performed on newborn pups ruled out any gross early postnatal skin barrier defects in Krt16−/− (Figure S1B). Newborn Krt16−/− mice weighed less than their littermates (Figure S1C) and subsequently continued to lag behind their littermates in size and in weight (Figure 1B–C). Such phenotypic changes coincided with the persistence of a markedly higher level of postnatal mortality, and over 60% of Krt16−/− mice died before weaning age. The Krt16−/− mice that did survive continued to grow and gained weight, but remained smaller and lighter than their littermate controls (data not shown).
Figure 1. Failure to thrive, high postnatal mortality, and posterior tongue lesions in Krt16−/− mice.
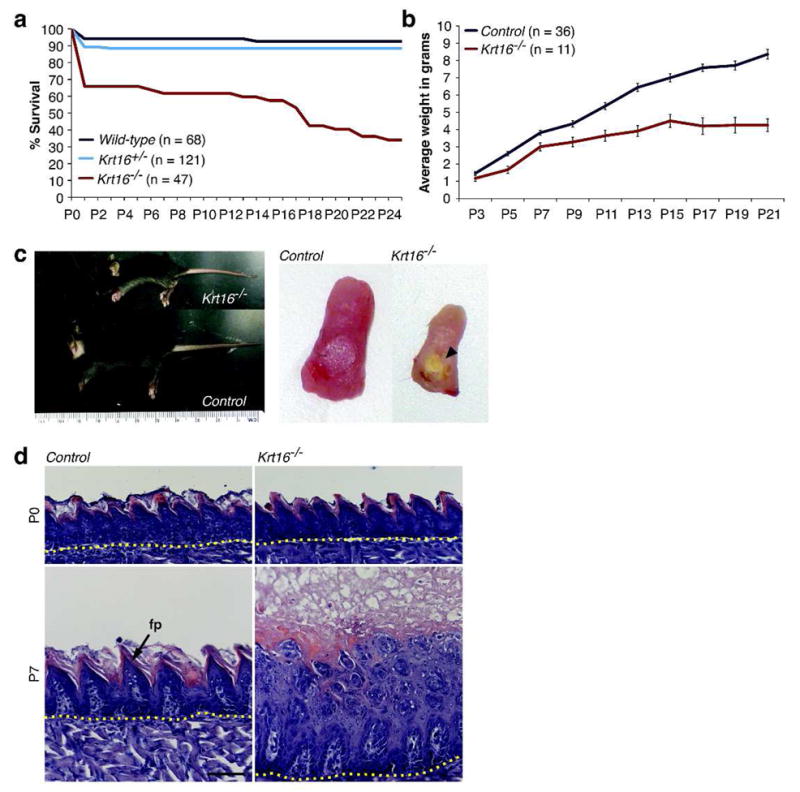
(a) High postnatal mortality in Krt16−/− mice. 34% of Krt16−/− mice die within 24 h after birth; only a third survive past weaning. (b) Krt16−/− mice weigh significantly less than control littermates. Error bars = SEM. (c) P17 Krt16−/− mice are smaller than littermates and a white-yellow plaque covers the posterior dorsal area of their tongues (arrowhead). (d) H&E-stained sections of posterior tongues at P0 and P7. Krt16−/− mice show no obvious defects at birth. By P7, the architecture of filiform papillae (fp) is markedly disrupted, and massive hyperkeratosis leads to macroscopically visible plaques. Note the paucity of cell lysis in the suprabasal layer. Dotted line = epithelial/muscle junction. Scale bar, 50μm.
Krt16 is essential for the structural integrity of dorsal tongue epithelium
Mice lacking the Krt16 binding partners, Krt6a and Krt6b, exhibit a more severe albeit similar growth retardation and early postnatal lethality phenotype (Wong et al, 2000; Wojcik et al, 2001). In Krt6a/b−/− mice, the formation of large hyperplastic lesions on the dorsal posterior tongue (akin to oral leukokeratosis, a PC symptom) is thought to occlude the laryngeal space, impair feeding, and lead to early death due to starvation. Such laryngeal obstruction, although extremely rare, has been reported in pediatric PC patients carrying KRT6a mutations (Smith et al, 2005; Haber and Drummond, 2011) and infants suffering from oral leukokeratosis often have trouble breastfeeding (Leachman et al, 2005).
When we examined the tongues of Krt16−/− mice at different ages, we also detected the presence of hyperplastic lesions, which are fully penetrant and become macroscopically and microscopically visible by P3 (Figure 1D). These lesions were confined to the dorsal midline area of the posterior tongue and were smaller than those observed in Krt6a/b−/− mice (Wong et al, 2000). Surviving Krt16−/− mice no longer had visible lesions, but the tongue architecture remained severely compromised, showing a thickened epithelium and loss of the characteristic filiform papillae morphology (Figure S2A). Although no epithelial fragility was detectable by routine histology in P0 Krt16−/− tongues (Figure 1D), transmission electron microscopy revealed early stages of cell lysis in a subset of posterior filiform papillae (Figure S2B). Interestingly, Krt17 protein and to a lesser extent Krt17 mRNA levels were significantly reduced in the tongue epithelium of Krt16−/− mice (Figure 2A–B), and immunofluorescence staining for Krt17 was consistently absent from the anterior column of Krt16−/− filiform papillae, but not the fungiform papillae (Figure 2A). The expression of Krt5, Krt6 and Krt10 was normal in Krt16−/− filiform papillae (Figure 2A–B), suggesting that the loss of Krt17 was not due to an overall change in the differentiation program in anterior column of filiform papillae. The constitutive as well as inducible expression of Krt17 in other tissues, including skin, was not affected (Figure 2A, Figure 4D). Our findings thus extend the notion that Krt6- and Krt16-containing filaments are essential for the maintenance of dorsal tongue epithelial integrity. Loss of Krt16 and concomitant reduction of Krt17 levels deplete most, but not all, of Krt6’s primary binding partners in tongue epithelium, leading to increased cell fragility and hyperplastic lesion formation, starvation, and a higher chance of postnatal death in Krt16−/− mice. Residual levels of Krt17 present in Krt16−/− tongue epithelium may explain the smaller lesions and lower number of early deaths in Krt16−/− mice as compared to Krt6a/b−/− mice (Wong et al, 2000; Wojcik et al, 2001). Despite their less severe nature, we cannot rule out the possibility that oral lesions in Krt16−/− mice are painful and thus impact feeding behavior.
Figure 2. Krt17 levels are selectively reduced in Krt16−/− filiform papillae.
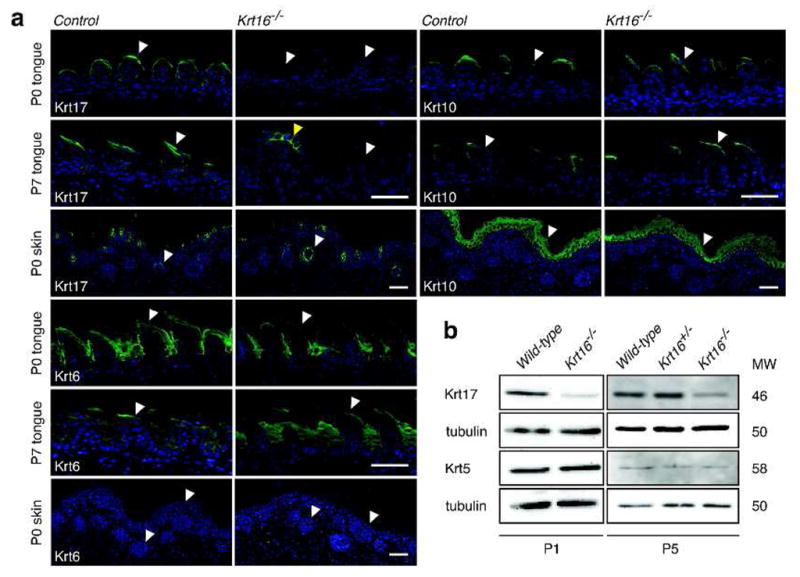
(a) Immunofluorescent stainings in P0 and P7 tongue cross-sections show a reduction of Krt17 in Krt16−/− filiform papillae (white arrowheads), but not in fungiform papillae (yellow arrowhead). Constitutive expression of Krt17 in hair follicles at P0 is not affected. Krt6 and Krt10 are unchanged in both filiform papillae in Krt16−/− tongues and skin (white arrowheads). Scale bars, 50μm. (b) Whole tongue lysates show a universal reduction in Krt17 expression in Krt16−/− tongue epithelium. Krt5 expression is unaltered. To a lesser extent, Krt17 mRNA levels are also reduced (data not shown).
Figure 4. Krt16−/− calluses - impaired mobility, hyperproliferation, and compromised epidermal barrier function.

(a) Krt16−/− mice are less active than controls in a behavioral assay (experimental time frame of 30 min). Asterisks = p-value < 0.002, Student’s T-test. (b) Toluidine Blue dye penetration in adult front paws highlights focal loss of epidermal barrier function in weight bearing areas (arrowheads). (c) Krt16−/− front paw calluses show a 2-fold increase in proliferation and a more modest increase in uninvolved areas adjacent to established calluses. Asterisk = p-value < 0.03, Student’s T-test. (d) Basal/suprabasal induction of Krt17 and marked reduction of barrier protein filaggrin in Krt16−/− front paw calluses. Krt1 expression is patchy in areas subject to barrier breach, but otherwise normal. Dotted line = epidermal/dermal junction. Scale bar, 50μm.
Adult Krt16−/− develop palmoplantar keratoderma
Starting at 4–6 weeks of age, surviving Krt16−/− mice developed prominent, hyperkeratotic calluses on the glabrous parts of both front and hind paws (Figure 3A–C). The boundaries of these calluses did not directly correlate with the loss of endogenous Krt16, which is expressed in a patchy fashion throughout paw pad epithelia including the nail hyponychium, as shown by whole-mount X-gal staining (Figure 3A). Instead, consistent with focal palmoplantar keratoderma (FPPK) in human PC patients, calluses in Krt16−/− mice were restricted to areas subject to physical pressure, i.e. the heel and the wrist (Figure 3A). Occasionally, we observed generalized hyperkeratosis on front paw pads as early as 3 weeks of age (Figure S3A), and focal hyperkeratosis near the base of the tail. Importantly, nail morphology was not affected in Krt16−/− mice (Figure S3B).
Figure 3. Krt16−/− mice develop palmoplantar keratoderma.

(a) Adult Krt16−/− mice develop PPK-like lesions in their front and hind paws. Callus formation is common in areas of high physical impact (arrowheads), despite the global Krt16 expression pattern in glabrous skin (see Xgal stain). Note the presence of focal hyperpigmentation in front paw calluses. (b) H&E-stained cross-sections of front paws, showing expansion of the epidermis and massive hyperkeratosis in Krt16−/− mice. Occasionally, focal suprabasal lysis is observed in Krt16−/− calluses (arrowheads). sc = stratum corneum, epi = epidermis. Scale bars, 100μm (calluses), 50μm (insets). (c) Hyperkeratosis without lysis in Krt16−/− hind paw epidermis. Note the change in shape and arrangement of suprabasal nuclei. sc = stratum corneum, sb = suprabasal layer, b = basal layer. Scale bar, 50μm.
The age of onset for callus formation on both front and hind paws was variable, and calluses differed in severity between animals (Figure 3A shows two representative examples of callus severity in Krt16−/−). 36% of Krt16−/− mice showed no alteration of their hind paw pads at all. Hind paw calluses also consistently formed after front paw pad calluses were already established. Visual changes in Krt16−/− front paw pads correlated with an expanded epidermal compartment (Figure 3B) that correlated with a 2-fold increase in proliferation (Figure 4C) in the absence of apoptotic cell death (Figure S3C). Areas adjacent to the callus showed a thickened epithelium, but no significant increase in proliferation (Figure 3B, Figure 4C). Paw pad epithelia in younger mice with no obvious signs of callus formation were indistinguishable from controls (data not shown). Since palmoplantar keratoderma in humans greatly impair mobility, we asked whether callus formation in Krt16−/− mice negatively impacts the animals’ activity level in a behavioral assay. We found that adult Krt16−/− mice are significantly less active than control animals and spent more time resting than walking (Figure 4A, Figure S3D). Several anomalies, including the patchy expression of Krt17 (which is absent from control mouse paw pad epidermis) and focal toluidine blue dye penetration (Figure 4B and 4D) suggested a disruption of the outside-in epidermal barrier in Krt16−/− front paw calluses, which may lead to secondary infections as implied by the presence of prominent hyper-pigmentation in those areas (Figure 3A). Krt17 expression is induced in response to barrier breach (DePianto et al, 2010; McGowan and Coulombe, 1998) and a mutation in the barrier protein filaggrin has been shown to intensify PC-related symptoms in a human patient with a coincident Krt16 mutation (Liao et al, 2007; Gruber et al, 2009). Filaggrin expression is normal in Krt16−/− paw pad epithelia, as reported for PC plantar lesions (Wollina et al, 1991), but is reduced in established front paw calluses (Figure 4D), confirming that Krt16−/− glabrous skin, while initially intact, is eventually unable to fend off the continuous physical pressure generated by walking and/or cleaning behavior, resulting in hyperproliferation and a focal loss of barrier protection. In humans, PPK are very painful. The cause of the pain is unknown, but clinical observations have noted the formation of blisters underneath or adjacent to calluses (Dahl et al, 1995), as well as secondary infections following fissuring of the hyperkeratotic tissue (Leachman et al, 2005). Alternatively, it is possible that PPK calluses exert an increased pressure on nerve endings in plantar skin, especially while walking. While an objective assessment of the level and type of pain or its source in Krt16−/− mice is beyond the scope of this study, we hypothesize that Krt16−/− mice experience substantial discomfort as a direct result of palmoplantar lesions and thus exhibit a significant decrease in their overall mobility.
Discussion
Here, we show that the loss of Krt16 function in mice causes the development of prominent calluses on the plantar side of front and hind paws, which significantly compromise mobility and eventually lead to overt loss of barrier properties. While the molecular mechanism of PPK pathogenesis in PC is still unclear, this symptom is currently thought to develop as a consequence of intermediate filament network disruption by dominantly-acting mutations in relevant keratins (Fu et al, 2011). To our surprise, deletion of Krt16 produced spontaneously arising PPK-like lesions in mice, suggesting that PPK pathogenesis in PC is more complex than previously appreciated and may represent, at least in part, a loss of function phenotype. Almost all human PC patients harboring KRT16 mutations report PPK (Leachman et a, 2005, Liao et al, 2007, Fu et al, 2011). However, many mutations in KRT16 only elicit milder overall PC phenotypes, often diagnosed as FPPK because of the limited and at times absent nail involvement (Liao et al, 2007; Shamsher et al, 1995; Smith et al, 2000; Smith et al, 2005). In particular, this is seen with small deletions in KRT16, which are thought to cause exclusion of the mutant form of KRT16 from filament assembly (Smith et al, 2000; Wilson et al, 2009; Cao et al, 2011). These findings together with our data suggest that both the dominant negative disruption of intermediate filaments as well as the exclusion of keratins from the network can contribute to PPK pathogenesis. Nail dystrophy, on the other hand, was not observed in Krt16−/− mice, and may depend more heavily on the dominant negative interference of keratin filaments. Accordingly, the deletion of individual or even multiple keratins leads to no or very minor nail defects in mice (Wong et al, 2000; Wojcik et al, 2001; Wong et al, 2005; this study), whereas a dominant mutation in Krt75 elicits the characteristic nail overgrowth in mouse (Chen et al, 2008). In humans, where all known PC-causative keratin mutations are dominant, nail involvement is highly penetrant. These observations in humans, mouse models, and now our findings in Krt16−/− mice showcase how a combination of loss and gain of function phenotypes can contribute to the complex overall clinical presentation of PC. Furthermore, our data substantiate the hypothesis that FPPK and PC can share a common pathogenesis (Wilson et al, 2009; Bowden 2010), since Krt16−/− mice developed FPPK along with oral lesions, another PC-like symptom. We also noticed differences in disease onset, phenotype severity, and change in barrier permeability in Krt16−/− mice, supporting the idea of keratin, non-keratin and/or environmental modifiers in PPK pathogenesis (Smith et al, 2000). It will be interesting to examine whether the mechanism(s) by which loss of Krt16 results in the development of PPK overlap(s) with the pathogenesis of other keratoderma, e.g. striate PPK (Desmoplakin) or diffuse PPK (KRT1, Desmoglein).
We only observed occasional and locally restricted areas of cell lysis in established Krt16−/− front paw calluses (Figure 3B, arrowhead and inset), but never in hind paw calluses or uninvolved skin, despite the global expression pattern of Krt16 in glabrous tissue. Expression of other structurally important keratins in the suprabasal layer, such as Krt1 and Krt10, was also generally intact (Figure 4D) and likely contributes to the maintenance of cellular integrity in the absence of Krt16. Thus, we speculate that Krt16 has an additional function in glabrous skin, separate from its role of structural support. Several studies have already implicated Krt16 and other type 1 keratins, such as Krt10 and Krt17, as significant players in epidermal homeostasis (Paladini et al, 1996; DePianto et al, 2010; Takahashi et al, 1994; Reichelt and Magin, 2002; Kim et al, 2006). A key feature of Krt16 expression in the skin is its selective induction following injury, UV exposure, or in chronic disease states (e.g. psoriasis) (Paladini et al, 1996; Del Bino et al, 2004; Leigh et al, 1995). Glabrous skin is a specialized tissue designed to withstand and adapt to significant mechanical trauma (Swensson et al, 1998; Bowden et al, 1987). It will be worth investigating the function of constitutive Krt16 expression in this unique environment, and whether the underlying mechanism bears any relationship to the role(s) fulfilled by Krt16 when induced by injury and other relevant stressors.
Materials and Methods
Generation of Krt16−/− mice
C57 Bl/6 ES cells in which the Krt16 coding sequence has been replaced with a lacZ-loxP-NeoR-loxP cassette were created by Velocigene using funds provided by the trans-NIH Knock-Out Mouse Project (KOMP), and obtained from the KOMP repository (supported by the NCRR-NIH). ES cells were injected into C57 Bl/6cBrd/cBrd albino blastocysts (distributed by the NCI, Frederick, MD). A high coat color male chimera was bred to C57 Bl/6cBrd/cBrd albino females and 100% germline transmission was observed. Krt16+/− F1 offspring were born at the expected 50:50 ratio and inter-crossed to generate Krt16−/− mice. All experiments involving mice were reviewed and approved by the Johns Hopkins Institutional Animal Care and Use Committee. Mouse lines were maintained under specific pathogen-free conditions (SPF), and fed chow and water ad libitum. All experiments were performed using littermate controls (wild-type or Krt16+/−).
Histopathology and immunofluorescence
Whole tongues and paws were fixed overnight in Bouin’s (Sigma) or 4% para-formaldehyde/PBS. Tissue was then either rinsed and embedded in Sakura Tissue-Tek O.C.T. (VWR, Radnor, PA), or dehydrated and processed for routine paraffin embedding. Sections were cut at 5μm and stained with hematoxylin/eosin (H&E) according to standard protocols. For immunofluorescence, frozen sections or rehydrated paraffin-embedded sections were washed in PBS, blocked in 5% NGS, 0.1% Triton-X100 for 1 h at room temperature, incubated in primary antibody solution (2.5% NGS, 0.1% Triton-X100) for 1 h at room temperature, washed in PBS, incubated in secondary antibody (2.5% NGS) for 1 h, counterstained with DAPI, mounted, and imaged using an inverted Zeiss fluorescence microscope with ApoTome attachment. Antibodies used were directed against: Krt17 (1:1000; McGowan and Coulombe, 1998), Krt6 (1:250; McGowan and Coulombe, 1998), Krt10 (1:500; Covance, Princeton, NJ), filaggrin (1:500; Covance, Princeton, NJ), Ki67 (Sp6 clone,1:200; ThermoFisher, Pittsburgh, PA), and AlexaFluor488 (1:1000; Invitrogen, Carlsbad, CA). TUNEL staining was performed as described (McGowan et al., 2002).
Whole-mount Xgal and barrier function assays
Whole paws were fixed for 1 h in 4% paraformaldehyde/PBS at 4°C, washed in PBS, and permeabilized via 3×15 min washes in PBS containing 2mM MgCl2, 0.01% sodium deoxycholate, and 0.02% NP-40. The tissue was then transferred to scintillation vials containing a 1mg/ml X-gal solution (30mM K4Fe(CN)6, 30mM K3Fe(CN)6, 2mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, PBS) for 30 min at 30°C. Samples were photographed immediately. Toluidine Blue dye staining, to assess outside-in barrier function, was carried out on newborn pups and adult paws as described (Hardman et al, 1998).
Behavioral assays to assess mobility
Individual mice were allowed to acclimate for 30 min and then placed into a rectangular plexiglas chamber with a stainless steel floor. To eliminate outside positional cues, the chamber was uniformly illuminated and its sides masked with cardboard. Mobility was monitored over a time frame of 30 min for infrared beam-breaking activity and the data was recorded using the Optimax software (Columbus Instruments, Columbus, OH). Results represent an average of three separate experiments.
Supplementary Material
Acknowledgments
The authors thank Dr. Michael Caterina for help with the behavioral assays, Janet Folmer for assistance with electron microscopy, and members of the Coulombe lab for discussion and critical reading of the manuscript. These studies were supported by grant AR44232 to P.A.C. from the National Institute of Arthritis, Musculoskeletal, and Skin Diseases (NIAMS) at the National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Bernot KM, Coulombe PA, McGowan KM. Keratin 16 expression defines a subset of epithelial cells during skin morphogenesis and the hair cycle. J Invest Dermatol. 2002;119:1137–1149. doi: 10.1046/j.1523-1747.2002.19518.x. [DOI] [PubMed] [Google Scholar]
- 2.Bowden PE. Mutations in a keratin 6 isomer (K6c) cause a type of focal palmoplantar keratoderma. J Invest Dermatol. 2010;130:336–338. doi: 10.1038/jid.2009.395. [DOI] [PubMed] [Google Scholar]
- 3.Bowden PE, Stark HJ, Breitkreutz D, et al. Expression and modification of keratins during terminal differentiation of mammalian epidermis. Curr Top Dev Biol. 1987;22:35–68. doi: 10.1016/s0070-2153(08)60098-8. [DOI] [PubMed] [Google Scholar]
- 4.Cao LH, Luo Y, Wen W, et al. A novel frameshift mutation in keratin 16 underlies Pachyonychia congenita with focal palmoplantar keratoderma. Br J Dermatol. 2011;165:1145–7. doi: 10.1111/j.1365-2133.2011.10450.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Jaeger K, Den Z, et al. Mice expressing a mutant Krt75 (K6hf) allele develop hair and nail defects resembling pachyonychia congenita. J Invest Dermatol. 2008;128:270–279. doi: 10.1038/sj.jid.5701038. [DOI] [PubMed] [Google Scholar]
- 6.Dahl PR, Daoud MS, Su WP. Jadassohn-Lewandowski syndrome (pachyonychia congenita) Semin Dermatol. 1995;14:129–134. doi: 10.1016/s1085-5629(05)80008-2. [DOI] [PubMed] [Google Scholar]
- 7.Del Bino S, Vioux C, Rossio-Pasquier P, et al. Ultraviolet B induces hyperproliferation and modification of epidermal differentiation in normal human skin grafted on to nude mice. Br J Dermatol. 2004;150:658–667. doi: 10.1111/j.0007-0963.2004.05886.x. [DOI] [PubMed] [Google Scholar]
- 8.Depianto D, Kerns ML, Dlugosz AA, et al. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat Genet. 2010;42:910–914. doi: 10.1038/ng.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025–1028. doi: 10.1038/jid.2010.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gruber R, Wilson NJ, Smith FJ, et al. Increased pachyonychia congenita severity in patients with concurrent keratin and filaggrin mutations. Br J Dermatol. 2009;161:1391–1395. doi: 10.1111/j.1365-2133.2009.09471.x. [DOI] [PubMed] [Google Scholar]
- 11.Haber RM, Drummond D. Pachyonychia congenita with laryngeal obstruction. Pediatr Dermatol. 2011;28:429–432. doi: 10.1111/j.1525-1470.2011.01232.x. [DOI] [PubMed] [Google Scholar]
- 12.Hardman MJ, Sisi P, Banbury DN, et al. Patterned acquisition of skin barrier function during development. Development. 1998;125:1541–1552. doi: 10.1242/dev.125.8.1541. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 14.Leachman SA, Kaspar RL, Fleckman P, et al. Clinical and pathological features of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:3–17. doi: 10.1111/j.1087-0024.2005.10202.x. [DOI] [PubMed] [Google Scholar]
- 15.Leigh IM, Navsaria H, Purkis PE, et al. Keratins (K16 and K17) as markers of keratinocyte hyperproliferation in psoriasis in vivo and in vitro. Br J Dermatol. 1995;133:501–511. doi: 10.1111/j.1365-2133.1995.tb02696.x. [DOI] [PubMed] [Google Scholar]
- 16.Liao H, Sayers JM, Wilson NJ, et al. A spectrum of mutations in keratins K6a, K16 and K17 causing pachyonychia congenita. J Dermatol Sci. 2007;48:199–205. doi: 10.1016/j.jdermsci.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 17.McGowan KM, Coulombe PA. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J Cell Biol. 1998;143:469–486. doi: 10.1083/jcb.143.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGowan KM, Tong X, Colucci-Guyon E, et al. Keratin 17 null mice exhibit age- and strain-dependent alopecia. Genes Dev. 2002;16:1412–1422. doi: 10.1101/gad.979502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLean WH, Rugg EL, Lunny DP, et al. Keratin 16 and keratin 17 mutations cause pachyonychia congenita. Nat Genet. 1995;9:273–278. doi: 10.1038/ng0395-273. [DOI] [PubMed] [Google Scholar]
- 20.Moll R, Franke WW, Volc-Platzer B, et al. Different keratin polypeptides in epidermis and other epithelia of human skin: a specific cytokeratin of molecular weight 46,000 in epithelia of the pilosebaceous tract and basal cell epitheliomas. J Cell Biol. 1982;95:285–295. doi: 10.1083/jcb.95.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paladini RD, Takahashi K, Bravo NS, et al. Onset of re-epithelialization after skin injury correlates with a reorganization of keratin filaments in wound edge keratinocytes: defining a potential role for keratin 16. J Cell Biol. 1996;132:381–397. doi: 10.1083/jcb.132.3.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reichelt J, Magin TM. Hyperproliferation, induction of c-Myc and 14-3-3sigma, but no cell fragility in keratin-10-null mice. J Cell Sci. 2002;115:2639–2650. doi: 10.1242/jcs.115.13.2639. [DOI] [PubMed] [Google Scholar]
- 23.Shamsher MK, Navsaria HA, Stevens HP, et al. Novel mutations in keratin 16 gene underly focal non-epidermolytic palmoplantar keratoderma (NEPPK) in two families. Hum Mol Genet. 1995;4:1875–1881. doi: 10.1093/hmg/4.10.1875. [DOI] [PubMed] [Google Scholar]
- 24.Smith FJ, Fisher MP, Healy E, et al. Novel keratin 16 mutations and protein expression studies in pachyonychia congenita type 1 and focal palmoplantar keratoderma. Exp Dermatol. 2000;9:170–177. doi: 10.1034/j.1600-0625.2000.009003170.x. [DOI] [PubMed] [Google Scholar]
- 25.Smith FJ, Liao H, Cassidy AJ, et al. The genetic basis of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:21–30. doi: 10.1111/j.1087-0024.2005.10204.x. [DOI] [PubMed] [Google Scholar]
- 26.Swensson O, Langbein L, McMillan JR, et al. Specialized keratin expression pattern in human ridged skin as an adaptation to high physical stress. Br J Dermatol. 1998;139:767–775. doi: 10.1046/j.1365-2133.1998.02499.x. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Folmer J, Coulombe PA. Increased expression of keratin 16 causes anomalies in cytoarchitecture and keratinization in transgenic mouse skin. J Cell Biol. 1994;127:505–520. doi: 10.1083/jcb.127.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson NJ, Messenger AG, Leachman SA, et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol. 2009;130:425–429. doi: 10.1038/jid.2009.215. [DOI] [PubMed] [Google Scholar]
- 29.Wojcik SM, Longley MA, Roop DR. Discovery of a novel murine keratin 6 (K6) isoform explains the absence of hair and nail defects in mice deficient for K6a and K6b. J Cell Biol. 2001;154:619–630. doi: 10.1083/jcb.200102079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wollina U, Schaarschmidt H, Funfstuck V, et al. Pachyonychia congenita. Immunohistologic findings. Zentralbl Pathol. 1991;137:372–375. [PubMed] [Google Scholar]
- 31.Wong P, Colucci-Guyon E, Takahashi K, et al. Introducing a null mutation in the mouse K6alpha and K6beta genes reveals their essential structural role in the oral mucosa. J Cell Biol. 2000;150:921–928. doi: 10.1083/jcb.150.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wong P, Domergue R, Coulombe PA. Overcoming functional redundancy to elicit pachyonychia congenita-like nail lesions in transgenic mice. Mol Cell Biol. 2005;25:197–205. doi: 10.1128/MCB.25.1.197-205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.