

Abstract
We report an infant with a unique combination of 22q11 deletion syndrome and 14q terminal deletion syndrome. The proband had clinical symptoms compatible with diagnosis of 22q11 deletion syndrome: microcephaly, micrognathia, high-arched palate, hypertelorism, short palpebral fissures, square nasal root, prominent tubular nose, hypoplastic nasal alae, bulbous nasal tip, dysplastic low-set ears, short philtrum, and heart defect, but no cell-mediated immunodeficiency typical for the syndrome. G-banding and fluorescence in situ hybridization analyses revealed a karyotype 45,XY,der(14)t(14;22)(q32.3;q11.2),-22.ish del(14)(q32.33)(D14S1420-),del(22)(q11.2q11.2)(N25-). Subsequent analyses disclosed a translocation between chromosomes 14 and 22 in the proband's mother with a deleted 14q telomere. Using comparative genome hybridization on oligonucleotide-based microarray (array-CGH), the deletion at 22q11.21 in the size of ∼4.25 Mb was revealed in the proband as well as the deletion of the telomeric area at 14q32.33qter (∼3.24 Mb) in the proband and his mother. However, both the proband and his mother showed mild symptoms (microcephaly, thin lips, carp-shaped mouth) typical for patients with the described terminal 14q deletion syndrome.
Key Words: 14qter microdeletion syndrome, Array-CGH, Deletion 14q, Deletion 22q11, DiGeorge syndrome, Velocardiofacial syndrome
Introduction
The 22q11 deletion syndrome (also known as DiGeorge/Velocardiofacial syndrome OMIM 188400/OMIM 192430) is a well-known, relatively common disorder, with a highly variable phenotype ranging from neonatal death to a normal phenotype with normal intellectual development [Ryan et al., 1997; Bartsch et al., 2003]. Common characteristics include cardiac defects, non-visible/hypoplastic thymus or infection problems, hypocalcemia, developmental delay, typical facial anomalies, and other malformations and deformities [Swillen et al., 2000; Óskardóttir et al., 2005]. Interstitial deletions at 22q11.2 have been recognized as the most common genetic aberration associated with this syndrome [Driscoll et al., 1992; Hall, 1993; Lindsay et al., 1995], with a total of 5 different 22q11.2 critical regions [Amati et al., 1999] and strong evidence for a major role of the TBX1 (Tbox 1) gene [Merscher et al., 2001]. Besides 22q11.2, a second region associated with DiGeorge syndrome was assigned to chromosome 10p, but the phenotype is more severe as developmental delay, deafness and renal malformations are typical [Schuffenhauer et al., 1995]. Although most 22q11.2 deletions occur sporadically, it is estimated that 8–28 % of deletions are inherited from a parental carrier [Driscoll et al., 1993; Hall, 1993; Ryan et al., 1997].
Terminal deletion of 14q32.3 is a rare entity with few published cases that have been defined as a specific phenotype [Ortigas et al., 1997]. Van Karnebeek et al. [2002] proposed a clinically recognizable terminal 14q microdeletion syndrome with the following characteristics: hypotonia, microcephaly, high and prominent forehead, blepharophimosis, epicanthus, short and bulbous nose, broad philtrum, thin upper lip and carp-shaped mouth, and developmental delay.
We report a male patient with an association of 22q11.2 and 14q32.33 deletions due to malsegregation of a 14;22 translocation in his mother. We also report on the mother with a terminal deletion of 14q and only mild symptoms typical for patients with this microdeletion syndrome.
Case Report
The male patient was born at 41 weeks gestation (weight = 3,750 g (75th percentile), length = 49 cm (<50th percentile), head circumference = 34 cm (10th percentile)) as the first child to a 20-year-old mother and a 22-year-old father, both healthy and non-consanguineous. During the pregnancy, one short hospitalization was required because of the risk of premature birth. A cesarean section was indicated due to amniotic fluid turbidity and fetal heart rate alterations. The Apgar score was 7-8-9 at 1-5-10 minutes, respectively. The infant was resuscitated due to mild respiratory distress with heart massage. Neonatal hypoglycemia and hypocalcemia were detected with a serum calcium concentration of 1.90–2.58 mmol/l (normal range 2.2–2.76 mmol/l). It resolved after initial calcium supplementation.
The infant was born with micrognathia, high-arched palate, hypertelorism, short palpebral fissures, square nasal root, prominent tubular nose, hypoplastic nasal alae, bulbous nasal tip, dysplastic low-set ears, thin lips, carp-shaped mouth, short philtrum, and short neck (fig. 1). Immunological studies showed hypogammaglobulinemia with no other significant abnormalities as well as no T cell deficit, but he demonstrated a higher incidence of respiratory infections. Subsequent cardiovascular examination showed only a right aortic arch with no other heart defect. Due to swallowing problems and congenital inspiratory stridor, laryngeal and esophageal endoscopy was performed. Cone epiglottis and elastic esophageal stenosis were detected. Examination in the cardiovascular center detected left-sided ligamentum arteriosum with a right aortic arch as the problem. The cardiosurgery resection of the ligamentum arteriosum was performed at the age of 2 years.
Fig. 1.
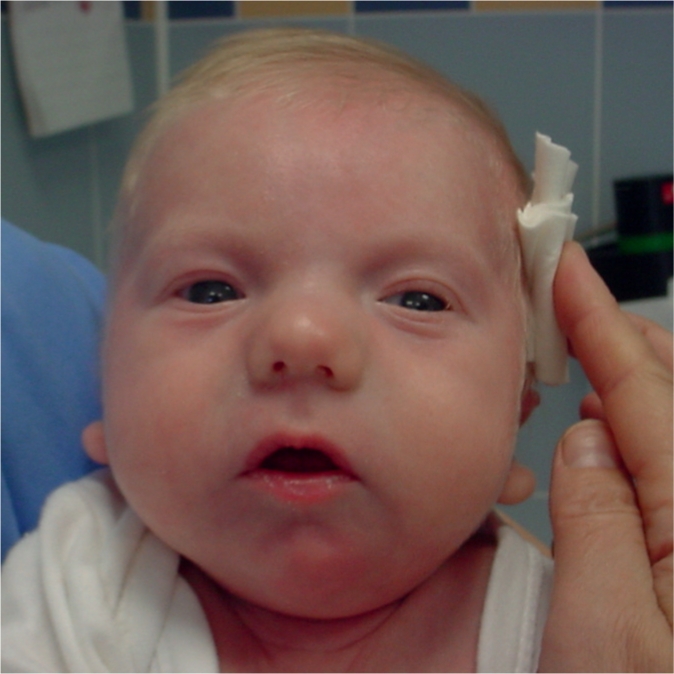
Photo of the proband at 2 months of age showing micrognathia, hypertelorism, prominent tubular nose, dysplastic low-set ears, thin lips, carp-shaped mouth, and a short neck.
At 27 months of age (weight = 10 kg (<2nd percentile), height = 80 cm (<2nd percentile), head circumference = 46 cm (<2nd percentile)), his mental-motor development was retarded by 1 year with practically no speech. X-ray examination showed mediastinal upside distension, most likely indicative of thymus hyperplasia. No hypoglycemia and hypocalcemia were detected.
At 38 months of age, the boy was hospitalized because of viral necrotizing laryngitis and died due to hypoxic encephalopathy by bronchopneumonia. The postmortem examination showed multiple pulmonary and gastric hemorrhages, and no thymus aplasia.
The mother was born as the first child from the first gravidity at term after an uncomplicated pregnancy. The family was unable to provide details on her weight, length, and head circumference at birth. There are no distinct dysmorphic features in a photography at about 4 months of age. At 10 years, she was monitored in neurology due to neuralgic pains. The brain examination showed no abnormalities. The proband's mother was firstly examined by a geneticist at 23 years of age (weight = 70 kg (75th–90th percentile), height = 158 cm (10th percentile), head circumference = 53 cm (2nd–10th percentile)). She was healthy and had a proportional stature with only mild craniofacial dysmorphic features: triangular face with high forehead and high hairline, short palpebral fissures, narrow nasal base, pointed nasal tip, short nose, short philtrum, thin upper lip, gothic palatum, and low-set ears with prominent lobes. Her mental-motor development was normal, and she achieved primary education. Except for the mother's uncle, who died from tetralogy of Fallot at 16 years of age (no evidence available), there was no family history of such or other pathologic phenotypes. The mother's parents and her younger sister are phenotypically normal with normal karyotypes.
Molecular Analysis a nd Results
First, FISH was performed in the proband, applying the LSI N25 SR/ARSA SG probe (Abbott Vysis, Inc., Downers Grove, Ill., USA) for the 22q11 region. A deletion of this region was found, but the control probe at 22q13 was translocated to an unknown acrocentric chromosome (fig. 2). G-banding analysis on chromosome metaphase spreads cultivated from peripheral blood lymphocytes showed an unbalanced rearrangement between chromosomes 14 and 22. Additional FISH analysis in the proband, applying the subtelomeric probe of chromosomes 14q (ToTel Vysion™ Probe Panel, Abbott Vysis), identified a deletion on chromosome 14q. The proband's karyotype was specified as 45,XY,der(14)t(14;22)(q32.3; q11.2),-22.ish del(14)(q32.33)(D14S1420-),ish del(22)(q11.2q11.2)(N25-).
Fig. 2.

FISH analysis using LSI N25 SR/ARSA SG probe proving the deletion of the 22q11 region (missing red signal at 22q11) with translocation of the remaining chromosome 22 to an unknown acrocentric chromosome (green signal for the gene ARSA at 22q13) in the proband.
The father's karyotype was normal, but chromosomal analysis of the mother's lymphocytes showed a translocation between chromosomes 14 and 22. FISH with the probe for DiGeorge syndrome (Abbott Vysis) showed 2 signals for the 22q11 region with 1 control probe translocated to chromosome 14. However, FISH for the subtelomeric regions of chromosomes 14 and 22 (Abbott Vysis) revealed a telomere 14q deletion in the proband's mother (fig. 3). According to these findings, the mother's karyotype was specified as 46,XX,t(14;22)(q32.3;q11.2).ish del(14)(q32.33)(D14S1420-),22q11.2(N25×2). To eliminate the possibility of a 3-chromosome-translocation, whole chromosome painting probes for chromosome 14 and 22 were used in the proband's mother (StarFISH Cambio, Cambridge, UK) with a negative result (fig. 4). MLPA testing of all telomeres was done in the proband and his mother to test if chromosome 14 has not received the telomeric end of another chromosome. MLPA using P036 and P070 kits (MRC Holland, Amsterdam, The Netherlands) showed a deletion of the telomeric part of chromosome 14q in both the proband and his mother and no change of the telomeric region of other chromosomes.
Fig. 3.

FISH analysis using a subtelomeric probe of chromosome 14q. The missing yellow signal at one chromosome 14 indicates a deletion of telomeric region 14q.
Fig. 4.

FISH whole chromosome painting (WCP) probes (red for chromosome 22 and green for chromosome 14) show t(14;22) and eliminate the possibility of a 3-chromosome translocation.
To determine the size of the deleted regions, oligonucleotide array-CGH using the Agilent Human Genome CGH 44K Oligo Microarray Kit containing over 43,000 probes (Agilent Technologies, Santa Clara, Calif., USA) was performed. In both the mother and the proband ∼3.24 Mb from 14q32.33 to the telomere of chromosome 14 were deleted. It comprised the region between 103,093,400–106,330,010 bp and included 59 oligonucleotide probes (fig. 5). The 4.25 Mb deletion of 22q11.21 was confirmed in the proband. It comprised the region between 14,433,273–18,691,904 bp and included 80 oligonucleotide probes (fig. 6). After combining the results of the G-banding, FISH and array-CGH experiments, we concluded that the proband had the karyotype 45,XY,der(14)t(14;22)(q32.3;q11.2),-22.ish del(22)(q11.2q11.2)(N25-),ish del(14)(q32.33)(D14S1420-). arr 14q32.33(103,093,400–106,330,010)×1, 22q11.1q11.21(14,433,273–18,691,904)×1 and his mother had 46,XX,der(14)t(14;22)(q32.3;q11.2).ish del(14)(q32.33)(D14S1420-), 22q11.2(N25×2). arr 14q32.33 (103,093,400–106,330,010)×1.
Fig. 5.

Array-CGH profile confirmed a 3.24 Mb deletion of 14q32.33 in the proband. The same deletion was also revealed in the mother.
Fig. 6.

Array-CGH profile confirmed a 4.25 Mb deletion of 22q11.21 in the proband.
Discussion
The patient described here is a unique case of 22q11 deletion syndrome in combination with 14q terminal deletion syndrome. This rare combination is the consequence of a maternal translocation between chromosomes 14 and 22.
The incidence of the 22q11.2 deletion is about 1/4,000 to 1/5,000 individuals [Devriendt et al., 1998] which places this disorder among the most common genetic syndromes. Recently, a phenotype-genotype correlation in patients with a linear 14q deletion or a deletion due to a ring chromosome led the investigators to the delineation of a recognizable 14q terminal deletion syndrome with specific clinical characteristics [van Karnebeek et al., 2002]. Besides the 14q terminal deletion syndrome (14q32.3qter region), a chromosome 14 interstitial deletion syndrome was also described [Gorski et al., 2005]; however, there are too few reported cases to assign specific features to specific chromosomal regions. The exact incidence of 14q terminal deletion syndrome was not established. However, the number of reported patients suggests that the frequency is very low. Considering this fact, the chance that the 22q11 deletion syndrome would occur concomitantly with 14q monosomy in a single individual is small, and their co-occurrence is at least noticeable.
The loss of the 22q11.2 region in our proband is probably the consequence of a maternal 14;22 translocation with loss of the very small derivative chromosome 22 during maternal meiosis. The maternal translocation was caused by a simple transfer of genetic material from chromosome 22 to chromosome 14. The proband's mother was healthy, with only mild visible phenotypic modifications and the examinations revealed a deletion of the 14q telomeric region.
The proband had many symptoms typical for the 22q11 deletion syndrome: micrognathia, hypertelorism, short palpebral fissures, dysplastic low-set ears, high arched palate, short philtrum, neonatal hypocalcemia, and only a right aortic arch, a congenital cardiac defect typical for this syndrome. In contrast, he had no cell-mediated immunodeficiency. Impaired T cell production due to thymus aplasia or hypoplasia and impaired immunologic function are common in patients with 22q11.2 deletions [Sullivan et al., 1998]. However, the clinical significance of these anomalies is uncertain; severe immune dysfunction is relatively rare. Some of the clinical findings that can also be attributed to the 14q monosomy are microcephaly, high-arched palate, thin upper lip, carp-shaped mouth, and developmental delay. Anomaly of the aortic arch was also reported in one case of a fetus with a 6 Mb deletion of chromosome 14q [de Pater et al., 2005]. However, most of these symptoms may be seen in both 14q monosomy and 22q11 deletion syndrome, and besides, the patient lacks the most typical symptoms characteristic for 14q terminal deletion syndrome as appointed by Ortigas et al. [1997] and summarized by van Karnebeek et al. [2002]; this would be, above all, hypotonia, high and prominent forehead, blepharophimosis, and epicanthus. These findings suggest that deletion of 14q in our patient did not have a great influence on his phenotype. This inference is supported by the fact that the proband's mother, who herself carried the deletion of the 14q region, is clinically normal, with only mild symptoms attributed to this deletion. As telomeres are extremely gene-rich [Saccone et al., 1992; Riethman et al., 2001], deletion or duplication of even small parts of chromosomal material in these regions may have severe phenotypic consequences. As shown by array-CGH, a relatively large area from 14q32.33 to 14qter of ∼3.24 Mb is deleted in the mother and the son. This region was shown to be deleted in many patients with 14q microdeletion syndrome with all the phenotypic consequences of the deletion.
The proband did not exhibit a cell-mediated immunodeficiency, and postmortem examination showed a normal development of his thymus. However, aplasia of the thymus is not only one of the most typical symptoms of 22q11 deletion syndrome, but de Pater et al. [2005] assumed that chromosome region 14q32qter also contains minimally 2 genes that are involved in thymus development. The first gene, SIVA1, operates in a caspase-dependent mitochondrial pathway of T cell apoptosis [Py et al., 2004], but no information regarding decreased expression of SIVA1 is currently available. The second gene, BCL11b, may be important in the development of thymocytes. It has been considered that in humans a hemizygous deletion of BCL11b may induce restricted development of the thymic cortical cellularity [Wakabayashi et al., 2003]. Nukina et al. [1989] described a patient with a dysmorphic face, micrognathia, cleft palate, and heart anomalies similar to DiGeorge syndrome, but who lacked aplasia of the thymus or hypoparathyroidism. The karyotype 45,XY,-14,-22,+der(14)(14pter-14q32.32::22q11.21-22qter) as the consequence of a maternal reciprocal translocation between chromosomes 14 and 22 was identified using high-resolution banding analysis. In this very similar case, the authors localized the gene responsible for the present DiGeorge syndrome to sub-band 22q11.1.
As clearly demonstrated in our case, a combination of different molecular cytogenetic methods allows a precise diagnosis of complex chromosomal rearrangements. Above all, array-based CGH opens new opportunities for high-resolution whole genome screening. It detects very small gene dosage alternations, is very useful in size mapping of the aberrations, which facilitates phenotype-genotype correlations, and saves multiple steps in the diagnostic process.
Acknowledgements
We thank the patient and his family for their cooperation. This study was supported by the MSM0021622415 from the Ministry of Education, Youth and Sports, Czech Republic.
References
- Amati F, Conti E, Novelli A, Bengala M, Digilio MC, et al. Atypical deletions suggest five 22q11.2 critical regions related to the DiGeorge/velo-cardio-facial syndrome. Eur J Hum Genet. 1999;7:903–909. doi: 10.1038/sj.ejhg.5200399. [DOI] [PubMed] [Google Scholar]
- Bartsch O, Nemeckova M, Kocarek E, Wagner A, Puchmajerova A, et al. DiGeorge/velocardiofacial syndrome: FISH studies of chromosomes 22q11 and 10p14, and clinical reports on the proximal 22q11 deletion. Am J Med Genet. 2003;117A:1–5. doi: 10.1002/ajmg.a.10914. [DOI] [PubMed] [Google Scholar]
- de Pater JM, Nikkels PG, Poot M, Eleveld MJ, Stigter RH, et al. Striking facial dysmorphisms and restricted thymic development in a fetus with a 6-megabase deletion of chromosome 14q. Pediat Develop Pathol. 2005;8:497–503. doi: 10.1007/s10024-005-0041-8. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Mortier G, Van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for Di-George syndrome: consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–933. [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, et al. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JL, Uhlmann WR, Glover TW. A child with multiple congenital anomalies and karyotype 46,XY,del(14)(q31q32.3): further delineation of chromosome 14 interstitial deletion syndrome. Am J Med Genet. 2005;37:471–474. doi: 10.1002/ajmg.1320370409. [DOI] [PubMed] [Google Scholar]
- Hall JG. CATCH 22. J Med Genet. 1993;30:801–802. doi: 10.1136/jmg.30.10.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay EA, Greenberg F, Shaffer LG, Shapira SK, Scambler PJ, Baldini A. Submicroscopic deletions at 22q11.2. Variability of the clinical picture and delineation of a commonly deleted region. Am J Med Genet. 1995;56:191–197. doi: 10.1002/ajmg.1320560216. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein GA, Heyer J, Puech A, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Nukina S, Nishimura Y, Kinugasa A, Sawada T, Hamaoka K, et al. A case of incomplete DiGeorge syndrome associated with partial monosomy 22q11.1 due to maternal 14;22 translocation. Jinrui Idengaku Zasshi. 1989;34:235–41. doi: 10.1007/BF01900727. [DOI] [PubMed] [Google Scholar]
- Ortigas AP, Stein CK, Thomson LL, Hoo JJ. Delineation of 14q32.3 deletion syndrome. J Med Genet. 1997;34:515–517. doi: 10.1136/jmg.34.6.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Óskardóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164:146–153. doi: 10.1007/s00431-004-1577-8. [DOI] [PubMed] [Google Scholar]
- Py B, Slomianny C, Auberger P, Petit PX, Benichou S. Siva-1 and an alternative splice form lacking the death domain, Siva-2, similarly induce apoptosis in T lymphocytes via a caspase-dependent mitochondrial pathway. J Immunol. 2004;172:4008–4017. doi: 10.4049/jimmunol.172.7.4008. [DOI] [PubMed] [Google Scholar]
- Riethman HC, Xiang Z, Paul S, Morse E, Hu XL, et al. Integration of telomere sequences with the draft human genome sequence. Nature. 2001;409:948–951. doi: 10.1038/35057180. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletion: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone S, De Sario A, Della Valle G, Bernardi G. The highest gene concentrations in the human genome are in the telomeric bands of metaphase chromosomes. Proc Natl Acad Sci USA. 1992;89:4913–4917. doi: 10.1073/pnas.89.11.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenhauer S, Seidel H, Oechsler H, Belohradsky B, Bernsau U, et al. DiGeorge syndrome and partial monosomy 10p: case report and review. Ann Genet. 1995;38:162–167. [PubMed] [Google Scholar]
- Sullivan KE, Jawad AF, Randall P, Driscoll DA, Emanuel BS, et al. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11.2 deletion syndromes. Clin Immunol Immunopathol. 1998;86:141–146. doi: 10.1006/clin.1997.4463. [DOI] [PubMed] [Google Scholar]
- Swillen A, Vogels A, Devriendt K, Fryns JP. Chromosome 22q11 deletion syndrome: update and review of the clinical features, cognitive behavioral spectrum, and psychiatric complications. Am J Med Genet. 2000;97:128–135. doi: 10.1002/1096-8628(200022)97:2<128::aid-ajmg4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- van Karnebeek CD, Quik S, Sluijter S, Hulsbeek MM, Hoovers JM, Hennekam RC. Further delineation of the chromosome 14q terminal deletion syndrome. Am J Med Genet. 2002;110:65–72. doi: 10.1002/ajmg.10207. [DOI] [PubMed] [Google Scholar]
- Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol. 2003;4:533–539. doi: 10.1038/ni927. [DOI] [PubMed] [Google Scholar]