

Abstract
Epidermal growth factor receptor (EGFR)-mediated cell signaling is critical for mammary epithelial cell growth and survival; however, targeting EGFR has shown no or only minimal therapeutic benefit in patients with breast cancer. Here, we report a novel regulatory mechanism of EGFR signaling that may explain the low response rates. We found that breast tumor kinase (Brk)/protein-tyrosine kinase 6 (PTK6), a nonreceptor protein tyrosine kinase highly expressed in most human breast tumors, interacted with EGFR and sustained ligand-induced EGFR signaling. We demonstrate that Brk inhibits ligand-induced EGFR degradation through uncoupling activated EGFR from Cbl-mediated EGFR ubiquitination. In addition, upon activation by EGFR, Brk directly phosphorylated Y845 in the EGFR kinase domain, thereby further potentiating EGFR kinase activity. Experimental elevation of Brk conferred resistance of breast cancer cells to cetuximab (an EGFR-blocking antibody)-induced inhibition of cell signaling and proliferation, whereas knockdown of Brk sensitized the cells to cetuximab by inducing apoptosis. Our findings reveal a previously unknown role of Brk in EGFR-targeted therapy.
Keywords: EGFR, Brk, Cbl, Src, breast cancer
Introduction
Epidermal growth factor (EGF) receptor (EGFR) has been implicated in breast tumorigenesis for more than two decades on the basis of numerous experimental, clinical, and epidemiological studies; EGFR expression was reported to correlate with breast cancer prognosis over 25 years ago (Sainsbury et al., 1985). The most recent studies show that EGFR expression is correlated with breast cancer relapse in selected patients (Rimawi et al., 2010). However, novel molecular therapeutic approaches targeting EGFR have shown no or limited efficacy in breast cancer clinical trials, including in patients with the basal-like breast cancer subtype, in which high EGFR expression is common (Normanno et al., 2005; Corkery et al., 2009). Since EGFR is critically important for mammary epithelial cell growth and survival (Taketani and Oka, 1983), we hypothesized that there exist mechanisms in breast cancer that can render breast cancer cells insensitive to pharmacological inhibition of EGFR; identification of such mechanisms may help to improve response to EGFR-targeted therapy in breast cancer.
Activation of EGFR by its ligands at the cell surface is followed by internalization of the ligand-receptor complex via receptor-mediated endocytosis (Sorkin and Von Zastrow, 2009). After internalization, EGFR traffics through a series of endosomal compartments and is either recycled back to the plasma membrane for continued signaling or routed to the lysosomes for degradation. Ligand-induced EGFR internalization followed by lysosomal degradation is an important negative feedback mechanism for regulating the amplitude and kinetics of EGFR signaling. Failure of this mechanism for EGFR downregulation can result in enhanced receptor signaling, which may lead to cell transformation and tumorigenesis (Marmor and Yarden, 2004; Polo et al., 2004).
Although not necessary for EGFR internalization, ubiquitination of EGFR is required for routing the receptor to sorting endosomes and, subsequently, to the lysosomal degradative compartment (Huang et al., 2007). Casitas B-lineage lymphoma (Cbl) is a ubiquitin ligase that mediates ubiquitination of several growth factor receptors, including EGFR (Levkowitz et al., 1998). Cbl targets activated EGFR for ubiquitination and subsequent endosomal sorting mainly by direct binding to Y1045-phosphorylated EGFR and additionally by indirect binding to the receptor through the Grb2 adaptor protein (Waterman et al., 2002; Grovdal et al., 2004).
Breast tumor kinase (Brk; also known as protein-tyrosine kinase 6/PTK6) is a nonreceptor tyrosine kinase originally identified in a screening of protein-tyrosine kinases expressed in human melanocytes (Lee et al., 1993). It was later cloned from a metastatic breast cancer cell line (Mitchell et al., 1994). Brk is overexpressed in the majority of breast tumors, particularly in advanced invasive and metastatic tumors, but its expression is either low or undetectable in normal breast epithelial cells (Mitchell et al., 1994; Barker et al., 1997; Born et al., 2005; Ostrander et al., 2007). High expression of Brk is also found in several other cancer types, including melanoma (Easty et al., 1997) and colon cancer (Llor et al., 1999). Originally classified as a Src-related kinase, Brk lacks the Src-characteristic N-terminal myristoylation consensus sequences necessary for fatty acid acylation and membrane association, and its SH2 and SH3 domains are atypical (Qiu and Miller, 2002; Qiu and Miller, 2004). Brk is now considered a member of a novel family of soluble protein-tyrosine kinases distantly related to c-Src (Serfas and Tyner, 2003). Brk is activated by EGF stimulation of cells, and experimental elevation of Brk sensitizes immortalized nonmalignant human mammary epithelial cells to EGF-induced mitogenic effects (Kamalati et al., 1996) and protects cells from anoikis (Irie et al., 2010). Brk may regulate clathrin-mediated EGFR endocytosis via phosphorylation of centaurin δ-2 (also known as ARAP1) (Kang et al., 2010). Brk interacts with EGFR, and Brk also interacts closely with other members of the EGFR family—HER2 (Born et al., 2005; Xiang et al., 2008) and HER3 (Kamalati et al., 2000)—to enhance HER2-induced activation of cell signaling and to mediate EGF-stimulated HER3 phosphorylation. Brk can also interact with tyrosine kinase receptor downstream substrates, such as Akt (Zhang et al., 2005) and insulin receptor substrate-4 (Qiu et al., 2005), regulate heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells (Ostrander et al., 2007), phosphorylate STAT3 (Liu et al., 2006) and STAT5b (Weaver and Silva, 2007), and phosphorylate paxillin (Chen et al., 2004) and p190RhoGAP-A (Shen et al., 2008) to promote breast carcinoma growth, migration, and invasion.
In the current work, we investigated the molecular mechanisms underlying the direct interaction between EGFR and Brk. Our results support a novel model for the interaction, which helps to unravel the role of Brk in mediating cell responses to EGF stimulation and may lead to improved EGFR-targeted therapy in breast cancer.
Results
Brk sensitizes cancer cells to EGF stimulation through inhibiting ligand-induced EGFR degradation
Brk is commonly expressed in human breast cancer (Mitchell et al., 1994; Born et al., 2005; Ostrander et al., 2007). Analysis of a panel of 16 breast cancer cell lines revealed Brk in 11 of the 16 cell lines, often at high levels (Figure 1a). Immunohistochemical staining showed positive Brk staining in 67% (36/54) of breast cancer specimens from patients (Figure S1). To investigate the role of Brk in regulating EGFR, we used SUM102 cells, which were derived from an intraductal breast adenocarcinoma and are basal-like (estrogen receptor and progesterone receptor negative, HER2 nonamplified, and CK5/6 and EGFR positive) (Sartor et al., 1997; Forozan et al., 1999; Hoadley et al., 2007; Eck et al., 2009). SUM102 cells had a relatively low level of Brk compared to the levels in other Brk-expressing breast cancer cell lines and a detectable level of EGFR (Figure 1a). We found that experimental elevation of Brk substantially sensitized cells (SUM102-Brk) to EGF-induced stimulation of cell proliferation, compared to control vector-transfected cells (SUM102-neo) (Figure 1b). Experimental elevation of Brk raised basal levels of activation-specific phosphorylation of Akt and Erk and sensitized the cells to EGF-induced increases in Akt and Erk phosphorylation (Figure 1c). Similar results were found in non-breast-cancer cell lines, such as A431 human vulvar squamous carcinoma cells, which express an unusually high level of EGFR (Figure S2a). Conversely, knockdown of Brk expression markedly reduced EGF-induced activation of cell signaling in SUM102 cells as well as in A431 cells (Figure 1d and Figure S2b). These results strongly suggested that Brk sensitizes cells to EGFR-mediated signal transduction.
Figure 1.

Brk sensitizes breast cancer cells to EGF stimulation by inhibiting ligand-induced EGFR degradation. (a) Brk was overexpressed more often than EGFR in breast cancer cell lines. Expression of Brk and EGFR in the indicated breast cancer cells was detected by Western blotting. β-actin was used as a loading control. (b) Brk sensitizes breast cancer cells to EGF-induced cell proliferation. SUM102-neo and SUM102-Brk cells were cultured in the presence or absence of the indicated concentrations of EGF in 0.5% FBS medium for 5 days. Relative cell growth and survival were determined by an MTT assay. (c) Brk enhances EGF-induced activation of cell signaling. Following transient transfection with Brk or vector for 24 h, SUM102 cells were treated with the indicated concentrations of EGF in 0.5% FBS medium for 10 min and then harvested for Western blotting with the indicated antibodies. (d) Knockdown of Brk expression decreases EGF-induced activation of cell signaling. Forty-eight hours after knockdown of Brk by siRNA, SUM102 cells were treated with the indicated concentrations of EGF in 0.5% FBS medium for 10 min and then harvested for Western blotting with the indicated antibodies. (e) Brk sustains activated EGFR level and signaling. SUM102 cells were treated with 10 μM cycloheximide for 30 min prior to incubation with 10 nM EGF in 0.5% FBS medium for the indicated time periods. Cell lysates were harvested for Western blotting with the indicated antibodies. EGFR-Yp = total phosphorylated EGFR. (f) Brk inhibits EGF-induced EGFR degradation. 35S-labeled methionine metabolic-labeled SUM102 cells were pulse-chased for up to 120 min in the presence or absence of 10 nM EGF in 0.5% FBS medium. The levels of EGFR immunoprecipitated at 30, 60, and 120 min were quantified by densitometry and plotted against the pulse-chase time period. Note: The ratios in this figure and in other figures represent quantitative analysis of densitometric values of specific band intensities normalized to the densitometric value of the leftmost lane in the same gel, which has a densitometric value and was arbitrarily set at 1. All of the experiments in this figure and in other figures were repeated at least once with similar findings.
We found that experimental elevation of Brk markedly inhibited downregulation of EGFR after EGF-induced activation of EGFR tyrosine kinase in SUM102 breast cancer cells (Figure 1e) as well as in A431 non-breast-cancer cells (Figure S2c). We then performed a pulse-label and chase experiment to determine the half-life of EGFR upon EGF stimulation in SUM102 and A431 cells with and without Brk overexpression. Compared with control vector-transfected cells, in which over 80% of EGFR was degraded by the end of the chase period, the Brk-transfected SUM102 and A431 cells exhibited almost no change in the level of EGFR (Figure 1f and Figure S2d). These results strongly supported the conclusion that overexpression of Brk inhibits ligand-induced degradation of EGFR.
Brk inhibits ligand-induced EGFR degradation through inhibiting EGFR-Cbl association and Cbl-mediated EGFR ubiquitination
To elucidate the mechanism by which Brk inhibits EGFR degradation, we compared the levels of EGFR ubiquitination and the association of EGFR with Cbl, an E3 ubiquitin ligase known for regulating EGFR degradation (Levkowitz et al., 1998), in SUM102-neo and SUM102-Brk cells after EGF stimulation. The association between EGFR and Cbl was markedly reduced in SUM102-Brk cells compared with SUM102-neo cells at 15, 30, and 90 min after EGF stimulation (Figure 2a). Consistent with the reduction in EGFR-Cbl association, the level of EGFR ubiquitination was lower in SUM102-Brk cells than in SUM102-neo cells (Figure 2a). Conversely, knockdown of endogenous Brk expression in SUM102 cells markedly increased the levels of EGFR-Cbl association and EGFR ubiquitination (Figure 2b). These results strongly indicated that Brk can inhibit EGF-induced EGFR-Cbl association and subsequent Cbl-mediated EGFR ubiquitination.
Figure 2.

Brk inhibits EGF-induced EGFR-Cbl association and Cbl-mediated EGFR ubiquitination. (a) Brk inhibits EGF-induced EGFR-Cbl association and EGFR ubiquitination. SUM102 cells were transiently transfected with wild-type Brk or neo control vector for 24 h. The cells were then stimulated with 10 nM EGF in 0.5% FBS medium for the indicated time periods. EGFR immunoprecipitates (I.P. α EGFR) or whole cell lysates were subjected to Western blotting with the indicated antibodies. (b) Knockdown of Brk expression enhances EGF-induced EGFR-Cbl association and EGFR ubiquitination. SUM102 cells were subjected to Brk knockdown or control siRNA treatment for 48 h. The cells were then processed and analyzed as described in (a). (c) Constitutively active Brk inhibits EGFR-Cbl association and EGFR ubiquitination. SUM102 cells were transiently transfected with wild-type Brk, constitutively active mutant Brk-Y447F, kinase-dead mutant Brk-K219M, or neo control vector for 24 h. EGFR immunoprecipitates (I.P. α EGFR) or whole cell lysates were subjected to Western blotting with the indicated antibodies.
To ascertain whether this Brk-mediated inhibition of EGF-induced EGFR-Cbl association and subsequent EGFR ubiquitination was directly linked to Brk kinase activity and expression, we examined the effects of expression of wild-type Brk, a constitutively active Brk mutant (Brk-Y447F), and a kinase-dead Brk mutant (Brk-K219M) on the levels of EGFR-Cbl association and EGFR ubiquitination in SUM102 cells (Figure 2c). To permit visualization of the basal levels of EGFR-Cbl association and EGFR ubiquitination in unstimulated SUM102 cells, the blots in Figure 2c were overexposed compared to those in Figures 2a and 2b. We found that the level of EGFR-Cbl association was reduced in the cells transfected with wild-type Brk, more profoundly reduced in the cells transfected with Brk-Y447F, and only minimally reduced in the cells transfected with Brk-K219M. The changes in the level of EGFR ubiquitination correlated well with the reductions in the level of EGFR-Cbl association. Similar results were found in A431 cells (Figure S3a). Inhibition of EGFR ubiquitination with overexpression of wild-type Brk was stronger in A431 cells than in SUM102 cells, likely because the wild-type Brk was activated by autocrine TGF-α, which is known to exist in A431 cells (Figure S3a). Indeed, similar to the findings after EGF treatment, TGF-α-induced EGFR-Cbl association and EGFR ubiquitination were also inhibited by Brk (Figure S3b).
Together, these findings indicated that Brk inhibits ligand-induced EGFR degradation after EGFR activation through interfering with EGFR-Cbl association and subsequent Cbl-mediated EGFR ubiquitination and that the kinase activity of Brk plays an important role in the process.
Brk inhibits Cbl-mediated EGFR ubiquitination through competing with Cbl for binding to the Y1045 phosphorylation site on EGFR
To elucidate the mechanism by which Brk inhibits Cbl-EGFR association, we first examined the dependence of EGFR-Brk association on EGFR activity. EGF-induced EGFR-Brk association was strongly inhibited by a small-molecule EGFR tyrosine kinase inhibitor, gefitinib, in SUM102-Brk cells (Figure 3a) as well as in A431-Brk cells (Figure S4), indicating that EGFR kinase activity is required for EGFR-Brk association. Next, we asked whether Brk kinase activity is also required for EGFR-Brk association. Because no Brk specific inhibitor is currently available, we used a genomic approach to coexpress wild-type EGFR and wild-type Brk or wild-type EGFR and kinase-dead Brk (Brk-K219M) in Chinese hamster ovary (CHO) cells, which have no detectable level of endogenous Brk or EGFR. Experimental elevation of EGFR can induce receptor activation due to receptor homodimerization, which led to a moderate EGFR-Brk association in Brk immunoprecipitates from unstimulated cells cotransfected with wild-type EGFR and wild-type Brk (Figure 3b); the association was markedly increased after EGF stimulation. In contrast, in cells cotransfected with wild-type EGFR and kinase-dead Brk, no EGFR-Brk association was seen in unstimulated cells, and only a minimal level of EGFR-Brk association was seen after EGF stimulation. Similar results were seen in EGFR immunoprecipitates (Figure 3b), although the difference was not as marked as in Brk immunoprecipitates, possibly because immunoprecipitation of myc-tagged EGFR with anti-myc antibody was less complete than immunoprecipitation of Brk with anti-Brk antibody. In any case, the data supported the conclusion that Brk kinase activity is required for EGFR-Brk association.
Figure 3.
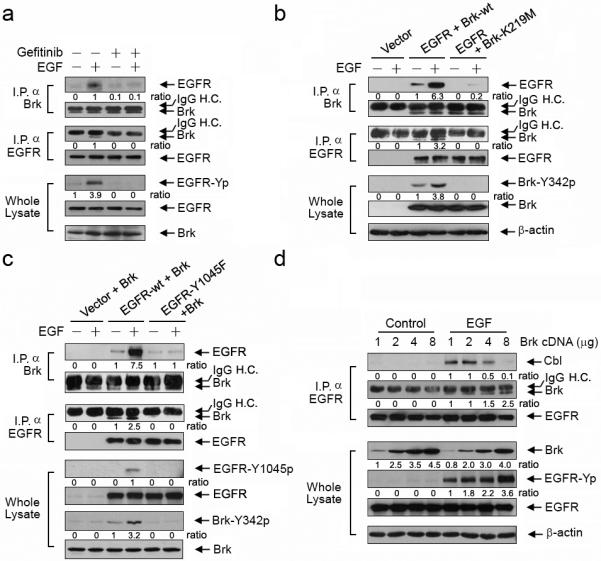
Brk binds to EGFR in both an EGFR kinase-dependent and a Brk kinase-dependent manner and competes with Cbl for binding to EGFR Y1045. (a) Brk binds to EGFR in an EGFR kinase-dependent manner. SUM102 cells were transiently transfected with wild-type Brk for 24 h and then exposed to 0.5 μM gefitinib or not for 16 h prior to 10 nM EGF in 0.5% FBS medium for 5 min as indicated. Brk immunoprecipitates (I.P. α Brk), EGFR immunoprecipitates (I.P. α EGFR), or whole cell lysates were subjected to Western blotting with the indicated antibodies. IgG H.C. = IgG heavy chain. (b) Brk binds to EGFR in a Brk kinase-dependent manner. CHO cells were transiently transfected with a control vector, cotransfected with wild-type EGFR and wild-type Brk, or cotransfected with wild-type EGFR and Brk-K219M for 24 h. The cells were then exposed to 10 nM EGF in 0.5% FBS medium for 5 min as indicated. Brk (I.P. α Brk) and EGFR immunoprecipitates (I.P. α EGFR) and whole cell lysates were subjected to Western blotting with the indicated antibodies. (c) Brk binds to EGFR and is activated by EGFR in an EGFR Y1045 phosphorylation-dependent manner. CHO cells were transiently cotransfected with wild-type Brk construct and one of neo vector, wild-type EGFR, or EGFR-Y1045F for 24 h. The cells were then exposed to 10 nM EGF in 0.5% FBS medium for 5 min as indicated. Brk (I.P. α Brk) and EGFR immunoprecipitates (I.P. α EGFR) and whole cell lysates were subjected to Western blotting with the indicated antibodies. (d) Brk competes with Cbl for binding to EGFR Y1045. CHO cells were transiently transfected with stepwise increasing DNA concentrations of Brk constructs for 24 h. The cells were stimulated with 10 nM EGF for 5 min. EGFR immunoprecipitates (I.P. α EGFR) and whole cell lysates were subjected to Western blotting with the indicated antibodies.
Because phosphorylated Y1045 on EGFR is a major docking site for Cbl (Levkowitz et al., 1998), we tested the hypothesis that Brk inhibits Cbl-mediated EGFR ubiquitination by competitively binding to Y1045-phosphorylated EGFR. We used CHO cells with coexpression of wild-type EGFR or EGFR-Y1045F mutant with Brk to eliminate potential interference from endogenous levels of related proteins. Using the same experimental approach as in Figure 3b, we found massive amounts of wild-type EGFR, but not EGFR-Y1045F, in Brk immunoprecipitates of cells after EGF stimulation (Figure 3c). Reciprocal EGFR immunoprecipitation showed that an increased amount of Brk coimmunoprecipitated with wild-type EGFR but not with EGFR-Y1045; however, as in Figure 3b, the difference was not as marked as in Brk immunoprecipitates. Furthermore, we found that activation-specific phosphorylation of Brk Y342 was increased in EGF-stimulated CHO cells cotransfected with Brk and wild-type EGFR but not in EGF-stimulated CHO cells cotransfected with Brk and mutant EGFR-Y1045F (Figure 3c). These findings supported a model wherein EGFR Y1045 phosphorylation is essential not only for EGFR-Brk association but also for activation of Brk after EGF treatment.
We next asked whether Brk and Cbl competitively bind to the same phosphorylated Y1045 site on EGFR. We correlated the level of EGFR-Cbl association with the level of EGFR-Brk association in CHO cells transfected with increasing amounts of Brk constructs. Figure 3d clearly shows an inverse relationship: with stepwise increases in the levels of Brk expression, the levels of EGFR-Brk association also increased, whereas the levels of EGFR-Cbl association correspondingly decreased. These findings indicated that Brk can compete with Cbl for binding to the phosphorylated Y1045 site on EGFR. Thus, Brk competes with Cbl for binding to Y1045-phosphorylated EGFR, thereby interfering with Cbl-mediated ubiquitination and subsequent degradation of EGFR.
In addition, we found a positive correlation between Brk cDNA dose-dependent expression and EGF-induced EGFR phosphorylation (Figure 3d, blot for EGFR-Yp), suggesting that Brk may have a direct role in regulating EGFR phosphorylation and functional consequences.
Brk phosphorylates EGFR on Y845 independently of Src or HER activity
As previously mentioned, experimental elevation of wild-type EGFR alone can induce EGFR autophosphorylation due to receptor homodimerization; however, we found that coexpression of EGFR with the constitutively active Brk mutant Brk-Y447F resulted in higher levels of total and site-specific EGFR tyrosine phosphorylation (particularly at Y845 and Y1045) than expression of EGFR alone or coexpression of EGFR with the kinase-dead Brk mutant Brk-K219M (Figure 4a; lanes 2-4, short exposures of blots of EGFR-Yp and EGFR-Y845p). This finding indicated that Brk-Y447 likely enhances EGFR autophosphorylation, directly phosphorylates EGFR, or both. Because wild-type Brk, when cotransfected with EGFR, can be moderately activated in CHO cells without EGF stimulation (Figure 3b, blot for Brk-Y342p), we used Brk-K219M instead of wild-type Brk as a negative control for Brk-Y447F in this experiment and the following experiments.
Figure 4.

Brk phosphorylates EGFR Y845 independently of the activities of the Src or HER families. (a) Brk-Y447F increases EGFR phosphorylation in an EGFR kinase-dependent manner but can increase EGFR Y845 phosphorylation independently of EGFR kinase activity. CHO cells were transiently cotransfected for 24 h with a construct containing neo control vector, wild-type EGFR (EGFR-wt), kinase-dead EGFR (EGFR-K721A), or EGFR-K721/Y845F double-mutant and a construct containing neo control vector, constitutively actively Brk (Brk-Y447F), or kinase-dead Brk (Brk-K219M) as indicated. Whole cell lysates were analyzed by Western blotting with the indicated antibodies. EGFR-Yp = total phosphorylated EGFR. Exp. = exposure time. (b and c) Brk-Y447F increases EGFR Y845 phosphorylation independently of Src or HER kinase activity. In (b), CHO cells were subjected to Src siRNA or control siRNA treatment for 48 h. The cells were then cotransfected for 24 h with EGFR-K721A and either Brk-Y447F or Brk-K219M. CHO cells transfected with a control vector or EGFR-wt were used as controls. In (c), CHO cells were cotransfected for 24 h with EGFR-K721A and control vector, Brk-Y447F, or Brk-K219M. The cells were then treated with 10 μM dasatinib or lapatinib or vehicle control (DMSO) as indicated for overnight. Whole cell lysates were analyzed by Western blotting with the indicated antibodies. (d) Brk directly phosphorylates Y845 of EGFR in vitro. GST-fusion proteins containing the kinase domain of EGFR-K721A or EGFR-K721A/Y845F were incubated with recombinant Brk in the presence of ATP for 30 min at 30°C. The products of this in vitro kinase reaction were analyzed by Western blotting with the indicated antibodies.
To determine whether EGFR can be a substrate for Brk, we used a kinase-dead EGFR mutant (EGFR-K721A), which produced no EGFR autophosphorylation when expressed alone (Figure 4a, lane 5). EGFR total and Y845 phosphorylation levels were higher when EGFR-K721A was coexpressed with Brk-Y447F than when EGFR-K721A was coexpressed with Brk-K219M (Figure 4a, lanes 5-7, long exposures of blots of EGFR-Yp and EGFR-Y845p; note: only the long-exposed films that had positive findings are shown). Although EGFR total and Y845 phosphorylation levels were low in the absence of EGFR kinase activity, this result clearly indicated that Brk-Y447F can phosphorylate EGFR Y845. No increased phosphorylation of EGFR on Y992, Y1045, Y1068, or Y1173 was detected by their respective phosphorylation site-specific antibodies (Yamaoka et al., 2011) when EGFR-K721A was coexpressed with Brk-Y447F (Figure 4a).
To further confirm that Y845 of EGFR can be phosphorylated by Brk-Y447F, we created an EGFR-K721A/Y845F double mutant. As expected, no Y845 phosphorylation was observed when EGFR-K721A/Y845F was coexpressed with Brk-Y447F in CHO cells; however, total tyrosine phosphorylation on EGFR-K721A/Y845F was not completely eliminated despite being clearly decreased (Figure 4a, lanes 8-10 of the short- and long-exposed EGFR-Y845p and EGFR-Yp blots). These findings indicated that Y845 of EGFR-K721A was phosphorylated when EGFR-K721A was coexpressed with Brk-Y447F but that additional tyrosine site(s) may also have been phosphorylated by Brk-Y447F.
Y845 of EGFR is a known Src phosphorylation site (Tice et al., 1999; Biscardi et al., 1999). We found that coexpression of EGFR-K721A and Brk-Y447F resulted in similar levels of Y845 phosphorylation on EGFR-K721A in cells with and without siRNA-mediated knockdown of Src (Figure 4b, lane 3 versus lane 5 of the EGFR-Y845p blot), indicating that Brk may phosphorylate EGFR Y845 independently of Src. Because the siRNA-mediated transient silencing of Src expression may be incomplete, and also because multiple members of the Src family, as well as the members of the HER family, may also phosphorylate Y845 of EGFR, we further treated cells with dasatinib (a Src family inhibitor) and lapatinib (a HER2/EGFR dual inhibitor) to confirm our findings. Figure 4c shows that, despite strong inhibition of Src by dasatinib (lanes 4-6 of the Src-Y416p blot), Y845 of EGFR-Y721A was still phosphorylated in cells cotransfected with EGFR-Y721A and Brk-Y447F (lane 5 of the EGFR-Y845p blot), whereas it was not phosphorylated in cells cotransfected with EGFR-Y721A and vector control (lane 4) or Brk-K219M (lane 6). There was a weak cross-inhibition of Brk-Y447F by dasatinib (lane 5 versus lane 2 or 8 of the Brk-Y342p blot; note: similar results were observed in cells treated with PP2, another Src inhibitor; data not shown). Thus, Y845 phosphorylation was less in dasatinib-treated cells (lane 5) than in vehicle-treated (lane 2) or lapatinib-treated cells (lane 8); however, the data clearly supported the conclusion that Y845 phosphorylation of EGFR in CHO cells cotransfected with Brk-Y447F and EGFR-Y721A is primarily Src independent.
Although CHO cells express HER2, we did not detect any phosphorylated HER2 when the cells were transfected with EGFR-Y721A and control vector, Brk-Y447F, or Brk-K219M (Figure 4c, blots of HER2 and HER2-p), nor did we detect any effect of lapatinib on Brk-Y447F-induced EGFR Y845 phosphorylation (lanes 7-9 of the EGFR-Y845p blot). This experiment rules out involvement of HER2 in Brk-Y447F-mediated Y845 phosphorylation of EGFR.
Lastly, to confirm that Brk can directly phosphorylate Y845 of EGFR, we conducted an in vitro Brk kinase assay by incubating GST fusion proteins containing the kinase domain of EGFR-K721A or EGFR-K721A/Y845F with a recombinant Brk protein in the presence or absence of ATP (Figure 4d). Consistent with the findings in Figure 4a, after incubation with recombinant Brk and ATP, Y845 EGFR phosphorylation was detected in the GST protein fused with EGFR-K721A kinase domain but not in the GST protein fused with EGFR-K721A/Y845F kinase domain, strongly indicating that Brk can directly phosphorylate Y845 of EGFR. Interestingly, the Y845-phosphorylated EGFR antibody also detected phosphorylated Brk, which was autophosphorylated in the presence of ATP. In vitro incubation of full-length EGFR-K721A and EGFR-K721A/Y845F proteins immunoprecipitated from CHO cells also confirmed phosphorylation of EGFR on Y845 as well as on some not-yet-identified sites by recombinant Brk (Figure S5); the additional phosphorylation sites will be determined in separate studies.
Brk phosphorylation of EGFR-Y845 potentiates EGFR functions
To investigate the role of Brk-induced EGFR Y845 phosphorylation in EGFR function, we analyzed EGF-induced association between EGFR and Brk in CHO cells cotransfected with wild-type Brk and either wild-type EGFR or EGFR-Y845F mutant. Figure 5a shows that the EGF-induced association between Brk and EGFR-Y845F was substantially less than the EGF-induced association between Brk and wild-type EGFR, suggesting that Brk-induced EGFR Y845 phosphorylation is important, although not essential, for EGFR-Brk association. Both Brk Y342 and EGFR Y1045 were phosphorylated following EGF stimulation of cells cotransfected with Brk and EGFR-Y845F mutant, but the levels were less than those in cells cotransfected with Brk and wild-type EGFR (Figure 5a).
Figure 5.

Brk promotes EGFR-Brk interaction through phosphorylating EGFR Y845. (a) Mutation of EGFR Y845 reduces EGF-induced EGFR-Brk association. CHO cells were transiently cotransfected with Brk and wild-type EGFR or EGFR-Y845F for 24 h and then treated with 10 nM EGF for 5 min or not. CHO cells transfected with control vector were used as controls. Brk and EGFR immunoprecipitates and whole cell lysates were subjected to Western blotting with the indicated antibodies. (b) Brk-Y447F induces EGFR Y1045 phosphorylation and promotes EGFR-Brk association through phosphorylating EGFR Y845. CHO cells were transiently cotransfected for 24 h with a construct containing neo vector, wild-type EGFR (EGFR-wt), EGFR-Y1045F, or EGFR-Y845F and a construct containing neo vector, Brk-Y447F, or Brk-K219M as indicated. Brk and EGFR immunoprecipitates and whole cell lysates were subjected to Western blotting with the indicated antibodies. (c) EGFR kinase activity is required for Brk-Y447F-induced EGFR Y1045 phosphorylation. CHO cells were cotransfected with wild-type Brk and control vector, Brk-Y447F, or Brk-K219M for 24 h. The cells were then treated with 0.5 μM gefitinib or vehicle control (DMSO) for 16 h. Whole cell lysates were subjected to Western blotting with the indicated antibodies.
Because EGF-induced association between EGFR and Brk is EGFR Y1045 phosphorylation dependent (Figure 3c), we next compared the levels of EGFR-Brk association in CHO cells expressing various combinations of EGFR constructs (wild type, EGFR-Y1045F, and EGFR-Y845F) and Brk constructs (Brk-Y447F and Brk-K219M) to further analyze the roles of Brk kinase activity and Brk-induced EGFR Y845 phosphorylation in EGFR-Brk association (Figure 5b). These experiments with various combinations of EGFR and Brk constructs produced three main findings. First, while there was only a minimal association between wild-type EGFR and kinase-dead Brk-K219M, there was a marked association between wild-type EGFR and constitutively active Brk-Y447F (Figure 5b, lanes 2-4 of the blots of EGFR for Brk immunoprecipitates [I.P. α Brk] and Brk for EGFR immunoprecipitates [I.P. α EGFR]), and phosphorylation of EGFR on both Y845 and Y1045 was higher with wild-type EGFR and Brk-Y447F than with wild-type EGFR and Brk-K219M (lanes 2-4 of the blots of EGFR-Y845p and EGFR-Y1045p). Second, mutation of EGFR Y1045 abolished the association between EGFR and Brk-Y447F (Figure 5b, lanes 5-7 versus lanes 2-4 of the blots of EGFR for Brk immunoprecipitates [I.P. α Brk] and vice versa) but did not affect Brk-Y447F-induced phosphorylation of EGFR Y845 (lane 3 versus lane 6 of the EGFR-Y845p blot). Compared with the result in Figure 3c, which showed that activation of wild-type Brk by EGF is EGFR Y1045 phosphorylation dependent, this finding with constitutively active Brk-Y447F indicated that, once activated, Brk can phosphorylate Y845 of EGFR independently of Y1045 phosphorylation. Third, mutation of EGFR Y845 markedly reduced the association between Brk-Y447F and EGFR (Figure 5b, lanes 8-10 versus lanes 2-4 of the blots of EGFR for Brk immunoprecipitates [I.P. α Brk] and vice versa) and also markedly reduced Brk-Y447F-induced phosphorylation of EGFR Y1045 (lane 3 versus lane 9 of the EGFR-Y1045p blot). These findings indicated that Brk-Y447F-induced EGFR Y1045 phosphorylation is dependent on prior Brk-Y447F-induced EGFR Y845 phosphorylation.
Our data in Figure 4 indicated that Brk-Y447F-induced EGFR Y1045 phosphorylation is EGFR kinase dependent because it was not seen with EGFR-K721A (Figure 4a, lanes 2-4 versus lanes 5-7 of the EGFR-Y1045p blot). In contrast, Brk-Y447F-induced EGFR Y845 phosphorylation was not EGFR kinase dependent because it was seen with EGFR-K721A (Figure 4a, lanes 5-7 of the long-exposed EGFR-Y845p blot). However, Brk-Y447F-induced EGFR Y845 phosphorylation was markedly higher on wild-type EGFR than on EGFR-K721A (Figure 4a, lane 3 versus lane 6 of the long-exposed EGFR-Y845p blot), indicating a role of EGFR kinase in Y845 phosphorylation. We thus further compared the levels of Brk-Y447F-induced EGFR Y845 and Y1045 phosphorylation in CHO cells with and without inhibition of EGFR kinase activity with gefitinib. Figure 5c shows that gefitinib treatment markedly decreased Brk-Y447F-induced EGFR Y845 phosphorylation, although EGFR Y845 phosphorylation could still be detected in gefitinib-treated cells (lane 6 versus lane 3 of the EGFR-Y845p blot). In contrast, Brk-Y447F-induced EGFR Y1045 phosphorylation was barely detected in the gefitinib-treated cells (lane 6 versus lane 3 of the EGFR-Y1045p blot). These findings confirmed that EGFR kinase activity is required for Brk-Y447F-induced EGFR Y1045 phosphorylation and indicated that EGFR kinase activity affects the level of but is not required for the occurrence of Brk-Y447F-induced EGFR Y845 phosphorylation. These findings also explained why Brk-Y447F can induce EGFR Y845 phosphorylation but the phosphorylation is much weaker in the kinase-dead EGFR-K721A than in the kinase-functional wild-type EGFR (Figure 4a). In addition, it appears that there was a slightly increased phosphorylation of EGFR Y1045 with Brk-K129M, supporting a previously described adaptor function for the kinase-dead protein (Harvey and Crompton, 2003; 2004).
Taken together, these findings indicated that Brk phosphorylates EGFR on Y845, which in turn enhances EGFR phosphorylation on Y1045, which is EGFR kinase dependent. Phosphorylation of EGFR Y1045 leads to further increase in EGFR-Brk association and Brk activation, which decreases EGFR-Cbl association and EGFR ubiquitination. Thus, the repetitive cycles between Brk activation and EGFR Y845/Y1045 phosphorylation constitute a positive feedback loop, through which EGFR activation-induced cell signaling is sustained and amplified.
Knockdown of Brk sensitizes breast cancer cells to EGFR-targeted therapy
The above-described novel functions of Brk suggested that Brk could sustain activated EGFR signaling and thus augment cell response to EGF stimulation and reduce the dependence of cells on new EGFR activation. To substantiate this critical role of Brk in breast cancer cells, we knocked down endogenous Brk in SUM102 breast cancer cells and confirmed that the knockdown did lower the level of EGF-induced EGFR phosphorylation at Y845, Y1045, Y1068 and Y1173 (Figure 6a). Knockdown of Brk sensitized SUM102 cells to cetuximab, an EGFR-blocking antibody that is approved for treatment of several types of human solid tumors, as shown by greater induction of apoptosis (measured by two independent apoptosis assays: DNA fragmentation and PARP cleavage) in cells treated with Brk siRNA and cetuximab than in cells treated with either treatment alone (Figure 6b). Conversely, experimental elevation of Brk lowered the levels of inhibition of Akt and Erk phosphorylation by cetuximab in SUM102 cells compared with the levels in control vector-transfected SUM102 cells (Figure 6c). Consistent with the results of Western blot analysis, the Brk-transfected SUM102 cells required higher doses of cetuximab to achieve the same level of growth inhibition as observed in the control vector-transfected cells, and this effect was more apparent in the cells transfected with constitutively active Brk-Y447F (Figure 6d). Similar results were found in A431 cells (Figure S6). Together, these data confirmed that knockdown of Brk can sensitize breast cancer cells to EGFR-targeted therapy whereas a high level of Brk expression can lower the sensitivity of cancer cells to EGFR-targeted therapy.
Figure 6.

Brk confers resistance of breast cancer cells to EGFR inhibition. (a) Knockdown of Brk reduces breast cancer cell response to EGF stimulation. SUM102 cells were subjected to treatment with Brk-specific siRNA or control siRNA for 48 h prior to serum starvation for overnight. The next day, the cells were treated with 10 nM EGF or not for 30 min and harvested for Western blotting with the indicated antibodies. (b) Knockdown of Brk sensitizes breast cancer cells to cetuximab. SUM102 cells were subjected to Brk knockdown as in (a) and then treated with 20 nM cetuximab or not for 24 h. Cell lysates were prepared for apoptosis enzyme-linked immunosorbent assay and Western blotting with the indicated antibodies. O.D., optical density. (c) Overexpression of Brk confers resistance to cetuximab-induced inhibition of cell signaling. SUM102 cells selected for overexpression of wild-type Brk and corresponding control cells were treated with the indicated concentrations of cetuximab in 0.5% FBS culture medium for 16 h. Whole cell lysates were subjected to Western blotting with the indicated antibodies. (d) Overexpression of Brk confers resistance to cetuximab-induced growth inhibition. SUM102 cells selected for overexpression of wild-type Brk or Brk-Y447F and corresponding control cells were treated with the indicated concentrations of cetuximab for 5 days. Cell survival and proliferation after cetuximab treatment were measured with an MTT assay and plotted as a percentage of the optical density at 570 nm of the untreated cells. ■ = Control vector-transfected cells; ● = wild-type-Brk-transfected cells; ▲ = Brk-Y447F-transfected cells.
Discussion
In this study, we found that Brk plays important roles in regulating EGFR function in both breast cancer cells and non-breast-cancer cells. Our data suggest a new model, depicted in Figure 7, through which Brk sustains activated EGFR signaling, sensitizes cancer cells to EGF stimulation, and renders Brk-overexpressing cells less sensitive to EGFR-blocking antibodies than cells with low or no Brk expression.
Figure 7.

Proposed model of novel roles of Brk in augmenting EGFR signaling. Upon EGF stimulation, several sites on EGFR, including Y1045, are phosphorylated (step 1). In cells with no or a low level of Brk expression (left panel), Cbl binds to phosphorylated Y1045 on EGFR (step 2), leading to EGFR ubiquitination and subsequent degradation. In cells with a high level of Brk expression (right panel), Brk competes with Cbl for binding to phosphorylated Y1045, preventing Cbl-mediated EGFR ubiquitination and degradation (step 2, see Figures 2 and 3). The association between EGFR and Brk leads to activation of Brk (step 3, see Figure 3), and the activated Brk can phosphorylate EGFR Y845 (step 4, see Figure 4), which increases EGFR Y1045 phosphorylation in an EGFR kinase-dependent manner and further potentiates the association between EGFR and Brk and activation of new Brk molecules (step 5, see Figure 5). Therefore, Brk inhibits EGFR-Cbl association and promotes EGFR-Brk association through this positive feedback loop between EGFR and Brk. P = phosphorylation; Ub = ubiquitination.
There are several key novel findings of this study. First, we demonstrated that the EGF-induced EGFR-Brk association is EGFR Y1045 phosphorylation dependent. One early study compared several growth factor-mediated cell signaling pathways, including those of fibroblast growth factors (acidic or basic), nerve growth factor, platelet-derived growth factor-BB, insulin, and macrophage colony-stimulating factor (Kamalati et al., 1996), and found that Brk enhanced the cellular response only to EGF stimulation and that Brk was activated only by EGF stimulation. Our current work now provides a mechanistic insight into this observation by demonstrating the unique requirement of EGFR Y1045 phosphorylation for EGFR-Brk association and Brk activation by EGFR. Second, we unraveled a novel interaction between EGFR, Brk, and Cbl. We showed that Brk can inhibit ligand-induced EGFR degradation after receptor activation. We demonstrated that binding of Brk to the Y1045 site of EGFR inhibits Cbl binding to the same site. The competition between Brk and Cbl for binding to EGFR constitutes a novel mechanism of uncoupling of activated EGFR from ubiquitination and degradation. One recent study showed that Brk can also inhibit EGFR endocytosis (Kang et al., 2010). Together, these findings indicate that Brk inhibits EGFR downregulation through multiple interacting mechanisms. Third, we showed that the Y845 site of EGFR can be directly phosphorylated by Brk. Brk-mediated EGFR Y845 phosphorylation further potentiates EGFR function and the interaction between EGFR and Brk. Lastly, we showed that knockdown of Brk sensitized breast cancer cells to cetuximab treatment whereas experimental elevation of Brk conferred resistance to cetuximab treatment.
The effect of nonreceptor protein-tyrosine kinases on uncoupling activated EGFR from endocytosis and degradation is emerging as an integral part of the oncogenic activation of EGFR, together with EGFR gene amplification and protein overexpression or mutation; however, such mechanisms have not yet been explored in the context of targeted therapy. Given that Brk is widely expressed in breast cancer (Mitchell et al., 1994; Barker et al., 1997; Ostrander et al., 2007), that EGFR plays critical roles in breast tumorigenesis, and that targeting EGFR has been disappointing overall in breast cancer, our findings justify the development of novel Brk inhibitors, which may be used alone and in combination with EGFR inhibitors for treating patients with breast cancer.
Materials and methods
Materials
EGF was obtained from Calbiochem. Cetuximab and gefitinib were obtained from ImClone Systems, Inc., and AstraZeneca Pharmaceuticals, respectively. The antibodies directed against EGFR, site-specific tyrosine-phosphorylated EGFR (Y845, Y992, Y1045, Y1068, and Y1173), total and S473-phosphorylated Akt, Erk2, T202/Y204-phosphorylated Erk, the Myc-tag (9B11), and ubiquitin (P4D1) were obtained from Cell Signaling Technology, Inc. Antibodies directed against total and Y342-phosphorylated Brk were obtained from Santa Cruz Biotechnology, Inc., and Millipore/Upstate Biotechnology, Inc., respectively. The anti-Cbl antibody was obtained from BD Biosciences/Transduction Laboratories. All other materials were purchased from Sigma-Aldrich, unless otherwise specified.
Cell lines and cell culture
All cell lines were purchased from American Type Culture Collection, except SUM102, SUM149, and SUM190 cells, which were kindly provided by Dr. Steven P. Ethier (Karmanos Cancer Institute). All the cells were maintained in DMEM culture medium supplemented with 10% fetal bovine serum (FBS). For treatment of cells with EGF, the cells were typically cultured in 0.5% FBS for overnight before stimulation with EGF.
cDNA constructs and transfection
The Brk constructs (wild-type, Y447F, and K219M) were kindly provided by Dr. Mark R. Crompton (Royal Holloway, University of London). Myc-tagged EGFR-K721A, EGFR-Y1045F, EGFR-Y845F, and EGFR-K721A/Y845F mutants were created with Stratagene’s site-directed mutagenesis kit following the instructions provided by the manufacturer. Brk siRNA oligonucleotides (targeting sequence AAGGTGATTTCTCGAGACAAC) were ordered from Dharmacon/Thermo Fisher Scientific. Transfection of these constructs was performed with Lipofectamine 2000 (Invitrogen) following the instructions provided by the manufacturer.
Immunoblotting and immunoprecipitation
Lysates from cultured cells were prepared with a lysis buffer as previously described (Li et al., 2008). Equal amounts of cell lysate were subjected to immunoprecipitation or immunoblot analysis as previously described (Li et al., 2008).
Cell survival and proliferation assays
Cell survival and proliferation assays were performed in 24-well culture plates with experimental conditions described in the figure legends. Upon completion of the desired treatments, the cells were exposed to 1 mg/ml 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (Sigma-Aldrich) in culture medium (500 μl/well) for 2 h at 37°C in a CO2 incubator and then lysed in 500 μl/well of lysis buffer containing 20% sodium dodecyl sulfate in dimethylformamide/H2O (1:1, v/v), pH 4.7, in a 37°C incubator for at least 6 h. The lysates’ optical density was determined with a microplate reader at a wavelength of 570 nm.
Apoptosis assays
Apoptosis was measured by using an enzyme-linked immunosorbent assay kit (Roche Diagnostics Corp., Indianapolis, IN) that quantitatively measures cytoplasmic histone-associated DNA fragments (mononucleosomes and oligonucleosomes) and by Western blotting with an antibody that recognizes both uncleaved and cleaved PARP after various treatments, as previously described (Li et al., 2008; Lu et al., 2010).
35S-Methionine metabolic-labeling and pulse-chase analyses
Cells were metabolically labeled with 100 μCi/ml 35S-labeled methionine (PerkinElmer) in methionine-free medium for 90 min. After removal of the 35S-labeled methionine-containing medium, the cells were chased with or without 10 nM EGF in culture medium for various time intervals prior to harvest. EGFR immunoprecipitates were washed and separated by SDS-PAGE for autoradiography.
In vitro Brk kinase assay
The cDNA of EGFR kinase domain (amino acid 689-955) for EGFRK721A and EGFRK721A/K845F was subcloned into the pGEX vector (GE Healthcare). GST fusion protein containing the kinase domain of EGFR-K721A or EGFR-K721A/K845F was induced using 1 mM IPTG in BL21 (DE3) cells at 4°C for 24 h and then purified using glutathione resin (GE Healthcare) following the manufacturer’s instructions. Brk in vitro kinase assay was carried out in a kinase buffer (25 mM Tris [pH 7.5], 5 mM β-glycerophosphate, 2 mM DTT, 0.1 mM Na3VO4, 10 mM MgCl2) with 2 μg of GST fusion protein, 0.1 μg of Brk recombinant protein (Millipore/Upstate Biotechnology), and 1 μl of 10 mM ATP (or not) in a total volume of 40 μl for 30 min at 30°C. After reaction, the reaction products were resolved by SDS-electrophoresis followed by immunoblot analysis with antibodies described in the figure legends.
Supplementary Material
Acknowledgments
We thank Dr. Mark R. Crompton (University of London) for providing the Brk DNA constructs, Dr. Steven P. Ethier (Karmanos Cancer Institute) for the SUM series of cell lines, and Stephanie Deming (Department of Scientific Publications, MD Anderson Cancer Center) for editing the manuscript. Funding: This work was supported in part by grants from the US Congressionally Directed Medical Research Programs of the Department of Defense (W81XWH-06-1-0544 and W81XWH-07-1-0526), the Breast Cancer Research Foundation, and the National Institutes of Health (5R01CA129036) and MD Anderson’s NIH Cancer Center Support Grant (CA016672).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Barker KT, Jackson LE, Crompton MR. BRK tyrosine kinase expression in a high proportion of human breast carcinomas. Oncogene. 1997;15:799–805. doi: 10.1038/sj.onc.1201241. [DOI] [PubMed] [Google Scholar]
- Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. 1999;274:8335–8343. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- Born M, Quintanilla-Fend L, Braselmann H, Reich U, Richter M, Hutzler P, et al. Simultaneous over-expression of the Her2/neu and PTK6 tyrosine kinases in archival invasive ductal breast carcinomas. J Pathol. 2005;205:592–596. doi: 10.1002/path.1720. [DOI] [PubMed] [Google Scholar]
- Chen HY, Shen CH, Tsai YT, Lin FC, Huang YP, Chen RH. Brk activates rac1 and promotes cell migration and invasion by phosphorylating paxillin. Mol Cell Biol. 2004;24:10558–10572. doi: 10.1128/MCB.24.24.10558-10572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkery B, Crown J, Clynes M, O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20:862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- Easty DJ, Mitchell PJ, Patel K, Florenes VA, Spritz RA, Bennett DC. Loss of expression of receptor tyrosine kinase family genes PTK7 and SEK in metastatic melanoma. Int J Cancer. 1997;71:1061–1065. doi: 10.1002/(sici)1097-0215(19970611)71:6<1061::aid-ijc24>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Eck SM, Cote AL, Winkelman WD, Brinckerhoff CE. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol Cancer Res. 2009;7:1033–1044. doi: 10.1158/1541-7786.MCR-09-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forozan F, Veldman R, Ammerman CA, Parsa NZ, Kallioniemi A, Kallioniemi OP, et al. Molecular cytogenetic analysis of 11 new breast cancer cell lines. Br J Cancer. 1999;81:1328–1334. doi: 10.1038/sj.bjc.6695007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grovdal LM, Stang E, Sorkin A, Madshus IH. Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res. 2004;300:388–395. doi: 10.1016/j.yexcr.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Harvey AJ, Crompton MR. Use of RNA interference to validate Brk as a novel therapeutic target in breast cancer: Brk promotes breast carcinoma cell proliferation. Oncogene. 2003;22:5006–5010. doi: 10.1038/sj.onc.1206577. [DOI] [PubMed] [Google Scholar]
- Harvey AJ, Crompton MR. The Brk protein tyrosine kinase as a therapeutic target in cancer: opportunities and challenges. Anticancer Drugs. 2004;15:107–111. doi: 10.1097/00001813-200402000-00002. [DOI] [PubMed] [Google Scholar]
- Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, et al. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Goh LK, Sorkin A. EGF receptor ubiquitination is not necessary for its internalization. Proc Natl Acad Sci U S A. 2007;104:16904–16909. doi: 10.1073/pnas.0707416104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie HY, Shrestha Y, Selfors LM, Frye F, Iida N, Wang Z, et al. PTK6 regulates IGF-1-induced anchorage-independent survival. PLoS One. 2010;5:e11729. doi: 10.1371/journal.pone.0011729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamalati T, Jolin HE, Fry MJ, Crompton MR. Expression of the BRK tyrosine kinase in mammary epithelial cells enhances the coupling of EGF signalling to PI 3-kinase and Akt, via erbB3 phosphorylation. Oncogene. 2000;19:5471–5476. doi: 10.1038/sj.onc.1203931. [DOI] [PubMed] [Google Scholar]
- Kamalati T, Jolin HE, Mitchell PJ, Barker KT, Jackson LE, Dean CJ, et al. Brk, a breast tumor-derived non-receptor protein-tyrosine kinase, sensitizes mammary epithelial cells to epidermal growth factor. J Biol Chem. 1996;271:30956–30963. doi: 10.1074/jbc.271.48.30956. [DOI] [PubMed] [Google Scholar]
- Kang SA, Lee ES, Yoon HY, Randazzo PA, Lee ST. PTK6 inhibits down-regulation of EGF receptor through phosphorylation of ARAP1. J Biol Chem. 2010;285:26013–26021. doi: 10.1074/jbc.M109.088971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Strunk KM, Spritz RA. A survey of protein tyrosine kinase mRNAs expressed in normal human melanocytes. Oncogene. 1993;8:3403–3410. [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, et al. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998;12:3663–3674. doi: 10.1101/gad.12.23.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lu Y, Liang K, Pan T, Mendelsohn J, Fan Z. Requirement of hypoxia-inducible factor-1alpha down-regulation in mediating the antitumor activity of the anti-epidermal growth factor receptor monoclonal antibody cetuximab. Mol Cancer Ther. 2008;7:1207–1217. doi: 10.1158/1535-7163.MCT-07-2187. [DOI] [PubMed] [Google Scholar]
- Liu L, Gao Y, Qiu H, Miller WT, Poli V, Reich NC. Identification of STAT3 as a specific substrate of breast tumor kinase. Oncogene. 2006;25:4904–4912. doi: 10.1038/sj.onc.1209501. [DOI] [PubMed] [Google Scholar]
- Llor X, Serfas MS, Bie W, Vasioukhin V, Polonskaia M, Derry J, et al. BRK/Sik expression in the gastrointestinal tract and in colon tumors. Clin Cancer Res. 1999;5:1767–1777. [PubMed] [Google Scholar]
- Lu Y, Li X, Lu H, Fan Z. 1, 9-Pyrazoloanthrones downregulate HIF-1alpha and sensitize cancer cells to cetuximab-mediated anti-EGFR therapy. PLoS One. 2010;5:e15823. doi: 10.1371/journal.pone.0015823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MD, Yarden Y. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 2004;23:2057–2070. doi: 10.1038/sj.onc.1207390. [DOI] [PubMed] [Google Scholar]
- Mitchell PJ, Barker KT, Martindale JE, Kamalati T, Lowe PN, Page MJ, et al. Cloning and characterisation of cDNAs encoding a novel non-receptor tyrosine kinase, brk, expressed in human breast tumours. Oncogene. 1994;9:2383–2390. [PubMed] [Google Scholar]
- Normanno N, De Luca A, Maiello MR, Mancino M, D’Antonio A, Macaluso M, et al. Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in breast cancer: current status and future development. Front Biosci. 2005;10:2611–2617. doi: 10.2741/1725. [DOI] [PubMed] [Google Scholar]
- Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA. Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells. Cancer Res. 2007;67:4199–4209. doi: 10.1158/0008-5472.CAN-06-3409. [DOI] [PubMed] [Google Scholar]
- Polo S, Pece S, Di Fiore PP. Endocytosis and cancer. Curr Opin Cell Biol. 2004;16:156–161. doi: 10.1016/j.ceb.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Qiu H, Miller WT. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J Biol Chem. 2002;277:34634–34641. doi: 10.1074/jbc.M203877200. [DOI] [PubMed] [Google Scholar]
- Qiu H, Miller WT. Role of the Brk SH3 domain in substrate recognition. Oncogene. 2004;23:2216–2223. doi: 10.1038/sj.onc.1207339. [DOI] [PubMed] [Google Scholar]
- Qiu H, Zappacosta F, Su W, Annan RS, Miller WT. Interaction between Brk kinase and insulin receptor substrate-4. Oncogene. 2005;24:5656–5664. doi: 10.1038/sj.onc.1208721. [DOI] [PubMed] [Google Scholar]
- Rimawi MF, Shetty PB, Weiss HL, Schiff R, Osborne CK, Chamness GC, et al. Epidermal growth factor receptor expression in breast cancer association with biologic phenotype and clinical outcomes. Cancer. 2010;116:1234–1242. doi: 10.1002/cncr.24816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury JR, Farndon JR, Sherbet GV, Harris AL. Epidermal-growth-factor receptors and oestrogen receptors in human breast cancer. Lancet. 1985;1:364–366. doi: 10.1016/s0140-6736(85)91385-6. [DOI] [PubMed] [Google Scholar]
- Sartor CI, Dziubinski ML, Yu CL, Jove R, Ethier SP. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Res. 1997;57:978–987. [PubMed] [Google Scholar]
- Serfas MS, Tyner AL. Brk, Srm, Frk, and Src42A form a distinct family of intracellular Src-like tyrosine kinases. Oncol Res. 2003;13:409–419. doi: 10.3727/096504003108748438. [DOI] [PubMed] [Google Scholar]
- Shen CH, Chen HY, Lin MS, Li FY, Chang CC, Kuo ML, et al. Breast tumor kinase phosphorylates p190RhoGAP to regulate rho and ras and promote breast carcinoma growth, migration, and invasion. Cancer Res. 2008;68:7779–7787. doi: 10.1158/0008-5472.CAN-08-0997. [DOI] [PubMed] [Google Scholar]
- Sorkin A, Von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketani Y, Oka T. Biological action of epidermal growth factor and its functional receptors in normal mammary epithelial cells. Proc Natl Acad Sci U S A. 1983;80:2647–2650. doi: 10.1073/pnas.80.9.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, et al. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002;21:303–313. doi: 10.1093/emboj/21.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver AM, Silva CM. Signal transducer and activator of transcription 5b: a new target of breast tumor kinase/protein tyrosine kinase 6. Breast Cancer Res. 2007;9:R79. doi: 10.1186/bcr1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang B, Chatti K, Qiu H, Lakshmi B, Krasnitz A, Hicks J, et al. Brk is coamplified with ErbB2 to promote proliferation in breast cancer. Proc Natl Acad Sci U S A. 2008;105:12463–12468. doi: 10.1073/pnas.0805009105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka T, Frey MR, Dise RS, Bernard JK, Polk DB. Specific epidermal growth factor receptor autophosphorylation sites promote mouse colon epithelial cell chemotaxis and restitution. Am J Physiol Gastrointest Liver Physiol. 2011;301:G368–G376. doi: 10.1152/ajpgi.00327.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Ostrander JH, Faivre EJ, Olsen A, Fitzsimmons D, Lange CA. Regulated association of protein kinase B/Akt with breast tumor kinase. J Biol Chem. 2005;280:1982–1991. doi: 10.1074/jbc.M412038200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.