

Abstract
The currently accepted scheme for reactive oxygen species production during ischemia/reperfusion injury is characterized by a deleterious mitochondria-derived burst of radical generation during reperfusion; however, recent examination of the penumbra suggests a central role for NADPH-oxidase (Nox)-mediated radical generation during the ischemic period. Therefore, we utilized a novel in vitro model of the penumbra to examine the free radical profile of ischemic murine hippocampal neurons using electron paramagnetic resonance spectroscopy, and also the role of Nox in this generation and in cell fate. We report that free radical production increased ~ 75% at 2-hrs of ischemia, and this increase was abolished by: (1) scavenging of extracellular free radicals with superoxide dismutase (SOD), (2) a general anion channel antagonist, or (3) the Nox inhibitor apocynin. Similarly, at 24-hrs of ischemia, [ATP] decreased > 95% and vital dye uptake increased 6-fold relative to controls; whereas apocynin, the Cl− channel antagonist 5-nitro-2-(3-phenylpropylamino)-benzoate (NPPB), or the free radical scavenger N-acetyl cysteine (NAC) each provided moderate neuroprotection, ameliorating 13–32% of [ATP]-depletion and 19–56% of vital dye uptake at 24-hrs. Our results support a cytotoxic role for Nox-mediated free radical production from penumbral neurons during the ischemic period.
Keywords: neuroprotection, membrane permeability, two-photon microscopy, anion channels
1. Introduction
Reactive oxygen species (ROS)1 are important second messengers that regulate a myriad of intra- and extracellular signaling pathways (Kakkar and Singh, 2007). In ischemic stroke pathology neuronal ROS production spikes during reperfusion, which damages DNA locally, and activates deleterious immune and cell death pathways in surrounding tissue (Barzilai, 2007; Flamm et al., 1978; Kakkar and Singh, 2007). In addition, ROS generated during ischemia are a key contributor to the permeabilization of the blood brain barrier (BBB), which leads to edema and contributes to the formation and expansion of the penumbra following stroke (Fraser, 2011). The penumbra is the hypoperfused region surrounding the ischemic core, and the relatively slow propagation of cell death in the penumbra makes this region an attractive target for clinical rescue, particularly as the majority of stroke-related morbidity and mortality is attributable to progressive expansion of the ischemic core into the penumbra (Lo, 2008). The mechanism(s) of cell death here are poorly understood, but are likely initiated by deleterious alterations of the local perfusate following the release of cytoplasmic contents from ruptured core cells (Lo, 2008; Yao et al., 2007a). Early BBB breakdown following ischemic stroke is a key factor in the development of the penumbra since this event contributes significantly to edema and cell swelling, which in turn underlies cell rupture (Fraser, 2011). Therefore, ischemic ROS generation plays a pivotal role in the initiation and spread of cell death via a variety of cellular and intercellular interactions.
Historically it has been suggested that the primary source of ROS generation is mitochondrial superoxide ( ) (Liu et al., 2002). Another important free radical source are plasma membrane-bound proteins such as the NADPH oxidase (Nox), which is a principle enzyme for the production of critical to intracellular signaling and cell death during ischemic stress, and which underlies ROS-mediated BBB breakdown following stroke (Fraser, 2011; Hong et al., 2006; Kahles et al., 2007). Indeed, neuronal Nox mRNA and protein expression are increased following middle-cerebral artery occlusion (MCAO), whereas BBB and cellular permeability, infarct spread, and neuronal apoptosis are all reduced or abolished by Nox inhibition with apocynin, or by knockout of Nox1, Nox4, or the Nox family subunit gp91phox (Hong et al., 2006; Kahles et al., 2007; Kahles et al., 2010; Tang et al., 2008; Vallet et al., 2005; Walder et al., 1997; Wang et al., 2006).
Regardless of the source of generation, intracellularly derived ROS gain access to the extracellular space primarily via plasmalemmal anion channels (Hawkins et al., 2007), and since elevated extracellular ROS can induce cell injury and death, it is reasonable to assume that preventing ROS efflux would limit the spread of cell death following insult. Indeed, numerous studies in a variety of pathological models have demonstrated protective effects of anion channel inhibition against ROS-mediated inflammation (Shimizu et al., 2004; Wang et al., 2005). Therefore, regulation of ROS transmission via blockade of anion channels is an attractive target for clinical intervention and may help limit the spread of cell death in the ischemic penumbra.
Previous examinations of ROS generation in ischemic brain have predominantly utilized acute models of focal ischemia that mimic the infarct core, where blood flow is occluded and cells are anoxic (Shi and Liu, 2007). Conversely, blood flow to the penumbral region is hypoperfused, but not blocked; and thus penumbral cells are hypoxic, and likely exhibit a different free radical profile (Shi and Liu, 2007). Due to technical limitations, ROS generation from penumbral neurons cannot be measured in vivo (Kahles et al., 2007); therefore, we used an electron paramagnetic resonance (EPR) spin-trapping assay to determine the effect(s) of a penumbral mimic (ischemic solution: IS (Yao et al., 2007a)) on ROS production in vitro. In addition, we tested the role of anion channels in ischemic ROS efflux using 4,4-diisothiocyanatostilbenedisulphonic acid (DIDS); since it is widely used as a general antagonist of anion channels (Hawkins et al., 2007), and because there is evidence that DIDS provides cytoprotection against ischemic insults via regulation of anion channels (Wang et al., 2005; Yao et al., 2009). Finally, we examined the cytoprotective effects of limiting plasmalemmal ROS generation during ischemic insult by anion channel or Nox antagonism, or by scavenging ROS with N-acetyl cysteine (NAC).
2. Results
2.1. Ischemia increases extracellular ROS accumulation
ROS generation in IS-challenged neurons increased ~ 75% relative to controls (EPR intensity (a.u.): 744.0 ± 82.0 vs. 427.6 ± 27.0, F(2,35) = 7.8, Fig. 1A&B), and computer simulations indicated that the observed signal was due to spin trapping of both hydroxyl and . To determine the relative contribution to the ischemic change in total radical production of intracellular ROS accumulation compared to extracellular ROS efflux, we treated samples with chromium oxalate (CrOx, 1.25 μg/μl) or superoxide dismutase (SOD, 100 U/ml), which dissipate only the resonance signal of, or scavenge, extracellular ROS, respectively (Han et al., 2001). In control experiments, CrOx or SOD decreased ROS generation 30–35%, indicating that at rest, intracellular versus extracellular ROS accumulation occurs at a 2:1 ratio (CrOx: 263.3 ± 44.4, F(2,26) = 4.7; SOD: 284.3 ± 12.6, F(2,23) = 4.18, Fig. 1). Conversely, either compound entirely abolished the ischemic increase in ROS generation (IS + CrOx: 273.3 ± 12.6, F(2,24) = 8.7, IS + SOD: 272.3 ± 35.7, F(2,22) = 4.8), and total ROS generation when CrOx or SOD were used to ablate the extracellular ROS signal was not different in IS relative to controls; indicating that the change in ROS generation during ischemia is attributable to extracellular accumulation of ROS.
Figure 1. IS increases free radical production from neurons.
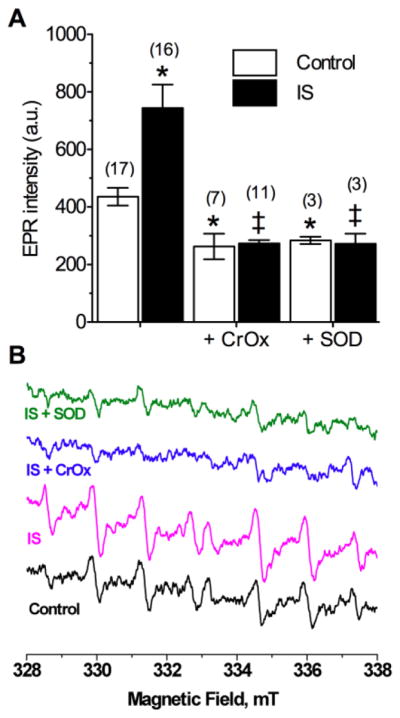
(A) Summary of EPR-revealed changes in superoxide production from neurons treated as indicated for 2-hrs. (B) Raw EPR traces from (A). Parentheses ( ) indicate n – values. Data are mean ± SEM. Asterisks (*) indicate significant difference from control; double daggers indicate significant difference from IS-alone (p < 0.05).
2.2. Anion channel-mediated efflux of Nox-derived radicals underlies ischemic ROS production
In controls, DIDS (400 μM) reduced ROS generation to the same degree as CrOx or SOD (290.0 ± 40.0, F(2,28) = 5.5, Fig. 2A&B), and DIDS in the presence of CrOx or SOD did not reduce ROS generation beyond the effect of any of these compounds alone (DIDS + CrOx: 279.6 ± 14.7, F(2,26) = 4.8; DIDS + SOD: 253.0 ± 3.5, F(2,22) = 4.2,). Similarly, during ischemia, DIDS-treatment reduced total radical generation to control levels (396.3 ± 26.7, F(2,31) = 7.9); however, generation remained greater than in normoxic DIDS-treated neurons. Therefore during ischemia, and with anion channels blocked, ROS accumulate in the cytosol. Similarly, the general Nox inhibitor apocynin (Apo: 10 μM) reduced the IS-mediated increase in ROS generation to control levels (420.0 ± 27.7, F(2,23) = 3.6, Fig. 2A&B), while co-treatment of DIDS with Apo did not further reduce ROS (362.0 ± 72.0).
Figure 2. DIDS or Apo prevents IS-mediated increase of ROS generation.
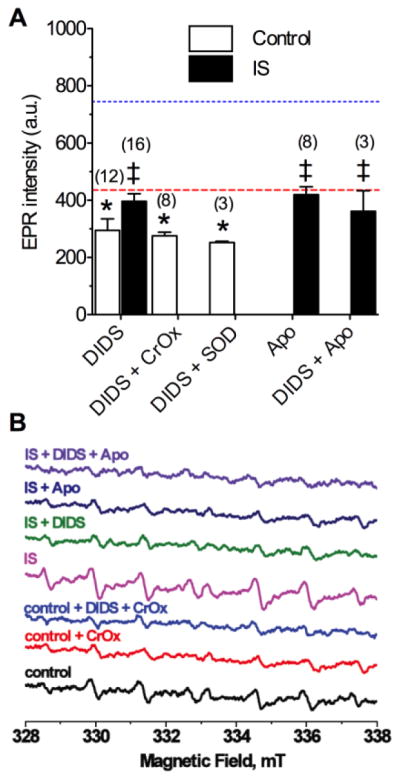
(A) Summary of EPR-revealed changes in superoxide production from neurons treated as indicated for 2-hrs. (B) Raw EPR traces from (A). Parentheses ( ) indicate n – values. Data are mean ± SEM. Dashed and dotted lines indicate normoxic and IS controls from Fig. 1A, respectively. Asterisks (*) indicate significant difference from control (Fig. 1A); double daggers indicate significant difference from IS-alone (Fig. 1A; p < 0.05).
2.3. DIDS does not permeate intact neurons
DIDS blocks ROS transmission across multiple lipid membranes (Beavis and Davatol-Hag, 1996; Hawkins et al., 2007); therefore, to assess the penetration of DIDS into neurons, we employed 2-photon confocal microscopy and took advantage of the natural fluorescence of DIDS molecules (Eisinger et al., 1982). In control experiments DIDS did not permeate plasma membranes through 2-hrs (Fig. 3A, n = 4 for each), whereas in IS, DIDS fluorescence was observed in some cells. To determine whether this penetration was due to membrane degradation, we also measured propidium iodide (PI) uptake. At 2-hrs, ~ 15% of neurons took up PI, and this uptake coincided with cellular DIDS fluorescence (Fig. 3B, n = 10–20 for each). Therefore, the accumulation of DIDS in some cells at 2-hrs is due to membrane degradation, and DIDS-mediated inhibition of ROS production from intact cells at this time-point is likely due to extracellular actions of DIDS.
Figure 3. DIDS penetrates damaged, but not intact neuronal membranes.

(A) Two-photon confocal DIC and Z-stack projection images (2 microns total) of neurons treated without DIDS, or with DIDS during normoxia or IS. Upper panel: DIC images. Lower panel: DIC images (grey) overlaid with PI (red) and DIDS (blue) fluorescent Z-stack projection images. Images are representative of 4 separate experiments for each treatment. (B) Summary of PI uptake. Data are mean ± SEM from 10–20 replicates per treatment. Asterisks (*) indicate significant difference from control (p < 0.05).
2.4. Nox inhibition or ROS scavenging provide moderate neuroprotection against ischemic insult
Next, we assessed the effects of IS ± DIDS ± Apo on neuronal viability. Relative to experimental onset (t = 0 hrs), control cells maintained > 85% of their [ATP] through 24-hrs; whereas [ATP] dropped progressively to 89.2 ± 0.1, 34.0 ± 3.5, and 3.9 ± 2.0% following 2-, 12-, and 24-hrs IS-treatment, respectively (Fig. 4A, n = 8–10 for each treatment). DIDS did not preserve [ATP] in IS-treated neurons at 24-hrs (Fig. 4C, n = 10 for each), likely due to secondary intracellular actions of DIDS on cells with damaged membranes at this time-point (Fig. 3B). However, another general Cl- channel antagonist, 5-nitro-1-(3-phenylpropylamino)benzoic acid (NPPB, 100 μM) increased [ATP] 3-fold relative to IS-alone and decreased IS-mediated PI uptake 18.8% (Figs. 3B,4C&D, n = 16 for each). Similarly, Apo or the ROS scavenger NAC had minimal effects on [ATP] in control experiments, but in ischemia increased [ATP] 7- and 10-fold, respectively, and ameliorated 25.7 ± 3.2 and 56.1 ± 4.5% of IS-mediated PI uptake relative to IS-alone (Fig. 4B–D, n = 3–10 for each [NAC]). When ischemic neurons were treated with both Apo and NAC simultaneously, their neuroprotective effects were not additive.
Figure 4. Nox inhibition or ROS scavenging is moderately protective against prolonged ischemic insult.

(A) Summary of [ATP] changes. (B) Dose-response curve of [ATP] vs. [NAC] at 24-hrs. (C) Summary of [ATP] changes from neurons treated as indicated for 24-hrs. Red and blue dashed lines represent baseline and ischemic controls, respectively, from Fig. 4A. (D) Summary of PI uptake by neurons treated as indicated for 24-hrs. Red and blue dashed lines represent baseline and ischemic controls, respectively, from Fig. 3B. Data are mean ± SEM from 8–10 replicates per treatment. Asterisks (*) indicate significant difference from control (Fig. 4A); double daggers indicate significant difference from IS (Fig. 4A; p < 0.05).
2.5. Ischemic Solution induces mixed modes of cell death in cultured neurons
In the infarct core, cell death proceeds primarily via necrosis due to excitotoxicity, mediated by over-activation of excitatory glutamatergic 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid and N-methyl-D-aspartic acid receptors (AMPARs, NMDARs) (Choi and Rothman, 1990). Conversely, in the penumbra, cell death is thought to occur via a mixture of cell death modalities, including necrosis, apoptosis, and autophagy (Lo, 2008; Rami, 2008; Rami and Kogel, 2008). In our 2-photon experiments, we observed moderate vital dye uptake at 2-hrs (Fig. 3A&B), consistent with the occurrence of rapid necrosis in some cells treated with this mimic solution. Therefore, we next examined the role of necrotic cell death in the IS model by antagonizing AMPARs and NMDARs with 6-cyano-7-nitroquinioxaline-2,3-dione (CNQX) and (2R)-amino-5-phosphonovaleric acid (APV), respectively; and also of apoptosis using the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK).
In vital dye exclusion assays, none of these treatments had a significant effect on cell viability during control experiments, whereas during IS-treatment, higher concentrations of CNQX + APV (50 μM each) reduced PI uptake ~ 20%, Z-VAD-FMK (5 or 50 μM) ameliorated ~ one third of IS-mediated PI uptake at 24-hrs (Fig. 5A, n = 16–32 for each). To confirm the occurrence of apoptosis in this model, we next examined the expression of Lamin A, a commonly studied marker of apoptosis whose cleavage is indicative of caspase 6 activation (Oberhammer et al., 1994). In Western blot analysis of Lamin A expression, we observed that IS treatment (6-hrs) increased Lamin A ~ 15-fold relative to controls, and that this increase was abolished by co-treatment with Z-VAD-FMK (Fig. 5B&C, n = 3).
Figure 5. Multiple cell death pathways are activated in IS-treated neurons.

(A) Summary of PI uptake by neurons treated as indicated for 24-hrs. Red and blue dashed lines represent baseline and ischemic controls, respectively, from Fig. 3B. Data are mean ± SEM from 16–32 replicates per treatment. (B) Summary of Lamin A expression in neurons treated as indicated for 6-hrs. Data are normalized to expression of α-actin on the same blot. (C) Sample paired Western blot images of Lamin A α-actin expression from (B). Images are representative of 3 separate experiments. (D) Summary of EPR-revealed changes in superoxide production from neurons treated as indicated for 2-hrs. Parentheses ( ) indicate n – values. Asterisks (*) indicate significant difference from control; double daggers indicate significant difference from IS (p < 0.05).
Finally, since glutamate receptor activation and associated Ca2+ influx has been linked to ROS and reactive nitrogen species production (Akaike et al., 1999), we examined the effect of AMPAR and NMDAR inhibition on ROS production. In normoxia, application of CNQX + APV (50 μM each) increased ROS generation ~ 14% from 466.5 ± 34.2 to 545.0 ± 28.7 at 2 hrs, while during IS-perfusion, CNQX + APV similarly increased ROS generation ~ 23%, from 655.2 ± 18.2 to 854.2 ± 48.1 (a.u., Fig. 5D, n = 4 for each).
3. Discussion
We demonstrate that deleterious ROS generation is increased in hippocampal neurons treated with an in vitro ischemic penumbral mimic perfusate, and that this increase is primarily due to efflux of Nox-derived ROS via plasma membrane anion channels. This conclusion is supported by our observations that anion channel or Nox antagonism, or ROS scavenging, each: (1) prevent IS-mediated increases in ROS generation, and (2) partially ameliorate IS-mediated neurotoxicity, in non-additive manners. In neurons, Nox subunits are found at various lumen membranes, and following prolonged ischemia, are translocated to plasma, mitochondrial, and endoplasmic reticulum membranes (Zhu et al., 2007), and therefore contribute to both cytosolic and extracellular ROS generation. Our results suggest a predominant role for cytosolic Nox-derived ROS production in the penumbra since co-antagonism of anion channels and Nox did not reduce EPR spectra beyond either treatment alone, as would be expected if ROS were produced by plasma membrane-bound Nox. Therefore, unlike in anoxic infarct core cells where ROS production is low during ischemia and cytotoxicity is predominately mediated by a deleterious burst of ROS during reperfusion (Shi and Liu, 2007), hypoperfused penumbral neurons maintain elevation of the generation, efflux, and extracellular accumulation of ROS, which is known to contribute to the spread of cell injury and death in the penumbra (Fraser, 2011).
ROS generation from penumbral neurons cannot presently be measured in vivo because tracers do not reach the ischemic tissue (Kahles et al., 2007); however, our results are consistent with ex vivo measurements from mouse brain following MCAO, where Nox-mediated production was increased in the penumbral region, but not in the core (Hong et al., 2006). Furthermore, our data are also consistent with measurements from penumbral epithelial cells and arteries in brain in which ROS production increases markedly during ischemia via a Nox-dependant mechanism (Kahles et al., 2007; Miller et al., 2009). Conversely, ROS production does not increase in ischemic core arteries. These results from three penumbral tissues further demonstrate that the profile of ROS generation in the penumbral region is markedly different from the infarct core, and in particular support a dominant contribution from Nox. Based on our present findings and these recent examinations, we conclude that Nox-mediated production is a key regulator of deleterious ROS signaling in the penumbral region, and that neuronal Nox contribute to this mechanism.
In the penumbra, oxidative phosphorylation persists due to a steady, albeit restricted supply of oxygen. This persistent reduced level of oxygen availability likely partially preserves cells’ ability to generate ATP, and thus opposes excitotoxic depolarization and early rapid necrotic cell death (Hochachka et al., 1996). However, this temporary preservation of ATP production allows for the induction of apoptosis and autophagy during prolonged insult (Leist et al., 1997), as occurs in the penumbra. As a result, a variety of cell death mechanisms appear to be concomitantly activated in this model (Lo, 2008; Rami, 2008; Rami and Kogel, 2008, present report), and this explains why cell viability was only partially preserved in our experiments by ROS scavenging, or by glutamate receptor or caspase antagonism.
In conclusion, we have used the present gold standard for free radical measurements (EPR), to record ROS production from live neurons treated with a novel ischemic penumbral mimic perfusate for the first time. We conclude that: (1) neuronal ROS production is increased during IS and this increase is manifested extracellularly, (2) this production is due to increased Nox-derived superoxide production, (3) the efflux of Nox-derived superoxide occurs via anion channels, (4) Nox inhibition provides mild protection against IS insult in neurons, and (5) neuronal cell death is mediated by a variety of pathways during IS insult.
Experimental Procedure
4.1. Cell cultures
HT22 mouse hippocampal neurons (a generous gift from Dr. Pam Maher, The Salk Institute, La Jolla, CA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, ATCC) supplemented with 10% bovine calf serum (Hyclone, Santa Clara, CA) and 100 U/ml penicillin/streptomycin (Invitrogen, Carlsbad, CA) and grown at 37°C in a 5% CO2 incubator. Cells were grown for 5–8 passages and split when they reached 60–80% confluence. For experiments, cells were seeded into 96-well microplates (Corning, Lowell, MA), glass-bottom 35-mm culture dishes (MatTek, Ashland, MA), or poly-L-lysine coated custom designed plastic coverslips precisely cut to the dimensions of the EPR tissue culture chamber (Fisher, Pittsburgh, PA). Cells were allowed to grow to ~ 80% confluence before experimentation and samples were treated as specified in the experimental design section (below). To reduce shear stress, cells seeded into 96-well microplates were gently washed with a TECAN PW96/384 Washer (TECAN, San Jose, CA) and then examined visually to ensure cells had not been washed away.
4.2. Experimental design
Samples were treated for 2, 6, 12 or 24-hrs (as indicated) in two treatment groups: cell death-negative control (DMEM/F12 media (Invitrogen), pH 7.4, gassed with 21% O2, 5% CO2, balance N2), or an ischemic penumbral perfusate mimic (IS, in mM: K+ 64, Na+ 51, Cl− 77.5, Ca2+ 0.13, Mg2+ 1.5, glucose 3, glutamate 0.1, [315 mOsM, pH 6.5, 1.5% O2, 15% CO2, balance N2]) (Yao et al., 2007a; Yao et al., 2007b). APV (5 or 50 μM), CNQX (5 or 50 μM), CrOx (1.25 μg/μl), NAC (0.1–1000 μM), and SOD (100 U/ml) were dissolved in water. SOD does not penetrate the cell membrane and therefore specifically scavenges extracellular superoxide (Mao and Poznansky, 1992). Apo (10 mM), DIDS (400 μM), NPPB (100 μM), and Z-VAD-FMK (5 or 50 μM) were dissolved in DMSO to a final bath [DMSO] < 0.01%, and all solutions were made fresh daily. Chemicals were purchased from Sigma unless otherwise indicated (Sigma-Aldrich, St. Louis, MO). It is important to note that atmospheric [O2] (21%) is hyperoxic relative to in situ tissue pO2. Nonetheless, 21% [O2] is commonly used as “normoxic” control in in vitro experimentation due to convenience and cost-related complications, and therefore we utilized 21% as our baseline control [O2] in order to be consistent with the majority of in vitro experiments previously published. Also, the in vivo ischemic penumbra is a heterogeneous milieu in which ionic, pH, metabolic substrate, and gaseous derangements vary considerably depending on the severity of insult and the time since insult-onset. Therefore, this region is difficult to exactly duplicate in vitro. IS was designed to incorporate and mimic key changes previously described in the literature from in vivo examinations of the penumbral rim (the region of tissue immediately adjacent to the infarct core, and thus most effected by local cell rupture), and the individual effects of each of these alterations on cell viability has been carefully examined elsewhere (Yao et al., 2007a; Yao et al., 2007b).
4.3. Determination of superoxide production in live neuronal cultures by electron paramagnetic resonance (EPR) spectroscopy
Measurement of production in live neuronal cultures was carried out by EPR spin trapping method using 70 mM of the spin trap agent DEPMPO (5-(diethylphosphoryl)-5-methyl-1-pyrroline-N-oxide). DEPMPO readily permeates mouse heart, liver, and brain tissues in vivo and is evenly distributed within 15-mins (Liu et al., 1999); and DEPMPO-spin adducts diffuse through lipid bilayers effectively (Anzai et al., 2003). Samples were treated for 2-hrs before transferring the coverslip to the EPR tissue culture chamber containing the spin trap agent in 40 μL of normoxic or IS gas-equilibrated media. The EPR cell was then tightly covered to avoid media leakage and inserted in the EPR cavity of a MiniScope MS200 Benchtop spectrometer maintained at 37°C. EPR spectra were recorded using the following spectral settings: microwave frequency, 9.45 GHz; microwave power, 5 mW; modulation amplitude, 0.2 mT; modulation frequency, 100 kHz; sweep width, 15 mT centered at 335 mT; scan rate, 0.75 mT s−1 and each spectrum was the average of 10 scans.
EPR traces were quantified by measuring the relative signal amplitudes detected automatically using the Magnettech Analysis Software (Version 2.02; Berlin, Germany). The average amplitudes of the largest four peaks in each spectrum were computed to minimize the random noise effects on the individual peaks. Computer simulation was carried out to identify the radical species enveloped in the signals using published hyperfine coupling constants (Chalier and Tordo, 2002; Frejaville et al., 1995; Stolze et al., 2000a; Stolze et al., 2000b). Published hyperfine parameters of DEPMPO-OOH and DEPMPO-OH radical adducts are inserted into the WinSim 2002 program (http://www.niehs.nih.gov/research/resources/software/tools/index.cfm) including initial guess of the percentage contribution from each species, and the program is allowed to produce the resultant spectrum, or to fit the experimental spectrum with the calculated one. This procedure was used to confirm the assignment of the radical species observed via representative spectra.
4.4. DIDS permeation imaging
Samples were grown in glass-bottom 35-mm culture dishes. Prior to experimentation, cells were rinsed once in serum-free DMEM and then incubated for 2-hrs with 0.2% DMSO or DIDS in DMEM or IS, and then rinsed once with DMEM or IS. DIDS fluorescence (Ex/Em: 342/418 nm in water, with an emission Red-shift to > 450 nM when bound to protein (Eisinger et al., 1982)) was visualized by a Zeiss LSM510 META confocal laser scanning 2-photon microscope using a 40 X C-Apochromat (NA 1.2) water immersion objective. Excitation was achieved by a Coherent Mira 900 laser tuning to 800 nm. Fluorescence shorter than 490 nm was reflected by a long pass filter (NFT 490), and then filtered by a short pass filter with cutoff at 685 nm (KP685). After taking DIDS and DIC images, 10 μg/ml of propidium iodide (PI, Sigma, Ex/Em: 514/590 nm) was added, and images from the same view were taken 5 to 8 mins afterwards (excited by laser line 514 nm, and filtered by a long pass filter (LP560). Z-stacks were taken at 0.4 μm intervals. Z-projections from 4 to 5 optical sections were created by averaging pixel intensity at each pixel position using Image J (NIH). Composite pictures were generated using Image J from DIC (grey), DIDS (blue), and PI (red).
4.5. ATP luciferase assay
Total ATP content [ATP] was assessed in solid-bottom, black 96-well microplates (Corning) using PerkinElmer ATPlite Luminescence Assay System kits as specified by the manufacturers protocol (PerkinElmer, MA, USA) and a Bio-Tek PowerWave 340 microplate spectrophotometer (Bio-Tek, Winooski, VT). Equal numbers of cells were seeded into each well and standard curves were generated using serial dilutions of a known ATP standard provided in each kit. The sensitivity of the detector was calibrated to the luminescence of the highest [ATP] standard in each experiment. DIDS quenched luminescence in a dose-dependent fashion and this effect was quantified in a separate experiment by adding serial dilutions of DIDS to serial dilutions of the ATP standard and subtracting the resulting luminescence from ATP standard luminescence measurements in the absence of DIDS on the same plate. Results were corrected for this factor and then normalized to ATP luminescence recorded from control cells assayed at t = 0 hours. Microplate ATP luciferase experiments were repeated 10 times and each plate contained 16 replicate wells of each treatment group. Blank wells and cell-free wells containing each treatment perfusate were also included on each plate, and the final data is corrected for these factors.
4.6. Protein extraction and Western blotting
Samples grown in 150 cm2 culture flasks were treated as indicated in the results section, and then rinsed twice with PBS and detached from the matrix with a cell scrapper into ice-cold PBS. The resulting cell suspensions were centrifuged at 250 × g for 5 mins at 4°C, the supernatant was aspirated away, and cells were re-suspended in cell lysis buffer. Samples were then homogenized by vortexing for 60 seconds and proteins were extracted by incubation in lysis buffer with mixing at 4°C for 45 mins, followed by centrifugation for 10 mins at 14,000 × g at 4°C. Supernatants were taken as whole cell lysates and protein concentration was measured using a bicinchoninic acid kit, according to the manufacturer’s instructions (Sigma).
Equal amounts of protein (40 μg/well) were separated on 4–12% precast NuPAGE bis-Tris SDS-PAGE gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Immobilin-P; Millipore, Bedford, MA). Western blots were performed with antibodies against α actin and cleaved Lamin A (1:2000, Cell Signaling, Danvers, MA). Specific bands were visualized after incubation with the appropriate secondary antibodies (Invitrogen) using enhanced chemiluminescence (GE Healthcare/Amersham Biosciences, Buckinghamshire, UK). Densitometry of Western blots from each experimental group were obtained (n = 3 for each), and absolute values were normalized to α-actin. Results were analyzed in arbitrary units, comparing each value with that obtained from each respective α-actin measurement on each blot.
4.7. Vital dye exclusion membrane viability assay
Membrane viability was assessed as the ability of cells to exclude the vital dye propidium iodide (PI). The dose-dependent response of DIDS on IS-induced PI uptake was assessed using a high-throughput 96-well microplate-based assay. PI uptake was assessed immediately following experimental treatment on a Bio-Tek PowerWave 340 microplate spectrophotometer (Bio-Tek, Winooski, VT, Ex/Em: 485/630 nm), and analyzed using Gen 5 software (Bio-Tek). Microplate PI experiments were repeated 3–5 times and each plate contained 16 replicate wells each of treatment groups. Blank wells and cell-free wells containing each treatment perfusate with PI were also included on each plate, and the final data is corrected for these factors.
Statistics
Data were analyzed using one-way analysis of variance (ANOVA), followed by Dunnet’s post-test. Significances were indicated if P < 0.05 assuming two groups had an equal variance. Statistical analysis was performed using Prism software (GraphPad, San Diego, CA, USA).
Highlights.
We model free radical production in the ischemic penumbra, which is poorly understood.
We utilized EPR, the gold standard to assay ROS in live cells.
Deleterious Nox-mediated superoxide is produced by ischemic penumbral neurons.
Increased free radical generation contributes to ischemic neurotoxicity.
Cl channel or NADPH oxidase antagonists block this increase and are neuroprotective.
Acknowledgments
This work was supported in part by NIH 5P01HD032573 to GGH, NIA 1K25AG026379 to SSA, and a National Sciences and Engineering Research Council of Canada fellowship to MEP. We would like to thank Orit Gavriolov and Jacinta Lucero for technical assistance.
Footnotes
Key abbreviations: AMPAR - 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid receptor/Apo – apocynin/APV - (2R)-amino-5-phosphonovaleric acid/BBB – blood brain barrier/CNQX - 6-cyano-7-nitroquinioxaline-2,3-dione/CrOx – chromium oxalate/DIDS - 4,4-diisothiocyanatostilbenedisulphonic acid/EPR – electron paramagnetic resonance/IS – ischemic solution/MCAO – middle cerebral artery occlusion/NAC – N-acetyl cysteine/NMDAR – N-methyl-D-aspartic acid receptor/Nox – nicotinamide adenine dinucleotide phosphate H oxidase/NPPB - 5-nitro-1-(3-phenylpropylamino)benzoic acid/ - superoxide anion/PI – propidium iodide/ROS – reactive oxygen species/SOD – superoxide dismutase/Z-VAD-FMK - carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akaike A, Katsuki H, Kume T, Maeda T. Reactive oxygen species in NMDA receptor-mediated glutamate neurotoxicity. Parkinsonism Relat Disord. 1999;5:203–7. doi: 10.1016/s1353-8020(99)00038-3. [DOI] [PubMed] [Google Scholar]
- Anzai K, Aikawa T, Furukawa Y, Matsushima Y, Urano S, Ozawa T. ESR measurement of rapid penetration of DMPO and DEPMPO spin traps through lipid bilayer membranes. Arch Biochem Biophys. 2003;415:251–6. doi: 10.1016/s0003-9861(03)00260-1. [DOI] [PubMed] [Google Scholar]
- Barzilai A. The contribution of the DNA damage response to neuronal viability. Antioxid Redox Signal. 2007;9:211–8. doi: 10.1089/ars.2007.9.211. [DOI] [PubMed] [Google Scholar]
- Beavis AD, Davatol-Hag H. The mitochondrial inner membrane anion channel is inhibited by DIDS. J Bioenerg Biomembr. 1996;28:207–14. doi: 10.1007/BF02110652. [DOI] [PubMed] [Google Scholar]
- Chalier F, Tordo P. 5-Diisopropoxyphosphoryl-5-methyl-1-pyrroline N-oxide, DIPPMPO, a crystalline analog of the nitrone DEPMPO: synthesis and spin trapping properties. J Chem Soc, Perkin Trans 2. 2002;12:2110–2117. [Google Scholar]
- Choi DW, Rothman SM. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu Rev Neurosci. 1990;13:171–82. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- Eisinger J, Flores J, Salhany JM. Association of cytosol hemoglobin with the membrane in intact erythrocytes. Proc Natl Acad Sci U S A. 1982;79:408–12. doi: 10.1073/pnas.79.2.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamm ES, Demopoulos HB, Seligman ML, Poser RG, Ransohoff J. Free radicals in cerebral ischemia. Stroke. 1978;9:445–7. doi: 10.1161/01.str.9.5.445. [DOI] [PubMed] [Google Scholar]
- Fraser PA. The role of free radical generation in increasing cerebrovascular permeability. Free Radic Biol Med. 2011;51:967–77. doi: 10.1016/j.freeradbiomed.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Frejaville C, Karoui H, Tuccio B, Le Moigne F, Culcasi M, Pietri S, Lauricella R, Tordo P. 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide: a new efficient phosphorylated nitrone for the in vitro and in vivo spin trapping of oxygen-centered radicals. J Med Chem. 1995;38:258–65. doi: 10.1021/jm00002a007. [DOI] [PubMed] [Google Scholar]
- Han D, Williams E, Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 2001;353:411–6. doi: 10.1042/0264-6021:3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell. 2007;18:2002–12. doi: 10.1091/mbc.E06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A. 1996;93:9493–8. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Zeng JS, Kreulen DL, Kaufman DI, Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol. 2006;291:H2210–5. doi: 10.1152/ajpheart.01270.2005. [DOI] [PubMed] [Google Scholar]
- Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–6. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- Kahles T, Kohnen A, Heumueller S, Rappert A, Bechmann I, Liebner S, Wittko IM, Neumann-Haefelin T, Steinmetz H, Schroeder K, Brandes RP. NADPH oxidase Nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiol Dis. 2010;40:185–92. doi: 10.1016/j.nbd.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Kakkar P, Singh BK. Mitochondria: a hub of redox activities and cellular distress control. Mol Cell Biochem. 2007;305:235–53. doi: 10.1007/s11010-007-9520-8. [DOI] [PubMed] [Google Scholar]
- Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KJ, Miyake M, Panz T, Swartz H. Evaluation of DEPMPO as a spin trapping agent in biological systems. Free Radic Biol Med. 1999;26:714–21. doi: 10.1016/s0891-5849(98)00251-2. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80:780–7. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500. doi: 10.1038/nm1735. [DOI] [PubMed] [Google Scholar]
- Mao GD, Poznansky MJ. Electron spin resonance study on the permeability of superoxide radicals in lipid bilayers and biological membranes. FEBS Lett. 1992;305:233–6. doi: 10.1016/0014-5793(92)80675-7. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, De Silva TM, Mast AE, Hickey H, Williams JP, Broughton BR, Sobey CG. NADPH oxidase activity is higher in cerebral versus systemic arteries of four animal species: role of Nox2. Am J Physiol Heart Circ Physiol. 2009;296:H220–5. doi: 10.1152/ajpheart.00987.2008. [DOI] [PubMed] [Google Scholar]
- Oberhammer FA, Hochegger K, Froschl G, Tiefenbacher R, Pavelka M. Chromatin condensation during apoptosis is accompanied by degradation of lamin A+B, without enhanced activation of cdc2 kinase. J Cell Biol. 1994;126:827–37. doi: 10.1083/jcb.126.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A. Upregulation of Beclin 1 in the ischemic penumbra. Autophagy. 2008;4:227–9. doi: 10.4161/auto.5339. [DOI] [PubMed] [Google Scholar]
- Rami A, Kogel D. Apoptosis meets autophagy-like cell death in the ischemic penumbra: Two sides of the same coin? Autophagy. 2008;4:422–6. doi: 10.4161/auto.5778. [DOI] [PubMed] [Google Scholar]
- Shi H, Liu KJ. Cerebral tissue oxygenation and oxidative brain injury during ischemia and reperfusion. Front Biosci. 2007;12:1318–28. doi: 10.2741/2150. [DOI] [PubMed] [Google Scholar]
- Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(−) channel. Proc Natl Acad Sci U S A. 2004;101:6770–3. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolze K, Udilova N, Nohl H. Spin trapping of lipid radicals with DEPMPO-derived spin traps: detection of superoxide, alkyl and alkoxyl radicals in aqueous and lipid phase. Free Radic Biol Med. 2000a;29:1005–14. doi: 10.1016/s0891-5849(00)00401-9. [DOI] [PubMed] [Google Scholar]
- Stolze K, Udilova N, Nohl H. Lipid radicals: properties and detection by spin trapping. Acta Biochim Pol. 2000b;47:923–30. [PubMed] [Google Scholar]
- Tang XN, Cairns B, Cairns N, Yenari MA. Apocynin improves outcome in experimental stroke with a narrow dose range. Neuroscience. 2008;154:556–62. doi: 10.1016/j.neuroscience.2008.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallet P, Charnay Y, Steger K, Ogier-Denis E, Kovari E, Herrmann F, Michel JP, Szanto I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience. 2005;132:233–8. doi: 10.1016/j.neuroscience.2004.12.038. [DOI] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–8. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–9. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Wang X, Takahashi N, Uramoto H, Okada Y. Chloride channel inhibition prevents ROS-dependent apoptosis induced by ischemia-reperfusion in mouse cardiomyocytes. Cell Physiol Biochem. 2005;16:147–54. doi: 10.1159/000089840. [DOI] [PubMed] [Google Scholar]
- Yao H, Shu Y, Wang J, Brinkman BC, Haddad GG. Factors influencing cell fate in the infarct rim. J Neurochem. 2007a;100:1224–33. doi: 10.1111/j.1471-4159.2006.04299.x. [DOI] [PubMed] [Google Scholar]
- Yao H, Sun X, Gu X, Wang J, Haddad GG. Cell death in an ischemic infarct rim model. J Neurochem. 2007b;103:1644–53. doi: 10.1111/j.1471-4159.2007.04879.x. [DOI] [PubMed] [Google Scholar]
- Yao H, Felfly H, Wang J, Zhou D, Haddad GG. DIDS protects against neuronal injury by blocking Toll-like receptor 2 activated-mechanisms. J Neurochem. 2009;108:835–46. doi: 10.1111/j.1471-4159.2008.05838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Fenik P, Zhan G, Mazza E, Kelz M, Aston-Jones G, Veasey SC. Selective loss of catecholaminergic wake active neurons in a murine sleep apnea model. J Neurosci. 2007;27:10060–71. doi: 10.1523/JNEUROSCI.0857-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]