

Abstract
Neurotrophins such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) act through the tropomyosin-related receptor tyrosine kinases (Trk) and the pan-neurotrophin receptor (p75) to regulate complex developmental and functional properties of neurons. While NGF activates both receptor types in sympathetic neurons, differential signaling through TrkA and p75 can result in widely divergent functional outputs for neuronal survival, growth, and synaptic function. Here we show that TrkA and p75 signaling pathways have opposing effects on the firing properties of sympathetic neurons, and define a mechanism whereby the relative level of signaling through these two receptors sets firing patterns via coordinate regulation of a set of ionic currents. We show that signaling through the p75 pathway causes sympathetic neurons to fire in a phasic pattern showing marked accommodation. Signaling through the NGF-specific TrkA, on the other hand, causes cells to fire tonically. Neurons switch rapidly between firing patterns, on the order of minutes to hours. We show that changes in firing patterns are caused by neurotrophin-dependent regulation of at least four voltage-gated currents: the sodium current and the M-type, delayed rectifier, and calcium-dependent potassium currents. Neurotrophin release, and thus receptor activation, varies among somatic tissues and physiological state. Thus, these data suggest that target-derived neurotrophins may be an important determinant of the characteristic electrical properties of sympathetic neurons and therefore regulate the functional output of the sympathetic nervous system.
Introduction
The physiological function of different neuron types is largely determined by their intrinsic membrane electrical properties. However, little is known about the molecular mechanisms controlling neuronal membrane electrical properties. The firing patterns of sympathetic neurons vary depending on the ganglion of origin and the identity of innervated tissue. Firing in these neurons range from a tonic to an accommodating, phasic pattern (Cassell et al., 1986; Wang and McKinnon, 1995; Jobling and Gibbins, 1999; Anderson et al., 2001). Voltage-gated currents are important determinants of a neuron's electrophysiological behavior (Llinás, 1988; Baranauskas, 2007), and regulation of currents likely underlies distinct sympathetic firing patterns. Consistent with this, sodium and calcium currents differ in sympathetic neurons projecting to different targets (Kukuła and Szulczyk, 1998; Kukwa et al., 2000; Rola and Szulczyk, 2000). Additionally, the expression of potassium currents differs between phasic and tonic neurons (Cassell et al., 1986; Wang and McKinnon, 1995). A possible explanation for these results is that target-specific firing properties are regulated by target-derived signals.
Neurotrophins are key target-derived developmental regulators, but also modulate ionic currents and firing pattern in adult neurons (McAllister et al., 1999; Chao, 2003). Neurotrophins regulate calcium, sodium, and potassium currents in sympathetic (Jia et al., 1999; Luther and Birren, 2006; Ford et al., 2008; Jia et al., 2008) and CNS neurons (Desai et al., 1999; Adamson et al., 2002; Wu and Yeh, 2005). Regulation of ionic currents may underlie neurotrophic regulation of firing patterns and function. For instance, NGF increases the excitability of rat sensory neurons via modulation of voltage-gated sodium and potassium currents and may underlie inflammation related nerve sensitization (Zhang et al., 2002, 2005, 2006a). Another example occurs in the cochlea where opposing gradients of BDNF and neurotrophin-3 (NT-3) maintain a spatial distribution of firing patterns in sensory neurons via regulation of potassium channel expression (Adamson et al., 2002). Thus, neurotrophic signaling has diverse and complex effects that may act to determine neuronal activity levels.
One way that targets could regulate sympathetic neuronal properties is through tissue-specific release of neurotrophins, or modulation of release from a single target. Indeed, different tissues release different levels and types of neurotrophins (Bierl et al., 2005; Randolph et al., 2007). Changes in neurotrophin production in vascular and heart muscle tissue, mast cells, and macrophages have been implicated in cardiovascular disease (Hasan et al., 2006; Hristova and Aloe, 2006). Injury also leads to increased production of NGF in peripheral tissues, which has been implicated in hypersensitization of sensory neurons in chronic pain (Zhang et al., 2002; Pezet and McMahon, 2006). While tissue-specific availability of neurotrophins has clear physiological relevance, less is known about how these signals regulate neuronal properties.
Here we demonstrate that activation of the neurotrophin receptors, p75 or TrkA, leads to expression of distinct firing patterns through differential regulation of sodium and potassium currents. This suggests that the relative level of signaling through these two receptors dynamically sets the firing properties and functional output of the sympathetic system.
Materials and Methods
Cell culture.
Cell culture was performed as previously described (Lockhart et al., 1997; Luther and Birren, 2006). The superior cervical sympathetic ganglia were removed from newborn rats (or mice in some experiments) and incubated for 1 h at 37°C in minimum essential medium (Invitrogen) containing 1.5 mg/ml collagenase (Worthington) and 5 mg/ml dispase (Invitrogen). Cells were dispersed by repeatedly passing the ganglia through fire-polished glass pipettes. Dispersed sympathetic neurons were transferred to an uncoated plastic culture dish and incubated for 2 h at 37°C to allow attachment of the more adherent Schwann cells and fibroblasts. Neurons were removed from the dish by rinsing with growth medium and counted. Neurons were plated at 13,000 cells per dish on glass-bottomed plates (MatTek Corporation) coated with collagen (50 μg/ml; BD Biosciences) and poly-d-lysine (0.5 mg/ml; BD Biosciences). Cultures were maintained in modified L15CO2 medium (Hawrot and Patterson, 1979; Lockhart et al., 1997), supplemented with 10% fetal bovine serum (Omega Scientific), 6 μg/ml dextrose, 2 mm glutamine (Invitrogen), 100 U/ml penicillin/100 μg/ml streptomycin (Invitrogen), 1 μg/ml 6,7,dimethyl-5,6,7,8-tetrahydropterine (DMPH4, EMD Biosciences), 5 μg/ml glutathione (Sigma), and 100 μg/ml l-ascorbic acid. NGF (5 ng/ml, Millipore) was added to support neuronal survival, and 1 μm cytosine arabinofuranoside (Ara-C, Sigma) was added to inhibit cell division. Dishes were used for recording on the second to fourth days after culturing at which time cells are not spontaneously active. Under these culture conditions neurons are predominantly noradrenergic, although they also have cholinergic synaptic properties in the presence of BDNF. Since the neurons are not spontaneously active, however, neurotransmitter release should be negligible or very low.
Electrophysiology.
Electrophysiological recordings were performed using an Axopatch 200B amplifier (Molecular Devices). Patch electrodes (2–6 MΩ) were pulled from borosilicate glass (Warner Instruments) on a Flaming/Brown horizontal micropipette puller, model P-97 (Sutter Instrument Co.). The mean series resistance of recordings was 16.6 ± 0.8 MΩ, which was routinely compensated by 95%. This resulted in calculated voltage errors below 2 mV at peak conductance (1.2 ± 0.03 mV for the voltage-gated potassium current and 1.6 ± 0.02 mV for sodium current). The mean whole cell capacitance was 20.4 ± 0.9 pF. Neither series resistance nor whole-cell capacitance showed any systematic relationship to firing pattern. Data traces were acquired using pClamp 8 and a Digidata 1320A digitizer board (Molecular Devices) and were digitized at 10 kHz and low-pass filtered at 2 kHz. All recordings were made at room temperature, and the bath was grounded using a 3 m KCl/agar salt bridge. Recordings were made in external saline consisting of the following (in mm): NaCl 150, KCl 3, MgCl2 2, Na HEPES 5, CaCl2 2, and dextrose 11; pH 7.4 and adjusted to 330 mOsm with sucrose. The pipette solution contained in mm: K gluconate 100, KCl 30, MgSO4 1, CaCl2 1, EGTA 10, HEPES 10, Na 3, and K2ATP 2; pH 7.4 and adjusted to 290 mOsm with sucrose. The junction potential under our conditions was ∼12.5 mV and the data were not corrected.
Firing patterns were examined in current clamp using series of current injections of increasing amplitudes and counting the resultant numbers of action potentials as previously described (Luther and Birren, 2006). The amplitudes of current injections were scaled to the threshold current (the smallest current that elicited an action potential in 5 of 5 trials) and ranged from −100% to 500% of threshold current. Pharmacological agents were applied to separate pools of neurons, which were recorded from between 15 min to up to 2 h after starting drug application. Multiple cells were recorded from in each culture dish. Recording in separate pools of neurons allowed us to compare drug effects at the same time point after the beginning of whole-cell recording. Voltage-clamp traces were leak subtracted by repeating protocols at 1/10 the test step amplitude, summing 10 trials, and subtracting the resulting current.
Quantification of firing patterns.
We observed two distinct firing patterns in our neurons: tonic and phasic. To objectively measure firing pattern and to allow statistical analysis of the pattern in a population of cells, we analyzed the percentage of spikes that occurred in the last half of the stimulus. Tonic cells fire multiple spikes at a constant interspike interval throughout a test step and have nearly 50% of spikes occurring in each half of the stimulus. Phasic cells, on the other hand fire a burst of one to several spikes at the beginning of the test step and have 0% of spikes in the second half and 100% of spikes in the first half of the stimulus (see Fig. 1). This measurement is independent of spike output; for example it will differentiate a tonic cell that fires 4 evenly timed spikes (50% in second half) from a phasic cell that fires a burst of 4 fast spikes at the beginning of the test step (0% in second half). We found this to be the simplest, most reliable way to differentiate objectively between firing patterns. The percentage of spikes occurring in the second half of the stimulus was calculated for each cell over four trials and mean values were used for statistical comparison between treatment groups.
Figure 1.

Phasic and tonic firing patterns in sympathetic neurons. A, Superimposed voltage traces recorded in current clamp from a sympathetic neuron firing in a phasic pattern (top). The neuron responded to 440 ms current pulses ranging from −100% of threshold current amplitude up to 500% of threshold current (shown below). This cell responded to all stimuli by firing one or two action potentials near the beginning of the pulse and then falling silent. B, The voltage response of the cell shown in A in response to a 500% of threshold current stimulus. C, Superimposed voltage traces recorded from a tonic cell (top) show that this cell fired repetitively throughout the duration of all stimuli above the threshold current. D, The voltage response of the cell shown in B in response to a 500% of threshold current stimulus.
Pharmacological agents.
NGF (Millipore) and BDNF (provided by Regeneron Pharmaceuticals) were aliquoted and frozen in L15 medium, and added directly to the external saline at a final concentration of 50 or 100 ng/ml, respectively. The compounds, K252a (Kamiya Biomedical), C2-ceramide, and C2-dihydroceramide (both from Biomol International), were aliquoted and stored frozen in DMSO before adding directly to the recording solution at 200 nm, and 25 μm final dilutions, respectively. We found no significant effect of the solvent on neuronal properties. Linopirdine dihydrochloride was obtained from Tocris Bioscience. TrkA IgG fusion protein was provided by Genentech and was applied at 2 μg/ml diluted in growth medium (Shelton et al., 1995). The REX anti-p75 function-blocking antibody was kindly provided by Dr. Louis Reichardt and was applied diluted 1:700 in external saline (Weskamp and Reichardt, 1991).
Statistical analysis.
Data records were analyzed using Clampfit 8 (pClamp 8, Molecular Devices). Data are expressed as means ± SEM and differences were considered statistically significant with p < 0.05. Statistical comparisons were made using SigmaStat 2.0 according to program defaults (Systat Software). Student's t test and ANOVA were used to compare two normally distributed data sets and three or more normally distributed sets, respectively. Nonparametric tests were used for non-normally distributed data sets. Normality was determined using the Kolmogorov–Smirnov test using SigmaStat 2.0.
Results
NGF regulates firing pattern in sympathetic neurons
The firing patterns of sympathetic neurons in response to long current pulses has been described as being either phasic or tonic (Cassell et al., 1986). We identified both phasic (Fig. 1A,B) and tonic (Fig. 1C,D) cells among our cultured superior cervical ganglia neurons. Firing patterns were examined in current clamp by injecting 440 ms square-wave current pulses scaled to the threshold current for each cell, ranging from −100% to 500% in 30% increments (Fig. 1A,C) [see Materials and Methods and Luther and Birren (2006)]. Phasic cells fired one or more spikes near the beginning of the current pulse and then became silent; a pattern that persisted with increasing stimulus strengths (Fig. 1A,B). Tonic cells fired continuously throughout the stimulus (Fig. 1C,D). Thus, the two patterns can be differentiated by analyzing the percentage of spikes occurring in the second half of the stimulus (see Materials and Methods). Tonic neurons fire ∼50% of spikes in each half of the stimulus (Fig. 1B) while a phasic neuron fires spikes only in the first half of the stimulus (Fig. 1D).
Under our baseline recording condition (saline) most cells fired in the tonic mode with only 12% of cells firing less than five spikes with 500% of threshold current stimuli. We found that application of 50 ng/ml nerve growth factor (NGF) to cultures resulted in a shift to lower spike output with 40% of cells firing fewer than five spikes with 500% amplitude stimuli; this shift was relatively rapid, occurring within 15 min of NGF exposure. NGF treatment (50 ng/ml) resulted in a reduction in the mean number of spikes fired across the range of stimulus amplitudes tested (Fig. 2A) (p < 0.001, n = 138 and n = 103 cells for the saline and NGF groups, respectively). This decrease in spiking resulted in an apparent bimodal distribution of spike output in NGF-treated cells, which may reflect differential responsiveness to NGF in subsets of cells (Fig. 2B). The NGF-mediated decrease in spike output was due to a shift to phasic firing in some cells. We can graphically depict the firing pattern of cells by plotting the time of action potential peaks relative to the onset of the stimulus as raster plots. This type of plot is shown for the 20% lowest spike output NGF cells in Figure 2C (21 cells, 4 trials for each cell). This plot shows that all of these cells fired 1–2 spikes and, most importantly, even cells that fired 2 spikes did so early in the stimulus, within 100 ms. In contrast, the raster plot shown in Figure 2D shows that the 20% highest-spiking NGF-treated cells fired spikes continuously throughout the stimulus (21 cells, 4 trials for each cell). The fact that NGF induced phasic firing only in some cells suggests that there are distinct subgroups that differ in their response to neurotrophin signaling.
Figure 2.

NGF induces phasic firing in a subpopulation of sympathetic neurons. A, A plot of the number of spikes fired against test step amplitude shows that bath-applied NGF (50 ng/ml; 15–120 min; see Materials and Methods) significantly decreased the mean spike output over the stimulus range tested (p < 0.001, two-way ANOVA followed by the Tukey test, n = 138 and n = 103 for saline and NGF, respectively). B, A histogram plot of spike output in response to a 500% of threshold stimulus shows that the mean population decrease in firing in NGF was due to a shift to lower spike output in a subpopulation of cells. C, A raster plot of spike times relative to stimulus onset elicited with a 500% of threshold stimulus shows that the 20% lowest spike output cells in NGF fired in a phasic pattern, i.e., with a burst of spikes near the beginning of the stimulus (21 cells, 4 trials each). D, A raster plot of spike times elicited with a 500% of threshold stimulus for the 20% highest spike NGF cells showing that these cells fired tonically (21 cells, 4 trials each).
NGF activation of TrkA and p75 differentially regulates firing pattern
Sympathetic neurons express two receptors that are activated by NGF: p75, a pan- neurotrophin receptor, and the specific NGF receptor, TrkA (Dixon and McKinnon, 1994; Wyatt and Davies, 1995; Bamji et al., 1998). TrkA and p75 receptors have different, and often opposing effects on neuron growth, survival, and neurotransmission (Rydén et al., 1997; Kohn et al., 1999; Yang et al., 2002). We therefore hypothesized that p75 and TrkA signaling promotes different firing patterns and that the relative strength of signaling through the two pathways determines the overall effect of NGF in particular cells. We tested this hypothesis by bath applying 50 ng/ml NGF along with blockers for either the TrkA or p75 receptor and examined firing patterns. Neurotrophins and/or other agents were bath applied for 15 min before recording and for up to 2 h before replacing the recorded culture with a new one from the incubator (Luther and Birren, 2006). To block TrkA signaling without affecting p75 we applied NGF together with 200 nm of the TrkA antagonist K252a in recording saline. We previously showed that this treatment blocks Trk signaling without affecting p75 activation and that, over the short time-frame of these acute experiments, it is not toxic to the neurons (Lockhart et al., 1997; Yang et al., 2002). Treatment with NGF and K252a significantly decreased spike output compared with saline or NGF applied alone (Fig. 3A) (n = 21, p < 0.01). This treatment also promoted phasic firing as can be seen in the spike time raster plot (Fig. 3B) (21 cells, 4 trials each, 500% stimulus). This plot shows that 85% of cells (18/21) fired 5 or fewer spikes and that the most spikes occurred within the first half of the stimulus: that is they fired in a phasic pattern.
Figure 3.

NGF activation of p75 and TrkA receptors differentially regulates firing patterns. NGF activates both p75 and TrkA receptors in sympathetic neurons. Coapplication of NGF with the TrkA blocker K252a or the p75-blocking antibody REX was used to selectively activate each signaling pathway. A, Plots of spike output versus stimulus amplitude show that NGF with the TrkA antagonist (200 nm K252a, filled circles) resulted in a significant decrease in spike output compared with saline (black line) or NGF alone (gray line; n = 22, p < 0.01, two-way ANOVA followed by the Tukey test). Application of NGF with the p75 function-blocking antibody (REX 1:700, open triangles) resulted in a significant increase in spike output compared with saline or NGF alone (n = 20, p < 0.01, two-way ANOVA followed by the Tukey test). The REX antibody applied alone had no significant effect (gray squares, n = 28). B, A raster plot of spike times elicited with a 500% of threshold stimulus shows that in NGF/K252a 85% of cells (18/21) fired five or fewer spikes occurring within the first half of the stimulus, showing a phasic pattern (n = 21, 4 trials each). C, A raster plot of spike times elicited with a 500% of threshold stimulus showing that NGF/REX promoted tonic firing (n = 20, 4 trials each). D, We used the percentage of spikes occurring in the second half of the stimulus to provide a measure of phasic and tonic firing. A bar plot shows the percentage of spikes occurring in the second half of the stimulus for the different treatment groups. Neurons in NGF and, to a greater extent, NGF/K252a fired fewer second-half spikes compared with saline (p < 0.05, all three groups different, Kruskal–Wallis ANOVA on ranks followed by Dunn's test). NGF/REX caused a significant increase in second-half spikes compared with saline and NGF alone (p < 0.05, all three groups different, Kruskal–Wallis ANOVA on ranks followed by Dunn's test). REX alone was not different from saline.
We selectively activated the TrkA receptor by applying NGF (50 ng/ml) along with the p75 function-blocking antibody REX (Weskamp and Reichardt, 1991). When applied with REX, NGF caused a significant increase in spike output compared with saline and NGF alone (Fig. 3A) (n = 20, p < 0.01). NGF applied with REX was also associated with tonic firing as can be seen in the spike time raster plot in Figure 3C (20 cells, 4 trials each, 500% stimulus). The REX antibody alone caused a trend toward an increase in spike output that did not reach statistical significance compared with saline, but was significantly different from REX/NGF. The absence of significant change in the presence of REX alone suggests that p75 is not strongly activated under baseline conditions in our cultures (Fig. 3A) (n = 11).
As mentioned above, we used the percentage of spikes a cell fired in the second half of a stimulus as an objective measure of how phasic or how tonic a cell was. This allowed us to perform statistical comparisons of firing patterns between treatment groups. We compared the number of spikes that occurred during the second half of a 440 ms, 500% of threshold current stimulus (see Materials and Methods). A phasic cell has 0% of spikes occurring in the second half (100% of spikes occurring in first half), while a tonic cell has ∼50% of spikes occurring during each half of the stimulus. This analysis is independent of actual spike number since it differentiates between a tonic cell that fires several evenly spaced spikes and a phasic cell that fires a burst of several spikes near the beginning of the test step. For a population of cells the closer the mean value is to 50% the more tonically firing cells exist and conversely, the closer the value is to 0% the more phasically firing cells there are. The plot shown in Figure 3D shows that cells fired significantly fewer spikes in the second half of the stimulus in NGF compared with saline, consistent with the shift of some neurons toward phasic firing under this condition (Fig. 2). Separating TrkA and p75 signaling demonstrated opposing effects on firing patterns. Neurons fired dramatically fewer spikes in the second half of the stimulus in NGF with K252a, compared with saline and NGF alone, suggesting that p75 activation promotes phasic firing (Fig. 3D) (23.6 ± 0.99%, 7.2 ± 1.60%, and 32.9 ± 0.64% for NGF, NGF/K252a, and saline, respectively; p < 0.05, all three groups different from each other). In contrast NGF/REX caused neurons to fire significantly more spikes in the second half compared with saline and NGF alone (Fig. 3D) (43.5 ± 0.38% vs 23.6 ± 0.99% and 32.9 ± 0.64% for NGF/REX, NGF, and saline, respectively; p < 0.05, all three groups different from each other). The REX antibody alone had no significant effect on second half spikes compared with saline (Fig. 3D). Together these data strongly suggest that NGF-dependent activation of p75 leads to a phasic firing pattern while NGF acting through TrkA promotes tonic firing.
Activation of p75 promotes phasic firing
In the previous section we found that short-term application of NGF with an antagonist for TrkA (K252a) leads to a decrease in spike output and a shift to phasic firing. While this suggests that NGF-dependent activation of the p75 receptor promotes phasic firing properties, we wished to rule out effects of K252a on additional non-Trk kinase pathways. To this end we examined firing patterns using two additional strategies to selectively manipulate p75 signaling.
While sympathetic neurons express p75 and TrkA, they do not express TrkB, the specific Trk receptor for BDNF (Dixon and McKinnon, 1994; Wyatt and Davies, 1995; Bamji et al., 1998). All neurotrophins bind to p75 with similar affinity, therefore, BDNF acts as a p75-specific ligand in these cells (Yang et al., 2002; Ivanisevic et al., 2003; Singh et al., 2008). Bath application of 100 ng/ml BDNF significantly decreased spike output compared with saline and NGF (Fig. 4A) (n = 47, p < 0.05). BDNF also caused cells to fire significantly fewer spikes in the second half of the stimulus compared with saline, suggesting an increase in phasic firing pattern (Fig. 4B) (17.2 ± 1.40% for BDNF and 32.9 ± 0.64% for saline, p < 0.05). The p75-blocking antibody REX blocked the effect of BDNF on decreasing spike output (Fig. 4A) and prevented the shift to phasic firing (Fig. 4B), suggesting that BDNF was in fact acting through p75 activation (n = 10).
Figure 4.
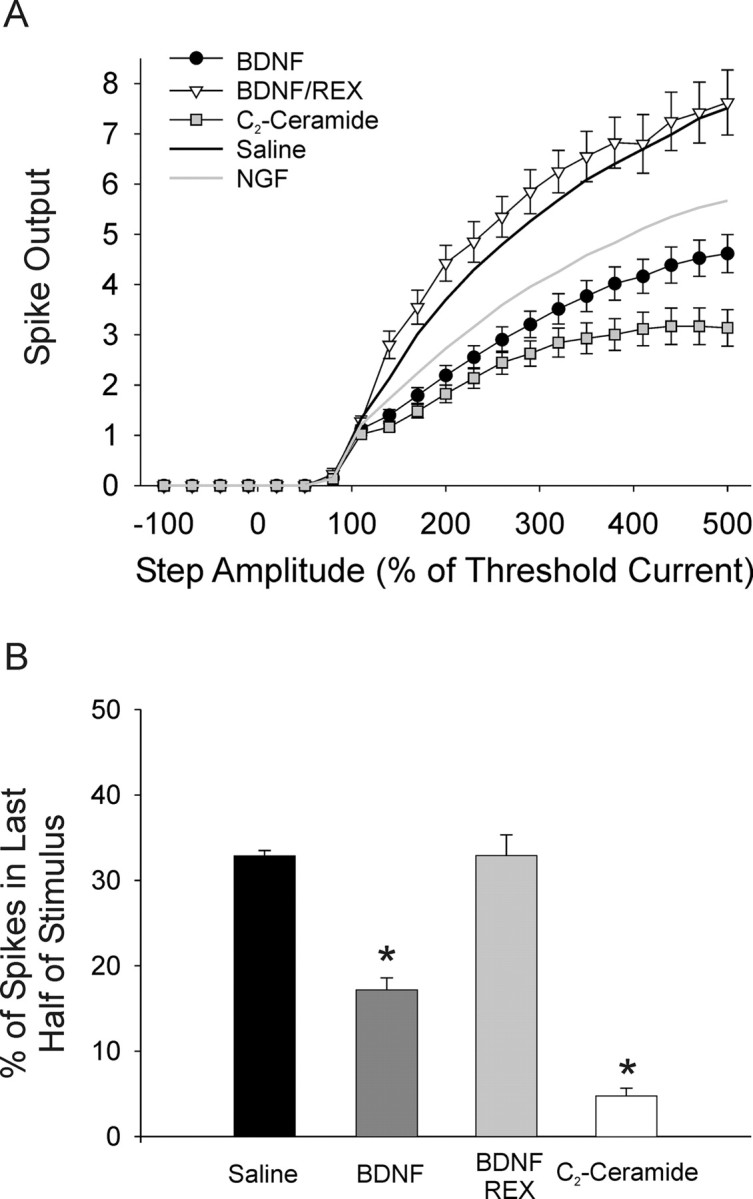
Activation of p75 promotes phasic firing. BDNF is a p75-specific ligand in sympathetic neurons and C2-ceramide mimics a second messenger generated by p75 activation. A, Plots of spike output versus stimulus amplitude show that bath application of 100 ng/ml BDNF (filled circles, n = 47) caused a significant decrease in spike output compared with saline (black line) or NGF alone (gray line; p < 0.001, two-way ANOVA followed by the Tukey test). The p75 antagonist, REX, blocked this effect (open triangles, n = 10). C2-ceramide, 25 μm, also significantly decreased spike output compared with both saline and NGF alone (gray squares, n = 47; p < 0.01, two-way ANOVA followed by the Tukey test). B, Bar plots of the percentage of spikes in the second half of the stimulus shows that both BDNF (dark gray bar) and C2-ceramide (open bar) significantly induced phasic firing in sympathetic neurons (p < 0.05, Kruskal–Wallis ANOVA on ranks followed by Dunn's test). The p75 antagonist, REX, blocked this effect of BDNF (light gray bar).
Additionally, we examined firing after bath application of 25 μm C2-ceramide to mimic activation of p75. Ceramide is a second messenger molecule generated following p75 activation (Dobrowsky et al., 1994), and treatment of sympathetic neurons with C2-ceramide is sufficient to mediate synaptic effects of p75 activation (Yang et al., 2002). Bath application of C2-ceramide resulted in a significant decrease in spike output over the range of test steps compared with both saline and NGF (Fig. 4A) (n = 47, p < 0.01). Application of C2-ceramide promoted phasic firing as evidenced by a decrease in the spikes fired in the second half of the stimulus compared with both saline and BDNF (Fig. 4B) (4.8 ± 0.91%, 17.2 ± 1.40%, and 32.9 ± 0.64% for C2-ceramide, BDNF, and saline, respectively; p < 0.05). No significant effect was seen with the inactive analog C2-dihydroceramide (25 μm; n = 21; data not shown). These data strongly suggest that activation of the p75 receptor promotes phasic firing in sympathetic neurons. While these experiments do not rule out the possibility of independent mechanisms for BDNF and C2-ceramide, the data are consistent with the idea that p75 activation promotes phasic firing through increased ceramide signaling.
BDNF and NGF differentially regulate firing in wild-type and p75 knock-out mouse neurons
We further investigated the separable effects of TrkA and p75 signaling by examining the effect of neurotrophin application on the firing properties of sympathetic neurons isolated from wild-type and p75 knock-out mice (Lee et al., 1992). In contrast to rat neurons, cultured wild-type mouse neurons tended to have higher spike output and tended not to fire in a phasic pattern. However, consistent with the results from rat neurons we found that 100 ng/ml BDNF (n = 14) resulted in a significant decrease in spike output in cultured mouse neurons. BDNF also caused a significant decrease in the percentage of spikes fired in the second half of the stimulus compared with saline and NGF (Fig. 5B) (29.5 ± 2.30%, 40.3 ± 1.10%, and 41.7 ± 0.67 for BDNF, saline, and NGF, respectively; p < 0.05). Unlike the rat neurons NGF (50 ng/ml; n = 12) had no significant effect on firing in wild-type mouse neurons (Fig. 5A,B).
Figure 5.

NGF and BDNF differentially regulate firing in wild-type and p75 knock-out mouse neurons. A, A plot of spike output versus stimulus strength for cultured wild-type mouse sympathetic neurons shows that, similar to rats, BDNF (gray squares, n = 14) significantly decreased spiking compared with saline (filled circles, n = 26; p < 0.001, two-way ANOVA followed by the Tukey test). Unlike in rat neurons NGF did not cause a significant decrease in spike output (open triangles). B, A bar plot of the percentage of spikes occurring in the second half of the stimulus shows that BDNF (gray bar) significantly promoted phasic firing in wild-type neurons (p < 0.05, Kruskal–Wallis ANOVA on ranks followed by Dunn's test), but NGF showed no effect (open bar). C, A plot of spike output versus stimulus amplitude in cultured p75 knock-out mouse neurons shows that in contrast to wild-type cells, BDNF (gray squares, n = 9) did not elicit a decrease in spiking compared with saline (filled circles, n = 16). NGF on the other hand, significantly increased spiking (open triangles, n = 13; p < 0.01, one-way ANOVA followed by the Tukey test), although there was no significant effect on the percentage of spikes occurring in the second half of the stimulus (D).
Application of NGF (n = 13) to cultures made from p75 deficient mice caused a significant increase in spike output (Fig. 5C) (n = 16; p < 0.01). This is consistent with the increase in spike output seen in cultured rat neurons treated with NGF and the p75 antagonist REX. While spike output increased with NGF there was no significant increase in spikes fired in the second half of the stimulus, perhaps consistent with the already strong tendency of mouse neurons to fire in the tonic mode under baseline conditions. Application of BDNF (n = 9) to p75 knock-out cultures did not elicit a decrease in spiking as seen in rat and wild-type mouse neurons. In fact BDNF caused a slight, but statistically significant increase in spiking, raising the possibility that in the absence of p75, BDNF had an effect at other Trk receptors (Fig. 5C) (p < 0.05). Indeed, the presence of p75 receptors has been shown to influence the specificity and affinity of Trk receptors, presumably through allosteric regulation of ligand binding sites (Bibel et al., 1999; Mischel et al., 2001; Zaccaro et al., 2001; Reichardt, 2006). BDNF had no significant effect on the percentage of spikes occurring in the second half of the stimulus (Fig. 5D), consistent with the lack of p75 signaling in these neurons.
Together these data suggest that TrkA and p75 have opposing effects on sympathetic neuronal firing patterns; activation of TrkA promotes tonic firing while activation of p75 promotes phasic firing. The exact firing pattern of an individual neuron may therefore be controlled by the ratio of activity in these two signaling pathways.
TrkA is activated under baseline, saline conditions
The finding that cells tend to fire tonically under baseline conditions raised the possibility that TrkA signaling could be present in our control saline condition. We include 5 ng/ml NGF (∼4 × 10−10 m) in our culture medium to support neuronal survival before electrophysiological experiments. This concentration of NGF preferentially activates the TrkA signaling pathway through high-affinity NGF binding sites (Maliartchouk and Saragovi, 1997; Ivanisevic et al., 2003). NGF is removed from the dish 15 min before recording, leaving the question of whether there is residual activation of neurotrophin signaling pathways in our baseline “saline” condition. If so, how much does this signaling contribute to the baseline firing pattern of sympathetic neurons? We addressed this question by measuring firing properties in cultures in which NGF had been removed from the culture medium for various periods before recording. We removed the NGF-containing medium and replaced it with NGF-free medium containing 2 μg/ml TrkA IgG fusion protein, which competitively binds residual NGF and prevents receptor interactions (Shelton et al., 1995). Cultures were incubated in this NGF-free, TrkA IgG medium for 1 (n = 18), 2 (n = 13), or 4 h (n = 10) before recording firing patterns in normal saline as already described.
We also recorded firing patterns from cultures treated with a bath-applied TrkA antagonist, 200 nm K252a, without prior removal of NGF or addition of TrkA IgG to the culture medium (n = 17, 15 min before recording and up to 2 h). We previously showed that the p75 antagonist, REX, had no significant effect when applied alone, suggesting that the p75 pathway is not significantly activated by ligand under baseline conditions (Fig. 3). Thus, acute application of K252a alone should affect firing patterns only if TrkA is activated under baseline conditions.
We found that spike output progressively decreased with the length of time in NGF-free medium or with acute application of K252a, as can be seen in plots of spike output versus stimulus strength (Fig. 6A) (all groups significantly different p < 0.05, except for 4 h vs K252a and 1 h vs 2 h; 2 h data not shown). Phasic firing also increased with length of time in NGF-free media as evidenced by the progressive decrease in the number of spikes occurring in the second half of the stimulus (Fig. 6B) (32.9 ± 0.64%, 18.9 ± 2.18%, 12.8 ± 2.47%, 1.8 ± 0.90%, for saline, 1 h, 2 h, and 4 h no NGF, respectively; saline different from all other groups and 1 h different from 4 h and K252a; p < 0.05). Bath application of K252a without prior NGF removal also caused a significant increase in phasic firing as shown by the decrease in the number of spikes in the second half of the stimulus (Fig. 6B) (5.5 ± 1.58%, different from saline and 1 h no NGF, p < 0.05).
Figure 6.

Baseline TrkA activity promotes tonic firing under control saline conditions. A, Growth medium contains a low concentration of NGF (5 ng/ml) needed for cell survival. A plot of spike output versus stimulus amplitude shows that removal of residual NGF with TrkA-Ig before recording progressively decreased spiking with length of time in no NGF (open triangles, 1 h no NGF, n = 18; gray squares, 4 h no NGF, n = 10; black line, saline without prior removal). Application of a TrkA antagonist (200 nm K252a, gray diamonds, n = 17) without depletion of residual NGF caused a decrease in spiking comparable to 4 h with no NGF (all groups different except 4 h no NGF vs K252a, p < 0.05, two-way ANOVA followed by the Tukey test). B, A bar plot of the percentage of spikes occurring in the second half of the stimulus shows that NGF withdrawal progressively induced phasic firing with increased time in no NGF. K252a applied without prior removal of residual NGF also significantly reduced second half spikes (saline different from all other groups, and 1 h different from 4 h and K252a, p < 0.05, Kruskal–Wallis ANOVA on ranks followed by Dunn's test).
These data suggest that TrkA is significantly activated under our culture conditions, contributing to tonic spike output in addition to promoting cell survival, and that this activity can last for up to 4 h after NGF withdrawal. Treatment with a much higher concentration of NGF (50 ng/ml) resulted in a shift of some cells to a phasic firing pattern (Fig. 2), suggesting that high concentrations of NGF activate the lower-affinity p75 receptor in addition to high-affinity sites.
We think that it is unlikely that these results occur secondary to apoptotic effects of NGF withdrawal. Cultured postnatal sympathetic neurons require NGF as a survival factor. However, neurons from animals of this age do not begin to die until 12 h following NGF withdrawal and can be rescued by readdition of NGF before that time (Deckwerth and Johnson, 1993). This suggests that even our 4 h NGF withdrawal would not result in apoptotic death. Furthermore, our cultured neurons appeared healthy with clearly identifiable nuclei and no blebbing of the cell membrane following 4 h in the NGF-free medium or a 2 h incubation in medium containing 200 nm K252a. Resting membrane potentials were similar for saline and 4 h NGF withdrawal and significantly hyperpolarized for acute K252a, also suggesting that neither treatment caused significant deterioration of cell health over the time course of the experiment (−54.9 ± 0.4 mV for no NGF withdrawal, −54.6 ± 1.2 mV for 4 h NGF withdrawal, and −62.4 ± 1.0 mV for K252a; K252a significantly different from both other groups, p < 0.001).
As predicted, application of NGF with K252a caused a significantly larger decrease in spike output compared with K252a alone consistent with predominant activation of p75 by NGF under these conditions (p < 0.01). Although significant, this difference between NGF/K252a and K252a alone was small and we saw no statistically significant difference in the number of spikes in the second half of the stimulus (compare Figs. 3, 6). This apparently small effect of p75 activation in the absence of TrkA signaling raises the possibility that p75 promotes phasic firing through negative regulation of the TrkA pathway. Indeed, p75 activation has been shown to negatively influence TrkA activation in both sympathetic and dorsal root ganglion neurons (Kimpinski et al., 1999; Hannila et al., 2004; Singh et al., 2008).
Differential regulation of voltage-gated currents by p75 and TrkA signaling
Our data suggest that activation of the p75 signaling pathway pushes sympathetic neuronal firing patterns toward the phasic mode and that TrkA signaling pushes firing toward the tonic mode. One mechanism that could allow these two neurotrophin receptors to mediate these effects would be via differential regulation of the voltage-gated currents that control neuronal firing properties. We therefore asked which currents differed between phasic and tonic cultured neonatal rat sympathetic neurons. We recorded from individual neurons to determine both the spike output and firing pattern as described above, and the amplitudes of four voltage-gated currents.
We examined the voltage-dependent sodium current and three potassium currents: the M-current, the delayed rectifier current, and the calcium-activated potassium current. We chose the M-current because it has previously been shown to differ between phasic and tonic sympathetic neurons and is modulated by NGF (Wang and McKinnon, 1995; Jia et al., 2008). We chose the sodium current, delayed rectifier potassium current, and calcium-dependent potassium current because previous studies have demonstrated modulation by NGF (Zhang et al., 2002; Luther and Birren, 2006). We chose to concentrate on these four currents because they are likely to be involved in regulating firing pattern. While additional currents are also modulated by neurotrophins and could potentially contribute to the regulation of firing properties, we focused on currents that could be isolated relatively well using two simple voltage-clamp protocols that could be rapidly performed, allowing us to obtain both voltage- and current-clamp information from the same neurons. This allowed us to obtain multiple electrophysiological measurements in the same cell and to relate them to the measured firing properties. To maximize the data obtained from each cell, we made most recordings in the absence of pharmacological channel blockers (unless otherwise stated). Therefore, the whole-cell currents we measure here show qualitative current dissections that describe key differences in voltage-gated current expression between phasic and tonic neurons.
The M-current was examined using a voltage-clamp protocol stepping from a holding potential of −30 mV to test steps ranging from −40 mV to −90 mV (Fig. 7A–C). Reported current amplitude values represent the average value over the last 25 ms of the test step. Holding the cell at −30 mV causes inactivation of some potassium currents and is near or below threshold for others (McFarlane and Cooper, 1992; Luther and Birren, 2006), resulting in relative isolation of the M-current (Wang and McKinnon, 1995). In support of this we found that the M-current antagonist muscarine chloride (30 μm) blocked nearly all of the current activated by this protocol above leak (Fig. 7B,C).
Figure 7.

Voltage-clamp protocols used to examine four currents in sympathetic neurons. A, Raw current traces recorded in voltage clamp of the non-inactivating M-type potassium current in the presence of 250 nm tetrodotoxin to block sodium current (top traces). The M-current was examined by holding cells at −30 mV and making hyperpolarizing test steps (−40 to −90 mV, bottom traces, 1500 ms, bottom). The outward current decreased according to the voltage dependence of the M-current channels and was measured over the last 25 ms of the test step (dashed lines, top). B, Current traces recorded in voltage clamp evoked with voltage steps from −30 mV to −50 mV show that 30 μm muscarine chloride, an M-current antagonist, reversibly inhibited current evoked by this protocol. C, Plots of M-current amplitude, measured as described for A, versus test step show that most (80–90%) of the current was blocked with 30 μm muscarine chloride (filled circles versus open triangles), suggesting that the M-current is relatively well isolated by this protocol. Leak current is shown (gray squares) determined by p/10 protocol. D, The delayed rectifier current (top traces) was examined by giving cells depolarizing steps (−50 to +20 mV, bottom traces, 500 ms) from a −60 mV holding potential. The delayed rectifier was measured over the last 25 ms of the test step (dashed lines, top). E, The inward sodium current, shown in inset, was activated using the same voltage protocol as in B. The sodium current was measured at peak for each test step. F, The calcium-activated potassium current was activated using the same voltage protocol shown in B. It was measured as a tail current at test step offset where indicated in the inset (dashed lines). Traces in D, E, and F were leak subtracted using p/10 protocols.
The sodium current, delayed rectifier current, and calcium-dependent potassium current were all examined using one protocol. Cells were stepped from a holding potential of −60 mV to test steps ranging from −50 mV to +20 mV (Fig. 7D, bottom). The delayed rectifier current was measured as the average current over the last 25 ms of the test step (Fig. 7D). The current referred to here as delayed rectifier likely represents contributions from the slowly inactivating and non-inactivating potassium current components described previously (McFarlane and Cooper, 1992; Luther and Birren, 2006). However, the rapidly inactivating A-type component is inactivated by holding the cell at −60 mV, and is thus not activated by this protocol (McFarlane and Cooper, 1992; Luther and Birren, 2006). The sodium current was measured as the peak inward current amplitude at each test step (Fig. 7E). Extrapolation of the average sodium current voltage plot for all cells gave an estimated reversal potential that agreed well with the calculated Nernst equilibrium potential of 98.9 mV (extrapolated reversal 99.0 mV), suggesting that our voltage clamp was at least sufficient to determine the peak amplitude of sodium current. The calcium-dependent potassium current was measured as the tail current occurring when cells were stepped back to the −60 mV holding potential following the test steps (−50 mV to +20 mV) as previously described (Fig. 7F) (Luther and Birren, 2006).
Phasic and tonic neurons show differential expression of four currents
We characterized these currents in phasic and tonic sympathetic neurons. Cells were acutely treated with 50 ng/ml NGF to activate both p75 and TrkA resulting in subpopulations of neurons that fired either tonically or phasically (Fig. 2). Neurons were first examined in current clamp to determine their firing pattern, as described in previous sections. We then switched to voltage clamp to measure the four voltage-gated currents in each cell as described above. We compared the values for the M-current, sodium current, delayed rectifier current, and the calcium-dependent potassium current between the 20% lowest spike output cells (phasic, 1.05 ± 0.03 spikes with a 500% threshold current stimulus; 10/48 cells) and the 20% highest spike output cells from the NGF treatment group (tonic, 9.33 ± 0.27 spikes with a 500% threshold current stimulus; 9/48 cells). The M-current was significantly larger in phasic neurons than tonic neurons (Fig. 8A) (p < 0.05). The delayed rectifier current was significantly smaller in phasic cells with test steps depolarized to 0 mV (Fig. 8B) (p < 0.05). Phasic neurons expressed a smaller amplitude sodium current than tonic neurons (Fig. 8C) (p < 0.01). We further examined the sodium currents by measuring the maximum rising slope of the action potential in the matched current clamp data for the same cells. The maximum rising slope is directly related to the maximum amplitude of the sodium current (Akopian et al., 1999; Renganathan et al., 2001). We found that phasic neurons expressed a significantly smaller maximum rise slope compared with tonic neurons, consistent with a smaller sodium current in those cells (134.05 ± 8.29 mV/ms vs 165.68 ± 9.83 mV/ms for phasic and tonic cells, respectively; p < 0.05). The calcium-dependent potassium current was also smaller in phasic cells compared with tonic cells (Fig. 8D) (p < 0.05). These findings suggest that the specific firing patterns of sympathetic neurons are correlated to specific expression patterns of at least four voltage-gated currents.
Figure 8.

Phasic and tonic neurons express different levels of four voltage-gated currents. Neurons were recorded from in NGF and identified as either tonic or phasic in current clamp before switching to voltage clamp to record four ionic currents. A, A plot of current amplitude versus voltage test step shows that the M-current was significantly smaller in tonic neurons (open triangles, n = 9) compared with phasic neurons (filled circles, n = 10) under identical experimental conditions (50 ng/ml NGF, p < 0.05, two-way ANOVA followed by the Tukey test). B, A current–voltage plot shows that the delayed rectifier current was significantly smaller in phasic neurons compared with tonic neurons for voltage steps depolarized to 0 mV (p < 0.05, Student's t test). C, A plot of sodium current amplitude versus voltage step shows that this current was smaller in phasic neurons (p < 0.01, two-way ANOVA followed by the Tukey test). D, A plot of the tail current amplitude, used to measure calcium-dependent potassium current, versus test step amplitude showing that this current was smaller in phasic compared with tonic neurons (p < 0.05, two-way ANOVA followed by the Tukey test).
Differential regulation of four voltage-gated currents by p75 and TrkA
The finding of systematic differences in four currents between phasic and tonic cells acutely treated with NGF suggested that tonic and phasic firing patterns are correlated with specific expression patterns of voltage-gated currents. We hypothesized that the relative activity in the p75 and TrkA signaling pathways determines firing pattern through regulation of these currents. In this model, high p75 signaling would increase M-current and decrease the sodium, delayed rectifier, and calcium-dependent potassium currents, leading to phasic firing. High relative TrkA signaling would lead to tonic firing and opposite regulation of the four voltage-gated currents. We tested this by examining spike output and the four voltage-gated currents under conditions mimicking p75 activation (C2-ceramide, 25 μm; n = 14 for all four currents). We compared this to saline alone (saline; n = 19), a condition in which TrkA is activated, but p75 is not (Fig. 6). Indeed, differences in receptor affinity predict that TrkA is selectively activated under this condition. Interaction of neurotrophins with their specific Trk receptors is of a relatively high affinity (Kd ∼10−11 m) compared with the lower-affinity p75 receptor (Kd ∼10−9 m) (Rodríguez-Tébar et al., 1990, 1992; Chao and Hempstead, 1995; Chao, 2003; Reichardt, 2006; Blöchl and Blöchl, 2007). We include NGF in our growth medium for cell survival at 5 ng/ml (∼4 × 10−10 m), a concentration that preferentially activates TrkA-high-affinity sites (Maliartchouk and Saragovi, 1997; Ivanisevic et al., 2003) and that results in sustained TrkA signaling over the time course of experiments (Fig. 6). Thus, analysis of C2-ceramide-treated cells compared with cells in saline provides a comparison between neurons with selective activation of a p75-mediated signaling pathway and neurons with predominant signaling through the TrkA pathway.
The C2-ceramide- and saline-treated cells showed systematic differences in the expression of voltage-gated currents that corresponded with those observed between phasic and tonic NGF-treated cells. The cells treated with C2-ceramide had a larger M-current than cells recorded from in saline (Fig. 9A) (p < 0.05). The delayed rectifier current was significantly smaller in C2-ceramide cells with test steps depolarized to −10 mV (Fig. 9B) (p < 0.05). Cells treated with C2-ceramide also had a smaller sodium current than saline-treated cells (Fig. 9C) (p < 0.01). The maximum rising slope of action potentials was also significantly smaller in C2-ceramide-treated cells, providing further support of decreased sodium current amplitude (103.63 ± 13.95 mV/ms vs 171.42 ± 9.73 mV/ms, for C2-ceramide versus saline, respectively; p < 0.001). Additionally, the C2-ceramide cells had a smaller calcium-dependent potassium current than saline-treated cells (Fig. 9D) (p < 0.01). Finally, we found that application of BDNF (100 ng/ml, n = 15) increased the M-current to similar levels as C2-ceramide application (data not shown, BDNF significantly different from saline but not C2-ceramide, p < 0.05) providing further confirmation that activation of the p75 signaling pathway is involved in regulation of ionic channels in these cells. Together, these results are consistent with our hypothesis that conditions of higher TrkA activation leads to tonic firing by decreasing the M-current and increasing sodium current, delayed rectifier current, and calcium-dependent potassium current, and that p75 signaling promotes phasic firing via opposite regulation of those four currents.
Figure 9.

Varying TrkA and p75 signaling differentially regulates four voltage-gated currents. A, A plot of current amplitude versus voltage test step shows that the M-type potassium current was larger under conditions of high p75 signaling (filled circles, C2-ceramide n = 14) compared with predominant TrkA activation (open triangles, saline, see Results, n = 19; p < 0.05, two-way ANOVA, followed by the Tukey test). B, A current–voltage plot showing that the delayed rectifier current was smaller in C2-ceramide compared with saline with voltage steps depolarized to −10 mV (p < 0.05, Student's t test). C, A current–voltage plot showing that neurons expressed a smaller voltage-gated sodium current in C2-ceramide compared with saline (p < 0.01, two-way ANOVA, followed by the Tukey test). D, A plot of current density versus voltage step shows that the tail current, representative of the calcium-dependent potassium current, was smaller in C2-ceramide compared with saline (p < 0.01, two-way ANOVA, followed by the Tukey test).
We further examined the differential effect of p75 and TrkA signaling on the M-current since it has been strongly implicated in controlling accommodation in multiple types of neurons including sympathetic neurons (Wang and McKinnon, 1995; Hernandez et al., 2008; Jia et al., 2008). We isolated the M-current using a voltage-clamp antagonist subtraction protocol. Example traces for test steps to −20 mV (black) and −40 mV (gray) are shown (Fig. 10A–C). Muscarine chloride is a potent antagonist of the M-current that acts by activating cholinergic muscarinic receptors (Brown and Adams, 1980; Owen et al., 1990). Voltage steps were performed in control saline, which activated multiple conductances, including the M-current (Fig. 10A). The same voltage steps were then repeated after application of 30 μm muscarine chloride (Fig. 10B). The difference current between these two conditions represents the current that was blocked by muscarine: the M-current (Fig. 10C,D) (Owen et al., 1990). We measured M-current (muscarine difference current) amplitude over the end of depolarizing steps from a holding potential of −60 mV (Fig. 10D, gray rectangle) and determined voltage dependence of activation by measuring tail-current amplitude following steps back to −30 mV, which normalized driving force (Fig. 10D, dashed line). We included 250 nm tetrodotoxin in the bath solution for these experiments to block voltage-gated sodium current.
Figure 10.
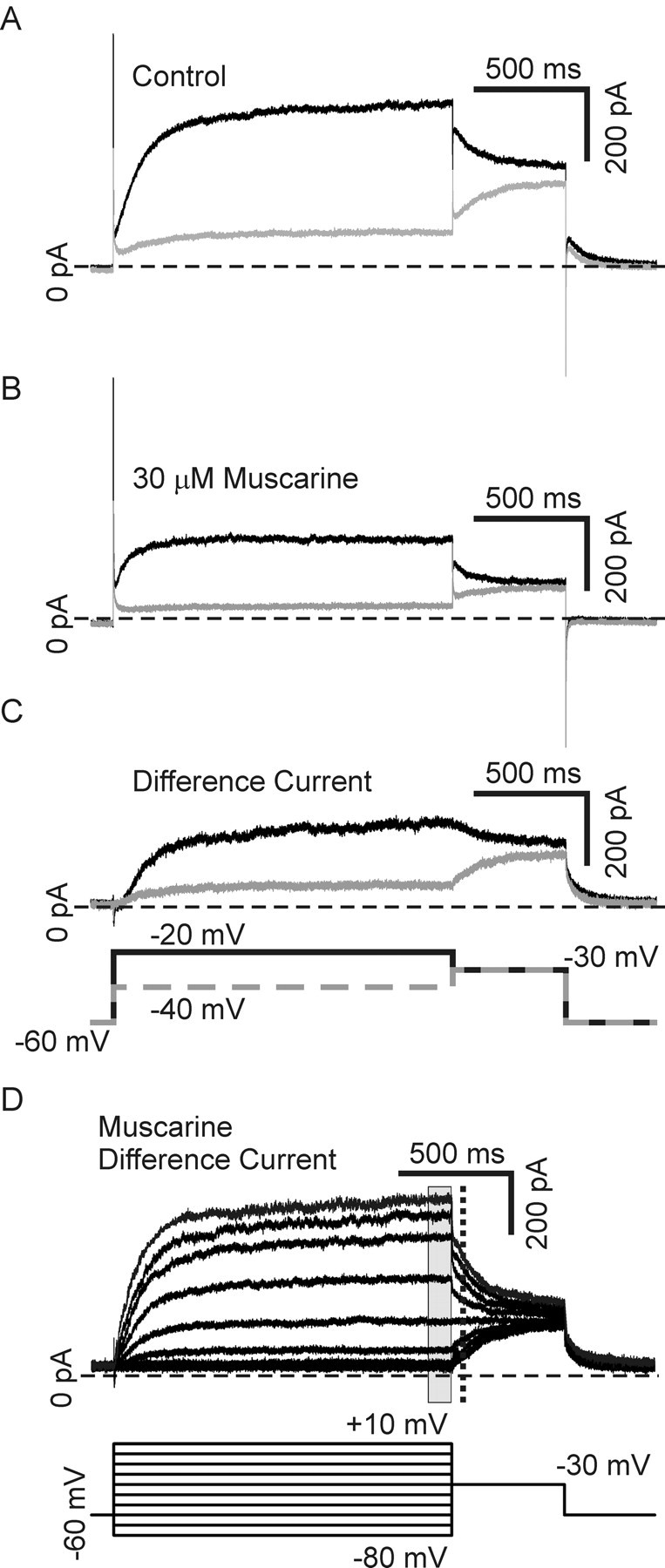
The M-current was isolated using a muscarine subtraction protocol. A, Depolarizing voltage-steps made from a holding potential of −60 mV in the presence of 250 nm tetrodotoxin to block sodium current, elicit mixed outward currents in our neurons. Steps to −40 mV (gray) and −20 mV (black) are shown. B, Application of the M-current antagonist, 30 μm muscarine chloride, blocks the M-current while having little or no effect on other potassium currents. Steps to −40 mV (gray) and −20 mV (black) in the same cell as in A after application of muscarine are shown. C, Subtraction of traces generated in muscarine from those generated in control saline gives a difference current representing the muscarine-sensitive current: the M-current. Muscarine difference currents for the cell depicted in A and B are shown for −40 mV (gray) and −20 mV (black) steps. D, A family of muscarine difference currents generated with test steps ranging from −80 mV to +10 mV made from −60 mV in the presence of 250 nm tetrodotoxin to block sodium current. The current–voltage dependence was determined by measuring the average current at the end of the test step (gray rectangle), and the voltage dependence of activation was determined by measuring tail currents upon stepping back to −30 mV (dashed line) to normalize for driving force.
We used the muscarine subtraction protocol to examine the M-current and compared data between saline (relative TrkA activation, n = 7) and in the presence of 100 ng/ml BDNF (p75 activation, n = 6). Figure 10D shows a family of muscarine difference currents elicited with steps from −60 mV to test steps ranging from −80 mV to +10 mV and then back to −30 mV. We used this protocol to look at both current amplitude and the current–voltage relationship. The current measured at the end of the test step (Fig. 10D, gray rectangle) was significantly larger for BDNF compared with saline over the test step range examined, consistent with our results without pharmacological subtraction (Figs. 9, 11A) (p < 0.05). The current–voltage relationship was examined by fitting Boltzmann functions to plots of the normalized tail current amplitude measured during the step back to −30 mV (Fig. 10A, dashed line). Neither the half-activation voltage nor the slope of current–voltage plots differed between saline and BDNF groups (Fig. 11B) (Boltzmann fit parameters, V½ −33.1 ± 1.2 mV vs −29.6 ± 2.6 mV and τ 7.2 ± 0.44 ms vs 8.6 ± 1.2 ms for saline versus BDNF, respectively). This suggests that p75 activation increases M-current amplitude through a mechanism that is independent of changes in activation voltage sensitivity. While we show the novel finding that p75 and TrkA oppositely regulate amplitude, these results agree with a previous report suggesting that NGF alters M-current amplitude by regulating channel open probability without affecting the voltage dependence (Jia et al., 2008).
Figure 11.

Activation of p75 increases M-current amplitude without altering voltage dependence of activation. A, Plots of muscarine difference current amplitude versus test step [measured over the end of the step (gray rectangle in Fig. 10D)] show that application of 100 ng/ml of the p75 agonist BDNF (open triangles, n = 6) caused an increase in M-current amplitude compared with saline (filled circles, n = 7; p < 0.05, two-way ANOVA, followed by the Tukey test). B, Plots of normalized tail current amplitude with steps back to −30 mV (measured at dashed line in Fig. 10D) show that the voltage dependence of activation of the M-current for cells recorded from in saline (n = 7) and BDNF (n = 6) did not differ (Boltzmann parameters, V½ −33.1 ± 1.2 mV vs −29.6 ± 2.3 mV and τ 7.2 ± 0.4 ms vs 8.6 ± 1.2 ms, for saline and BDNF, respectively). Boltzmann fits are shown graphed as solid lines.
We also examined muscarine difference currents for changes in the kinetics of activation and deactivation. Single exponential functions were fitted to muscarine subtraction current traces generated by depolarizing voltage steps from −60 mV for activation, and with voltage steps from −30 mV down to a range of hyperpolarizing test steps for deactivation (Fig. 12A,C). While there was a trend toward faster kinetics for both activation and deactivation of the M-current for BDNF-treated cells (n = 6) compared with saline (n = 7), this did not reach statistical significance (Fig. 12B,D). The observed trend toward altered M-current kinetics is interesting however, in light of a report showing that M-current in phasic sympathetic neurons possess faster activation and deactivation kinetics than tonic neurons (Jia et al., 2008).
Figure 12.

The kinetics of activation and deactivation were not significantly altered by p75 activation. Single exponential functions (solid lines) fitted to muscarine difference currents elicited by depolarizing voltage steps to −30 mV from −60 mV for cells recorded from in saline or BDNF. B, A plot of the activation time constant (determined from exponential fits) shows no significant difference between saline (filled circles, n = 7) and BDNF (open triangles, n = 6) over test steps ranging from −40 mV to −20 mV. C, Single exponential functions (solid lines) fitted to muscarine difference currents generated with a hyperpolarizing pulse to −60 mV made from a holding potential of −30 mV for cells recorded from in saline (black) or BDNF (gray). D, A plot of the time constant of deactivation shows no significant difference between saline (filled circles, n = 7) and BDNF (open triangles, n = 6) with steps ranging from −40 mV to −90 mV.
Induction of phasic firing by C2-ceramide is blocked by the M-current antagonist, linopirdine
Our finding that p75 signaling regulates M-current amplitude provides a link between neurotrophic regulation of firing properties and regulation of channel properties. We directly tested the hypothesis that an increase of the M-current via p75 signaling contributes to phasic firing by applying the specific M-current blocker linopirdine dihydrochloride (Aiken et al., 1995; Schnee and Brown, 1998) while activating the p75 signaling pathway with C2-ceramide. As expected, application of linopirdine (12.5 μm) caused a decrease in the M-current (Fig. 13A) (12.5 μm, 57.5 ± 8.5% decrease with steps from −30 mV to −50 mV, n = 4, p < 0.01) that was partially reversible. Block of the M-current by linopirdine measured with this protocol is qualitatively similar to the block by muscarine chloride (compare Fig. 7) suggesting that linopirdine is indeed acting via M-current blockade.
Figure 13.
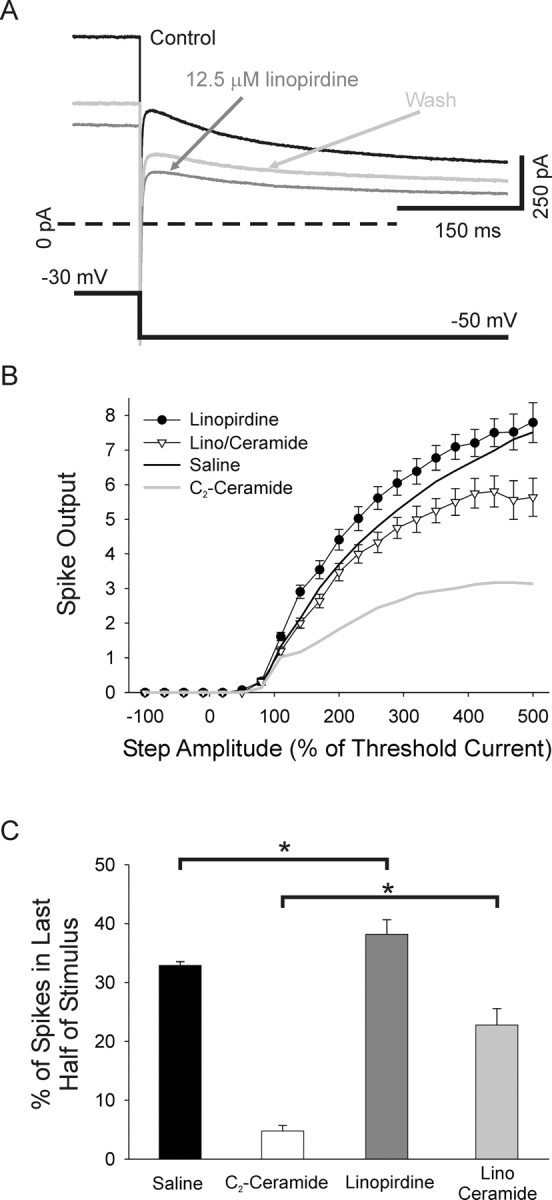
M-current blockade occludes the change in firing pattern induced by p75 activation. A, Superimposed raw current traces recorded in voltage clamp from a cultured sympathetic neuron in response to steps from −30 mV to −50 mV showing that application of 12.5 μm linopirdine decreased the M-type current (black trace versus dark gray trace). This decrease partially reversed after a 15 min wash (light gray trace). B, A plot of spike output versus test step amplitude shows that application of the M-current blocker, 12.5 μm linopirdine with C2-ceramide (open triangles, n = 9), blocked the C2-ceramide-induced decrease in spike output (gray line; p < 0.01, two-way ANOVA, followed by the Tukey test). Application of linopirdine alone (filled circles) caused a trend toward increased spike output compared with saline (black line), which did not reach statistical significance. C, Bar plots of the percentage of spikes occurring in the second half of the stimulus shows that linopirdine alone significantly promoted tonic firing (filled bar versus dark gray bar; p < 0.05, Kruskal–Wallis ANOVA on ranks, followed by Dunn's test). Linopirdine decreased the ability of ceramide to promote phasic firing (ceramide, open bar compared with linopirdine/ceramide, gray bar; p < 0.05, Kruskal–Wallis ANOVA on ranks, followed by Dunn's test).
We found that linopirdine occluded the ability of 25 μm C2-ceramide to reduce spike output (Fig. 13B) (n = 9; lino/ceramide is different from ceramide, linopirdine alone, n = 11 and saline; p < 0.01). Furthermore, linopirdine reduced the ability of C2-ceramide to induce a phasic firing pattern as evidenced by the increase in the number of cells firing action potentials in the second half of a 500% of threshold stimulus in cells treated with linopirdine and C2-ceramide compared with cells treated with C2-ceramide alone (Fig. 13C) (4.78 ± 0.91 vs 22.8 ± 2.81 for ceramide alone and lino/ceramide; respectively, p < 0.05). Linopirdine alone promoted tonic firing as evidenced by the increase in second half spikes compared with saline (Fig. 13C) (32.9 ± 0.64 vs 38.2 ± 2.45 for saline and linopirdine alone, respectively, p < 0.05). Linopirdine alone had no significant effect on spike output compared with saline. There was, however, a trend to higher spike output with linopirdine, consistent with the M-current acting to promote phasic firing and consistent with previous results in rat sympathetic neurons (Jia et al., 2008). These results demonstrate that neurotrophin-dependent modulation of sympathetic neuronal firing patterns is dependent on the modulation of the M-current and suggests that neurotrophins regulate neuronal functional properties through TrkA and p75 regulation of an array of voltage-gated currents.
Discussion
We have shown that neurotrophin-dependent p75 and TrkA signaling contributes to setting the characteristic firing patterns of sympathetic neurons. Cultured rat SCG neurons exhibited a range of firing patterns under baseline conditions with most cells firing tonically. We found that the number of phasic cells increased, and spike output decreased following activation of p75. Conversely, increased activation of TrkA promoted tonic firing and increased spike output. We demonstrated that differences in the specific firing pattern of sympathetic neurons are related to amplitudes of four voltage-gated currents. Phasic cells expressed a larger M-current and smaller sodium current, delayed rectifier current, and calcium-dependent potassium current than tonic neurons. Signaling through p75 and TrkA receptors oppositely regulated the amplitude of these currents, consistent with the idea that neurotrophic regulation of multiple currents contributes to the regulation of firing properties. We found that p75 increased the amplitude of the M-current without influencing voltage dependence or kinetics. Furthermore, application of an M-current antagonist blocked p75-induced phasic firing, showing that p75 acts, at least in part, via regulation of the M-current to direct changes in firing pattern. This suggests that differential neurotrophin signaling within the sympathetic ganglia and from target tissue may regulate sympathetic tone in vivo by influencing the intrinsic excitability of sympathetic neurons.
The action potential waveform and repetitive firing characteristics of neurons are dependent on expressed voltage-gated currents. Here we show that under identical experimental conditions (bath-applied NGF), phasic and tonic firing was associated with different expression patterns of four voltage-gated currents. The M-type potassium current was larger in phasic than tonic neurons, consistent with the prolonged, relatively slow membrane hyperpolarization caused by activation of this current, which leads to spike accommodation (Wang and McKinnon, 1995; Jia et al., 2008). We also found that both delayed rectifier and calcium-activated potassium currents were smaller in phasic than tonic neurons. This is consistent with previous studies suggesting that a decrease in these currents decreases tonic firing, presumably due to a slowing of membrane repolarization following action potentials (Muller and Yool, 1998; Muller et al., 2000; Malin and Nerbonne, 2002; Gu et al., 2007). Voltage-gated sodium current was also decreased in phasic compared with tonic neurons, consistent with its role in action potential generation. Indeed, pharmacological manipulations that increase sodium current promote tonic firing and blockers decrease firing in Purkinje neurons (Khaliq and Raman, 2006). Our findings implicate specific levels of four voltage-gated currents with determining specific firing patterns in sympathetic neurons.
While solid evidence links changes in membrane currents with changes in firing patterns, less is known of how currents are regulated to control spiking. Here we show that in sympathetic neurons, NGF coordinately influences ionic conductances via TrkA and p75 to determine spiking pattern. We found opposite regulation of four voltage-gated currents by the two receptors. Cells acutely treated with the sympathetic p75 agonist BDNF or C2-ceramide, a second messenger generated by p75 activation, expressed a larger M-current. C2-ceramide treatment also resulted in smaller sodium, delayed rectifier, and calcium-dependent potassium currents than in cells under conditions where TrkA signaling predominated (Fig. 6). Cells recorded from in BDNF or C2-ceramide tended to fire phasically while cells recorded from in saline fired tonically (Fig. 4). Furthermore, we found that the M-current blocker linopirdine blocked C2-ceramide-dependent induction of phasic firing, demonstrating a causal relationship between changes in firing pattern and neurotrophin-dependent regulation of ionic currents.
The effects of neurotrophin receptor signaling on the currents that control neuronal firing may occur via changes in channel phosphorylation, or production of intracellular signaling molecules. These molecular changes link neurotrophin signaling to ion channel regulation. This has been reported for molecules such as phosphatidylinositol-bis-phosphate (PIP2) that alter channel conductance or gating (Zhang et al., 2003; Jia et al., 2007, 2008) or induce insertion of additional channels (Zhang et al., 2005). Activation of TrkA potentiates sodium current via phosphoinositide-3 kinase signaling and potentiates calcium current via ras-MAP kinase in bullfrog sympathetic neurons (Lei et al., 1997, 2001; Ford et al., 2008). Activation of p75 causes a ceramide-dependent regulation of sodium and potassium currents in rat sensory neurons via activation of G-protein-coupled receptors (Zhang et al., 2002, 2006b). While the proximal signaling molecules that regulate ion channel properties in our experimental system have not yet been identified, our experiments suggest that these signals at least regulate the level of conductance through the M-type channel. We found, using a muscarine subtraction protocol, that the M-current amplitude was increased by p75 activation, but that neither voltage dependence nor kinetics were affected. This finding is consistent with a previous report that NGF regulates M-current in sympathetic neurons by altering channel open probability without affecting the voltage dependence of activation (Jia et al., 2008). Our results suggest that p75 and TrkA oppositely regulate conductance through M-type channels already existing in the membrane (possibly via regulation of open probability) or influence channel trafficking and membrane insertion.
Our data suggest that p75 and TrkA signaling pathways oppositely regulate firing and four voltage-gated currents. One possibility is that functional interactions between the receptors accounts for these observations. Many studies suggest that p75 and Trk signaling pathways show complex, cell specific functional interactions. For example, p75 activation potentiates TrkA signaling (Epa et al., 2004) and TrkA activation inhibits p75-dependent ceramide accumulation through separate pathways in PC12 cells (Dobrowsky et al., 1995; Bilderback et al., 2001). These findings raise the possibility that the bidirectional regulation of firing patterns we observe in sympathetic neurons could be due to similar functional interactions. The qualitative similarity we find between blocking TrkA (Fig. 6) and activating p75 (Fig. 4) suggests that p75 may induce phasic firing via negative regulation of TrkA. Indeed p75 activation was found to induce axon pruning in mouse sympathetic neurons (Singh et al., 2008) and adult sensory neurons (Kimpinski et al., 1999) via suppression of TrkA signaling. Our data suggest that TrkA promotes tonic firing via regulation of four voltage-gated currents and that p75 promotes phasic firing either by inhibiting those currents or by inhibiting TrkA signaling through a ceramide-dependent mechanism. This model is supported by the similar effects of C2-ceramide and the sympathetic p75-specific agonist BDNF in our cells, although we cannot rule out the possibility that p75 acts via an alternative mechanism and that ceramide has similar, but independent physiological effects.
We found that neurons in low NGF fired tonically and that increasing NGF led to some cells switching to phasic firing while other cells remained tonic (Fig. 2), suggesting that different subpopulations of neurons have distinct responses to neurotrophins. This could be due to variable p75 and TrkA expression in subpopulations of cells. Quantification of staining intensities in sympathetic cultures double labeled with p75 and TrkA antibodies showed considerable variability of expression in individual neurons, with some neurons showing high TrkA and low p75 expression and vice versa, but with many cells expressing each receptor approximately equally (our unpublished results). Although we do not see an obviously bimodal distribution of p75 and TrkA receptor expression, the bimodal distribution of firing properties seen in NGF may be due to allosteric interactions between p75 and Trk receptors, or to downstream interactions between their signaling pathways. Coexpression of p75 is required for high-affinity activation of TrkA by NGF (Hempstead et al., 1991; Kaplan et al., 1991), and the precise ratio of p75:TrkA expression has been found to exquisitely regulate TrkA activation by NGF in PC12 and transfected Chinese hamster ovary cells (Benedetti et al., 1993; Twiss et al., 1998). Evidence suggests that p75 and TrkA receptor expression and the extent of receptor signaling are related in a nonlinear manner, which could produce a bimodal distribution of firing properties in a population of neurons with variable expression of the receptors.
Our findings contribute to a growing body of evidence linking neurotrophin signaling to regulation of voltage-gated currents (Lei et al., 1997; Fjell et al., 1999) and provides evidence of regulation by target-derived factors. While NGF is a target-derived factor for sympathetic neurons, BDNF is made within the sympathetic ganglion itself (Causing et al., 1997). Since BDNF is a p75-specific ligand for these neurons, this suggests that the balance of local and target-dependent neurotrophin signaling could drive the firing rate of the neurons. This is consistent with the finding that sympathetic neurons tend to fire phasically in isolated SCG where access to BDNF would be greater than access to target-derived NGF (Wang and McKinnon, 1995). Thus, our work may provide insight for understanding how neurotrophin availability in vivo contributes to changes in electrophysiological properties and functional output. For example, heart attack changes sympathetic output and cardiac innervation coincident with changes in tissue neurotrophin levels (Kaye et al., 2000; Hiltunen et al., 2001; Qin et al., 2002). Thus, neurotrophin signaling through multiple receptor pathways in vivo could regulate functional neuronal properties and contribute to major disease states when disrupted. Thus, therapies that modulate p75 and Trk signaling may provide treatments for important human diseases.
Footnotes
This work was supported by National Institutes of Health Grants HD042716 and P30NS45713 for Core Facilities for Neurobiology at Brandeis University. We thank Dr. Louis Reichardt for generously providing the TrkA antibody used for immunocytochemistry and the REX antibody used as a functional p75 blocker. We also thank Dr. Adam Taylor for critical reading of this manuscript.
References
- Adamson CL, Reid MA, Davis RL. Opposite actions of brain-derived neurotrophic factor and neurotrophin-3 on firing features and ion channel composition of murine spiral ganglion neurons. J Neurosci. 2002;22:1385–1396. doi: 10.1523/JNEUROSCI.22-04-01385.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Anderson RL, Jobling P, Gibbins IL. Development of electrophysiological and morphological diversity in autonomic neurons. J Neurophysiol. 2001;86:1237–1251. doi: 10.1152/jn.2001.86.3.1237. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–923. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranauskas G. Ionic channel function in action potential generation: current perspective. Mol Neurobiol. 2007;35:129–150. doi: 10.1007/s12035-007-8001-0. [DOI] [PubMed] [Google Scholar]
- Benedetti M, Levi A, Chao MV. Differential expression of nerve growth factor receptors leads to altered binding affinity and neurotrophin responsiveness. Proc Natl Acad Sci U S A. 1993;90:7859–7863. doi: 10.1073/pnas.90.16.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999;18:616–622. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierl MA, Jones EE, Crutcher KA, Isaacson LG. ‘Mature’ nerve growth factor is a minor species in most peripheral tissues. Neurosci Lett. 2005;380:133–137. doi: 10.1016/j.neulet.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Bilderback TR, Gazula VR, Dobrowsky RT. Phosphoinositide 3-kinase regulates crosstalk between Trk A tyrosine kinase and p75(NTR)-dependent sphingolipid signaling pathways. J Neurochem. 2001;76:1540–1551. doi: 10.1046/j.1471-4159.2001.00171.x. [DOI] [PubMed] [Google Scholar]
- Blöchl A, Blöchl R. A cell-biological model of p75NTR signaling. J Neurochem. 2007;102:289–305. doi: 10.1111/j.1471-4159.2007.04496.x. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Cassell JF, Clark AL, McLachlan EM. Characteristics of phasic and tonic sympathetic ganglion cells of the guinea-pig. J Physiol. 1986;372:457–483. doi: 10.1113/jphysiol.1986.sp016020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Chao MV, Hempstead BL. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- Dixon JE, McKinnon D. Expression of the trk gene family of neurotrophin receptors in prevertebral sympathetic ganglia. Brain Res Dev Brain Res. 1994;77:177–182. doi: 10.1016/0165-3806(94)90194-5. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Jenkins GM, Hannun YA. Neurotrophins induce sphingomyelin hydrolysis. Modulation by co-expression of p75NTR with Trk receptors. J Biol Chem. 1995;270:22135–22142. doi: 10.1074/jbc.270.38.22135. [DOI] [PubMed] [Google Scholar]
- Epa WR, Markovska K, Barrett GL. The p75 neurotrophin receptor enhances TrkA signalling by binding to Shc and augmenting its phosphorylation. J Neurochem. 2004;89:344–353. doi: 10.1111/j.1471-4159.2004.02344.x. [DOI] [PubMed] [Google Scholar]
- Fjell J, Cummins TR, Fried K, Black JA, Waxman SG. In vivo NGF deprivation reduces SNS expression and TTX-R sodium currents in IB4-negative DRG neurons. J Neurophysiol. 1999;81:803–810. doi: 10.1152/jn.1999.81.2.803. [DOI] [PubMed] [Google Scholar]
- Ford CP, Wong KV, Lu VB, Posse de Chaves E, Smith PA. Differential neurotrophic regulation of sodium and calcium channels in an adult sympathetic neuron. J Neurophysiol. 2008;99:1319–1332. doi: 10.1152/jn.00966.2007. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol. 2007;580:859–882. doi: 10.1113/jphysiol.2006.126367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannila SS, Lawrance GM, Ross GM, Kawaja MD. TrkA and mitogen-activated protein kinase phosphorylation are enhanced in sympathetic neurons lacking functional p75 neurotrophin receptor expression. Eur J Neurosci. 2004;19:2903–2908. doi: 10.1111/j.0953-816X.2004.03381.x. [DOI] [PubMed] [Google Scholar]
- Hasan W, Jama A, Donohue T, Wernli G, Onyszchuk G, Al-Hafez B, Bilgen M, Smith PG. Sympathetic hyperinnervation and inflammatory cell NGF synthesis following myocardial infarction in rats. Brain Res. 2006;1124:142–154. doi: 10.1016/j.brainres.2006.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawrot E, Patterson PH. Long-term culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen JO, Laurikainen A, Väkevä A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–253. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- Hristova M, Aloe L. Metabolic syndrome—neurotrophic hypothesis. Med Hypotheses. 2006;66:545–549. doi: 10.1016/j.mehy.2005.08.055. [DOI] [PubMed] [Google Scholar]
- Ivanisevic L, Banerjee K, Saragovi HU. Differential cross-regulation of TrkA and TrkC tyrosine kinase receptors with p75. Oncogene. 2003;22:5677–5685. doi: 10.1038/sj.onc.1206864. [DOI] [PubMed] [Google Scholar]
- Jia M, Li M, Liu XW, Jiang H, Nelson PG, Guroff G. Voltage-sensitive calcium currents are acutely increased by nerve growth factor in PC12 cells. J Neurophysiol. 1999;82:2847–2852. doi: 10.1152/jn.1999.82.6.2847. [DOI] [PubMed] [Google Scholar]
- Jia Q, Jia Z, Zhao Z, Liu B, Liang H, Zhang H. Activation of epidermal growth factor receptor inhibits KCNQ2/3 current through two distinct pathways: membrane PtdIns(4,5)P2 hydrolysis and channel phosphorylation. J Neurosci. 2007;27:2503–2512. doi: 10.1523/JNEUROSCI.2911-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Bei J, Rodat-Despoix L, Liu B, Jia Q, Delmas P, Zhang H. NGF inhibits M/KCNQ currents and selectively alters neuronal excitability in subsets of sympathetic neurons depending on their M/KCNQ current background. J Gen Physiol. 2008;131:575–587. doi: 10.1085/jgp.200709924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling P, Gibbins IL. Electrophysiological and morphological diversity of mouse sympathetic neurons. J Neurophysiol. 1999;82:2747–2764. doi: 10.1152/jn.1999.82.5.2747. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res. 2000;86:E80–E84. doi: 10.1161/01.res.86.7.e80. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Raman IM. Relative contributions of axonal and somatic Na channels to action potential initiation in cerebellar Purkinje neurons. J Neurosci. 2006;26:1935–1944. doi: 10.1523/JNEUROSCI.4664-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimpinski K, Jelinski S, Mearow K. The anti-p75 antibody, MC192, and brain-derived neurotrophic factor inhibit nerve growth factor-dependent neurite growth from adult sensory neurons. Neuroscience. 1999;93:253–263. doi: 10.1016/s0306-4522(99)00156-6. [DOI] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD. Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci. 1999;19:5393–5408. doi: 10.1523/JNEUROSCI.19-13-05393.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukuła K, Szulczyk PJ. Properties of Na+ currents in putative submandibular and cardiac sympathetic postganglionic neurones. J Auton Nerv Syst. 1998;69:12–20. doi: 10.1016/s0165-1838(98)00003-4. [DOI] [PubMed] [Google Scholar]
- Kukwa W, Macioch T, Rola R, Szulczyk P. Kinetic and pharmacological properties of Ca(2+) currents in postganglionic sympathetic neurones projecting to muscular and cutaneous effectors. Brain Res. 2000;873:173–180. doi: 10.1016/s0006-8993(00)02552-x. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Lei S, Dryden WF, Smith PA. Regulation of N- and L-type Ca2+ channels in adult frog sympathetic ganglion B cells by nerve growth factor in vitro and in vivo. J Neurophysiol. 1997;78:3359–3370. doi: 10.1152/jn.1997.78.6.3359. [DOI] [PubMed] [Google Scholar]
- Lei S, Dryden WF, Smith PA. Nerve growth factor regulates sodium but not potassium channel currents in sympathetic B neurons of adult bullfrogs. J Neurophysiol. 2001;86:641–650. doi: 10.1152/jn.2001.86.2.641. [DOI] [PubMed] [Google Scholar]
- Llinás RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Lockhart ST, Turrigiano GG, Birren SJ. Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci. 1997;17:9573–9582. doi: 10.1523/JNEUROSCI.17-24-09573.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Birren SJ. Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol. 2006;96:946–958. doi: 10.1152/jn.01078.2005. [DOI] [PubMed] [Google Scholar]
- Maliartchouk S, Saragovi HU. Optimal nerve growth factor trophic signals mediated by synergy of TrkA and p75 receptor-specific ligands. J Neurosci. 1997;17:6031–6037. doi: 10.1523/JNEUROSCI.17-16-06031.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Nerbonne JM. Delayed rectifier K+ currents, IK, are encoded by Kv2α-subunits and regulate tonic firing in mammalian sympathetic neurons. J Neurosci. 2002;22:10094–10105. doi: 10.1523/JNEUROSCI.22-23-10094.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McFarlane S, Cooper E. Postnatal development of voltage-gated K currents on rat sympathetic neurons. J Neurophysiol. 1992;67:1291–1300. doi: 10.1152/jn.1992.67.5.1291. [DOI] [PubMed] [Google Scholar]
- Mischel PS, Smith SG, Vining ER, Valletta JS, Mobley WC, Reichardt LF. The extracellular domain of p75NTR is necessary to inhibit neurotrophin-3 signaling through TrkA. J Biol Chem. 2001;276:11294–11301. doi: 10.1074/jbc.M005132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YL, Yool AJ. Increased calcium-dependent K+ channel activity contributes to the maturation of cellular firing patterns in developing cerebellar Purkinje neurons. Brain Res Dev Brain Res. 1998;108:193–203. doi: 10.1016/s0165-3806(98)00049-2. [DOI] [PubMed] [Google Scholar]
- Muller YL, Reitstetter R, Yool AJ. Antisense knockdown of calcium-dependent K+ channels in developing cerebellar Purkinje neurons. Brain Res Dev Brain Res. 2000;120:135–140. doi: 10.1016/s0165-3806(00)00004-3. [DOI] [PubMed] [Google Scholar]
- Owen DG, Marsh SJ, Brown DA. M-current noise and putative M-channels in cultured rat sympathetic ganglion cells. J Physiol. 1990;431:269–290. doi: 10.1113/jphysiol.1990.sp018330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Qin F, Vulapalli RS, Stevens SY, Liang CS. Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am J Physiol Heart Circ Physiol. 2002;282:H363–H371. doi: 10.1152/ajpheart.00319.2001. [DOI] [PubMed] [Google Scholar]
- Randolph CL, Bierl MA, Isaacson LG. Regulation of NGF and NT-3 protein expression in peripheral targets by sympathetic input. Brain Res. 2007;1144:59–69. doi: 10.1016/j.brainres.2007.01.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tébar A, Dechant G, Barde YA. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Tébar A, Dechant G, Götz R, Barde YA. Binding of neurotrophin-3 to its neuronal receptors and interactions with nerve growth factor and brain-derived neurotrophic factor. EMBO J. 1992;11:917–922. doi: 10.1002/j.1460-2075.1992.tb05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rola R, Szulczyk P. Quantitative differences between kinetic properties of Na(+) currents in postganglionic sympathetic neurones projecting to muscular and cutaneous effectors. Brain Res. 2000;857:327–336. doi: 10.1016/s0006-8993(99)02318-5. [DOI] [PubMed] [Google Scholar]
- Rydén M, Hempstead B, Ibáñez CF. Differential modulation of neuron survival during development by nerve growth factor binding to the p75 neurotrophin receptor. J Biol Chem. 1997;272:16322–16328. doi: 10.1074/jbc.272.26.16322. [DOI] [PubMed] [Google Scholar]
- Schnee ME, Brown BS. Selectivity of linopirdine (DuP 996), a neurotransmitter release enhancer, in blocking voltage-dependent and calcium-activated potassium currents in hippocampal neurons. J Pharmacol Exp Ther. 1998;286:709–717. [PubMed] [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK, Park KJ, Hong EJ, Kramer BM, Greenberg ME, Kaplan DR, Miller FD. Developmental axon pruning mediated by BDNF-p75NTR-dependent axon degeneration. Nat Neurosci. 2008;11:649–658. doi: 10.1038/nn.2114. [DOI] [PubMed] [Google Scholar]
- Twiss JL, Wada HG, Fok KS, Chan SD, Verity AN, Baxter GT, Shooter EM, Sussman HH. Duration and magnitude of nerve growth factor signaling depend on the ratio of p75LNTR to TrkA. J Neurosci Res. 1998;51:442–453. doi: 10.1002/(SICI)1097-4547(19980215)51:4<442::AID-JNR4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Wang HS, McKinnon D. Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J Physiol. 1995;485:319–335. doi: 10.1113/jphysiol.1995.sp020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weskamp G, Reichardt LF. Evidence that biological activity of NGF is mediated through a novel subclass of high affinity receptors. Neuron. 1991;6:649–663. doi: 10.1016/0896-6273(91)90067-a. [DOI] [PubMed] [Google Scholar]
- Wu CW, Yeh HH. Nerve growth factor rapidly increases muscarinic tone in mouse medial septum/diagonal band of Broca. J Neurosci. 2005;25:4232–4242. doi: 10.1523/JNEUROSCI.4957-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt S, Davies AM. Regulation of nerve growth factor receptor gene expression in sympathetic neurons during development. J Cell Biol. 1995;130:1435–1446. doi: 10.1083/jcb.130.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Slonimsky JD, Birren SJ. A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci. 2002;5:539–545. doi: 10.1038/nn0602-853. [DOI] [PubMed] [Google Scholar]
- Zaccaro MC, Ivanisevic L, Perez P, Meakin SO, Saragovi HU. p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J Biol Chem. 2001;276:31023–31029. doi: 10.1074/jbc.M104630200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohács T, Lopes CM, Jin T, Logothetis DE. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na(+) current and delayed rectifier K(+) current in rat sensory neurons. J Physiol. 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J Physiol. 2006a;575:101–113. doi: 10.1113/jphysiol.2006.111575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol. 2006b;96:1042–1052. doi: 10.1152/jn.00120.2006. [DOI] [PubMed] [Google Scholar]