

Abstract
Hyperthermophilic archaeal viruses including Sulfolobus spindle-shaped viruses (SSVs) such as SSV-1 and SSV-Ragged Hills exhibit remarkable morphology and genetic diversity. However, they remain poorly understood, in part because their genomes exhibit limited or unrecognizable sequence similarity to genes with known function. Here we report structural and functional studies of E73, a 73-residue homodimeric protein encoded within the SSV-Ragged Hills genome. Despite lacking significant sequence similarity, the NMR structure reveals clear similarity to ribbon-helix-helix (RHH) domains present in numerous proteins involved in transcriptional regulation. In vitro dsDNA binding experiments confirm the ability of E73 to bind dsDNA in a non-specific manner with micromolar affinity, and characterization of the K11E variant confirms the location of the predicted DNA binding surface. E73 is distinct, however, from known RHHs. The RHH motif is elaborated upon by the insertion of a third helix that is tightly integrated into the structural domain, giving rise to the “RH3” fold. Within the homodimer, this helix results in the formation of a conserved, symmetric cleft distal to the DNA binding surface, where it may mediate protein-protein interactions, or contribute to the high thermal stability of E73. Analysis of backbone amide dynamics by NMR provides evidence for a rigid core, and fast ps-ns timescale NH bond vector motions for residues located within the antiparallel β-sheet region of the proposed DNA-binding surface, and slower μs to ms timescale motions for residues in the α1-α2 loop. The role of E73 and its SSV homologs in the viral life cycle are discussed.
The last decade has seen resurgent interest in viruses outside traditional agricultural and medical interests. Reasons include growing appreciation for their enormous abundance, their huge impact on global carbon and nitrogen cycles, the recognition that viruses represent the greatest reservoir of genetic diversity on the planet, and their central role in evolution, where viruses clearly play a major role in horizontal gene transfer (1–3). In addition, roles for viruses in several major evolutionary transitions have also been proposed, including facilitating the development of DNA and DNA replication mechanisms (4) and the origin of the eukaryotic nucleus (5). Finally, there is also considerable interest in viral genesis and evolution in and of itself (3). In order to evaluate broad evolutionary hypotheses and relationships between viruses, knowledge of viruses infecting the Archaea, the third domain of life, is clearly essential.
Remarkably, the virosphere has now been shown to extend to almost every known environment on earth, including the extreme acidic, thermal and saline environments where archaeal organisms may dominate (6, 7). It is from these environments that the best studied archaeal viruses have been isolated. However, relative to the bacterial and eukaryotic domains, where more than 5,000 viruses have been studied in detail, archaeal viruses are vastly understudied, with fewer than 50 viral species described in any detail (3).
The Crenarchaea and Euryarchaea are the most intensively studied archaeal phyla. Euryarchaeal viruses are most frequently head-and-tail phages (8), although there are notable exceptions (3, 9, 10). In contrast, viruses infecting the Crenarchaea exhibit remarkably unusual and diverse morphotypes (3, 6, 7, 11). On the basis of these unusual morphologies and their genetic diversity, crenarchaeal DNA viruses have been classified into 10 viral families, with additional viruses awaiting assignment (3).
The Fuselloviridae (12–14), or Spindle-shaped viruses (SSV), which are ubiquitous in acidic hot springs around the world, were among the first crenarchaeal viral families to be recognized, and are important model systems for archaeal viruses. SSVs are generally characterized by ~ 60 × 100 nm lemon-shaped virions, with tail fibers emanating from one end, although some members show more pleomorphic morphologies, and package circular double stranded DNA (dsDNA) genomes approximately 16 kbp in size (14). The genomic sequence for SSV1 (15), the type virus for the fuselloviridae, contains 34 putative open reading frames (ORFs). The virion has been shown to contain 5 proteins: VP1, VP2 and VP3, and small amounts of D244 and C792 (16, 17). VP1 and VP3 are putative capsid proteins, while VP2 is a small DNA binding protein thought to be involved in packaging viral DNA.
Importantly, only a few ORFs in these viral genomes show significant identity to genes with known function. For SSV1, ORF D335 encodes an integrase (18) and has been experimentally characterized (19–22), while B251 shows weak homology to DNaA (23). More recently, improved bioinformatics approaches have also identified putative zinc finger motifs in B129, C102a and A79, and suggest the presence of ribbon-helix-helix motifs in C80 and E51 (17, 24, 25). Despite such predictions, bioinformatics approaches are currently unable to provide functional predictions for a large majority of the SSV1, and other crenarchaeal viral proteomes.
However, because 3D structural similarities persist longer than similarities in genomic or amino acid sequence (11, 26, 27), structural studies can identify distant evolutionary relationships, or other structural features that, in-turn, suggest functional roles for these proteins (11, 17, 27–35). Combined structural and biochemical work on fuselloviral proteins have thus identified D63 as a potential ROP-like adaptor protein (11, 28), F93 and F112 as winged-helix DNA binding proteins (17, 27), and SSV1 D244 and SSVRH D212 as nucleases with similarity to Holliday-junction cleavage enzymes (34).
Here, we turn our structural focus to ORF E73 from SSV-RH (14). Though not among the current set of 13 core genes common to all fuselloviruses (13), E73 and its orthologs are found in six of the eight SSV genomes currently in the public data bases (13, 36), where 86% of the residues in SSV-RH E73 are strictly conserved in a multiple sequence alignment with SSV2 79a, SSV4 73, SSV5 GP23 and SSV6 GP17. Identity falls to 31% between E73 and SSV1 E51, which lacks a C-terminal extension present in other SSV orthologs, but gene synteny is conserved. Similarity to E51 is also noteworthy due to the work of Frols et al. (24), who found that UV irradiation of the infected host results in a chronologically regulated SSV1 transcription cycle. This begins with a small UV inducible immediate early transcript, and ensuing expression of the 3 early transcripts, including T5 which encodes E51. This is followed by late transcripts and subsequent viral production.
CD (37) and BLAST (38) searches with E73 do not identify conserved domains or significant similarities to proteins of known function. However, more recent profile-profile search methods like HHpred (39–41) suggest distant similarity to the ribbon-helix-helix DNA binding motif within the N-terminus of E73 (17, 24, 25), and a C-terminal extension of unknown function. Specifically, 4 of the top 5 hits from HHpred search (pdb70_2Mar12 database) map the first 50 residues of E73 to proteins containing the RHH motif, with e-values of 7.4•10−3 for ParG (42), 5.5•10−2 for CcdA (43), 0.92 for Shigella flexneri yiiF (Northeast Structural Genomics Consortium, unpublished) and 17 for PutA (44). Here we report the NMR based structure and backbone dynamics of E73 that reveal an interesting elaboration on a well known ribbon-helix-helix motif that we term the RH3 domain, complemented with biochemical work that demonstrates a role for the E73 RH3 domain in DNA recognition and confirms the predicted DNA binding surface. Potential roles for E73 as a transcriptional regulator early in the SSV life cycle are discussed.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Cloning, expression, and purification were carried out as previously described (45). For uniformly 15N or tandem 15N and 13C labeling, transformed BL21(DE3) (Invitrogen) bacteria were grown in minimal media supplemented with (1 g 15NH4Cl and 2.5 g [13C]-glycerol)/L, respectively, as described by Studier et al. (46).The vector containing the E73 K11E mutant was generated using the QuickChangeII kit (Agilent). Expression and purification of the variant protein was as described above.
Chemical and Thermal Stability
Guanidine hydrochloride (GdnHCl)-induced equilibrium unfolding was carried out by monitoring the molar ellipticity at 222 nm on a Jasco J-810 spectropolarimeter (Jasco Inc., Easten, MD) at a concentration of 10 μM (monomer concentration as assessed by absorbance measurements at 280 nm (OD280 nm readings) in a buffer of 0.1 M KCl, 0.05 M K2HPO4, pH 7.0 at 25°C. Similarly, thermal denaturation was followed by the change in molar ellipticity at 222 nm from 4 °C to 98 °C in a 1 cm path length cell with increasing temperature at 1 °C/min. For both GdnHCl and temperature denaturation experiments, ellipticity readings were normalized to the fraction of protein folded (ff) or unfolded (fu) using the standard equation ff=([θ]- [θ]u/[θ]n-[θ]u) and fu=(1-ff); where [θ]n and [θ]u represent the ellipticity for the fully folded and fully unfolded species, respectively; [θ] is the observed ellipticity at 222 nm at a given GdnHCl concentration or temperature. Data were analyzed assuming a two-state transition of a folded dimer to two unfolded monomers without intermediates, consistent with denaturation profiles reported for the extremely thermostable RHH gene product ORF56 from the Sulfolobus islandicus plasmid pRN1 (47). ΔGu (H2O) and dissociation constant Kd were calculated as described in Zeeb et al. (48) and Chao et al. (49).
NMR Spectroscopy
All protein spectra were recorded on a Bruker DRX 600 spectrometer at 312 K (39 °C) in a 50 mM potassium phosphate, 1 mM EDTA, 0.1 mM PMSF, 0.01% sodium azide buffer (pH 5.0) containing 5% (v/v) D2O or 100% D2O (for acquisition of 3D 13C-edited 1H-1H-TOCSY, 1H-1H-NOESY and 2D 1H-13C-CT-HSQC spectra) on E73 protein samples of 1 mM dimer concentration (as determined by Bradford assays and OD280 nm readings). Chemical shifts of backbone and side chain atoms were extracted from a series of double and triple resonance NMR experiments including HNCA (50), HNCACB (51), CBCA(CO)NH (52), C(CO)NH (53), HBHA(CO)NH (54), HCC(CO)NH (53), 1H-13C-CT-HSQC (55) and HCCH-TOCSY (56) as reported in (45). 1H, 13C, and 15N chemical shifts were indirectly referenced to DSS. 1H/2H solvent exchange experiments were performed by first lyophilizing 500 μl of a 1 mM (dimer) 15N-labeled E73 protein in sample buffer followed by resuspension of the lyophilized protein in the same volume of 100% D2O, and acquisition of a series of 2D 1H-15N-HSQC correlation spectra at subsequent time points following protein resolubilization in D2O. A control lyophilization experiment was done prior to the H2O/D2O exchange whereby an 2D 1H-15N-HSQC of the protein was recorded following lyophilization and resolubilization in H2O and was found to be identical to the 2D 1H-15N-HSQC spectrum recorded in H2O/buffer of unlyophilized E73 indicating that the protein structure is unaffected by the lyophilization process.
NMR Structure Calculations
Structure calculations were performed using the CNS v1.2 (57) ARIA v2.2 (58) software programs with non crystallographic C2 symmetry (ncs.def) restraints to account for the fact that E73 is a symmetric homodimer (45). The initial structure models were guided by 23 experimentally identified intermolecular 1H-1H-nOes (based on a hypothetical model of the E73 RHH core domain) combined with 61 dihedral angle constraints generated by TALOS based on chemical shift data (59). These models permitted the manual assignment of 527 interproton nOes. The assignment of ambiguous 1H-1H-nOes was performed with ARIA. ARIA calculations proceeded in nine iterations of spectra calibration and assignment of ambiguous and unambiguous 1H-1H nOes and estimates of resulting inter-proton distance restraints followed by calculation of an ensemble of lowest conformational energy structures. Values for the molecular dynamics and simulated annealing protocol (anneal.inp) and the non-crystallographic symmetry (ncs.def) rotation matrix are reported in Supplementary Table S0, and available as Supplementary Information. Following each computational cycle, resulting structures were manually inspected and any restraint violations analyzed and corrected. The ARIA calculations were repeated using NMR assignments generated from previous ARIA iterations until convergence was achieved. For the final set of calculations, 100 structures were generated and the 20 lowest energy structures were selected for analysis. In total, 2,095 nOe-derived interproton distance restraints, 244 dihedral angle restraints, and 100 hydrogen bond distance restraints (i.e. 2 distance restraints were assigned per NH identified as being protected from H2O/D2O exchange) were used as input for ARIA/CNS (Table 1).
Table 1.
Experimental restraints and structural statistics for an ensemble of 20 lowest energy accepted NMR E73 structures
| Number of NMR restraints | ||||||
| Total nOe distance restraints | 2095 | RMSD (Å) | ||||
| Intraresidue | 453 × 2 | Bonds | 0.00300 ± 0.00005 | |||
| Sequential | 239 × 2 | nOes | 0.0258 ± 0.0007 | |||
| Short-range | 24 × 2 | Impropers | 0.324 ± 0.010 | |||
| Medium-range | 191 × 2 | Dihedrals | 0.367 ± 0.029 | |||
| Long-range | 46 × 2 | Backbone atomsa, b | 0.50 ± 0.09 | |||
| Inter-protomer | 189 | Heavy atomsa,c | 1.02 ± 0.14 | |||
| H-bond distance restraintsd | 50 × 2 | Ramachandran analysis from Procheck_NMRa | ||||
| Dihedral angle restraints | 122 × 2 | Most favored regions | 86.6% | |||
| Additionally favored regions | 11.7% | |||||
| Number of NMR restraints violations | Generously allowed regions | 1.7% | ||||
| nOe > 0.5 Å | 0 | Disallowed regions | 0% | |||
| Dihedrals > 0.3 Å | 0 | |||||
| Energiese | Ramachandran Plot Summary from Molprobityf | |||||
| Etotal | 487.1 ± 8.4 | EnOe | 109.2 ± 4.8 | Eimp 19.8 ± 1.2 | Most favored regions | 94.1% |
| Ebond | 22.7 ± 0.8 | Ecdih | 4.0 ± 0.7 | Allowed regions | 4.3% | |
| Eangle | 158.8 ± 2.5 | Evwd | 172.6 ± 4.2 | Disallowed regions | 1.7% | |
| Global Quality Scores | ||||||
| Programs: | Verify3D | ProsaII | Procheckf (phi-psi) | |||
| Raw Score | 0.23 | 0.55 | −0.18 | |||
Root mean square deviation (RMSD) and Ramachandran plot analysis calculated for residue stretch 10–70 & derived from Procheck_NMR
Backbone atoms refer to N, Cα, C′, and O atoms of the protein polypeptide chain
Heavy atoms refer to all atoms except hydrogen atoms
Two H-bond distance restraints were assigned per amide hydrogen protected from H/D exchange
Refers to average energies (kcal/mol) for the 20 structures of E73 in the ensemble calculated from CNSv1.2
Molprobity analysis for selected residue ranges: 10–70 (protomer A) and 10′-70′ (protomer B)
The stereochemistry and quality of these structures was analyzed with PROCHECK-NMR (60), MolProbity (61), Verify3D (62), ProsaII (63), and electrostatic surfaces calculated with the APBS program (64). An ensemble of twenty lowest energy conformers of E73 has been deposited in the protein data bank, PDB ID 4aai, and code r4aaimr for the NMR restraints. Figures were prepared with PYMOL (65). Structural homology was identified with the DALI (66) SSM (67) web servers; superposition utilized the Least Square Alignment protocol of Kabsch et al. (68) as implemented in LSQMAN (69).
15N NMR Relaxation Experiments
Backbone amide 15N NMR relaxation experiments including measurements of 15N-T1, 15N-T2, and heteronuclear 15N-[1H]-nOe were conducted in duplicate on a Bruker DRX 600 MHz solution NMR spectrometer using standard NMR pulse sequences (70–72) at 312 K and a pH of 5.0, in a manner analogous to our recent work with tryptophan repressor variants (73, 74). Briefly, 15N-T1 relaxation profiles were sampled at eight different relaxation delay time points of 40 ms, 96 ms, 200 ms, 400 ms, 600 ms, 1,000 ms, and 1,200 ms. 15N-T2 relaxation profiles were sampled at eight different relaxation delay periods of 8 ms, 16 ms, 32 ms, 40 ms, 64 ms, 80 ms, 104 ms, and 152 ms, with the delay between the series of 15N 180 degree pulses applied in the CPMG sequence set to 0.5 ms (75, 76). The 15N-T1 and 15N-T2 data were collected using 512 complex points in the 1H acquisition time dimension (t2) and 256 complex data points in the 15N (t1) indirect time evolution dimension, using a WALTZ-16 15N-decoupling scheme during data acquisition. 15N-T1 and 15N-T2 were calculated from the series of NMR spectra using a two-parameter single exponential decay function:
| (1) |
Where I(t) is the peak height after a delay time t and Io is the signal height at time t = 0.
Heteronuclear 15N-[1H]-nOes were measured using a water-flip-back 2D heteronuclear 1H/15N nOe pulse sequence and the results corrected for the finite repetition delay according to the method of Grzesiek and Bax (71). For each amide, the 15N-[1H]-nOe was established as the ratio of peak intensities (I/Io) from NMR data sets acquired with (I) or without (Io) 1H presaturation. 15N-[1H]-nOe spectra were recorded using 1024 and 256 complex data points in t2 and t1, respectively, using 48 scans per t1 increment. The 15N-[1H]-nOe experiments were recorded using a delay of 4.5 sec between scans to minimize the introduction of systematic errors in measured 15N-[1H]-nOes that could arise from incomplete signal recovery and H2O saturation (71). For the datasets acquired with 1H saturation, the 4.5 sec recovery delay incorporated a 1H presaturation pulse sequence whereby a series of 90 μsec pulses interspersed with a 50 msec delay were applied at an radiofrequency field strength of 5 kHz for a time duration of ~ 4 sec.
To minimize the impact of magnetic field drift, 15N-T2 and 15N-[1H]-nOe data were collected in an interleaved manner, while 2D 15N-T1 datasets were acquired consecutively using a list of shuffled relaxation delay time points.
All relaxation data were processed using NMRPipe, NMRDraw and Sparky (77, 78). The resulting 15N-T1, 15N-T2, and 15N-[1H]-nOe data were then analyzed in terms of spectral density functions as described by Bracken et al. (79) and more recently in Tripet et al. (74). The data were also analyzed using the FastModelFree program (80) which interfaces with the ModelFree 4.0.1 program of Palmer and colleagues (81) and is based on the extended model-free formalism developed by Lipari and Szabo (82, 83). The 15N NMR relaxation data have been deposited in the BioMagResBank (BMRB) under accession number 17069 and are also reported in Supplementary Tables S1–S3.
Electromobility Shift Assays (EMSA)
A library of nineteen 1,000 bp DNA fragments covering the entire SSV-RH genome was generated by PCR and used to screen for E73 DNA recognition. To eliminate the possibility of missing a binding site at the junctions, each 1,000 bp fragment (except for 1) overlapped the previous fragment by 100 bp. The electrophoretic mobility of each DNA fragment in the presence of E73 was analyzed on agarose gels. Each DNA fragment (100–200 ng) was incubated in 20 μl of binding buffer (10 mM Tris-HCl, pH 8.0, 10 mM HEPES, 1 mM EDTA, 50 mM KCl, 50 μg/ml bovine serum albumin, and 1,000 ng of unrelated DNA (300 and 600 bp fragments generated by PCR from the Staphylococcus aureus IsdB gene) for 30 min at 48 °C with increasing amounts of E73 (from 0 to 20μM). Loading buffer (5 μl of 20 mM Tris-HCl, 10 mM EDTA, 50% glycerol) was added, and 25 μl of the sample mixture analyzed on a 1.2% agarose gel with 0.5X TBE running buffer. Samples were run at 135 V for 10 min, followed by 1 hour gel migration at 100 V, and visualized under UV light following sypro-gold (Invitrogen, Inc.) staining.
RESULTS
Thermostable Dimer
Previously reported biophysical characterization utilizing size exclusion chromatography, analytical ultracentrifugation, dynamic light scattering and 2D 1H-15N NMR spectroscopy indicated that E73 is a homodimer in solution with a molecular mass of 20 kDa (45). We have now also assessed the stability of the E73 protein to chemical and temperature denaturation. Chemical denaturation of E73 with guanidinium hydrochloride follows a two-state unfolding process with a relatively high unfolding midpoint at 4.8 M (Fig. 1). Consistent with these data, thermal denaturation analysis also showed the dimeric E73 protein to be extremely thermostable with a Tm ≥ 98°C (Fig. 1B insert). Calculation of the free energy change for unfolding of E73 dimer (i.e. ΔGu (H2O)) yielded a value of 26.5 kcal/mole with an m value of −4.3 kcal mol−1 M−1, and a Kd estimate for protomer dissociation from the E73 dimer of less than one picomolar.
Figure 1.
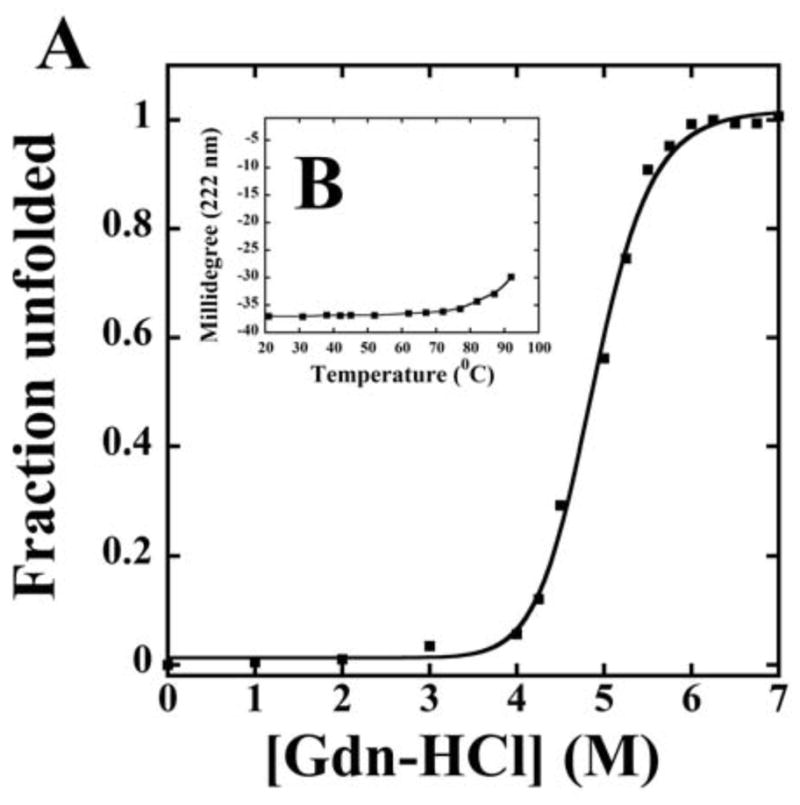
(A) GdnHCl-induced unfolding of E73 monitored by CD at 222 nm at 25°C in 0.1 M KCl, 0.05M PO4 (pH 7) buffer with varying concentrations of GdnHCl. The fraction of protein unfolded (fu) was calculated as described in experimental procedures. E73 protein concentration was 10 μM. Insert (B) Temperature-induced unfolding of the E73 protein monitored by CD at 222 nm in a 0.1 M KCl, 0.05 M PO4, pH 7 buffer. Protein concentration was 10 μM.
Structure of E73
To determine the solution NMR structure of E73, sequential assignments of 1H, 13C and 15N resonances of backbone and side-chain atoms were performed using well-established heteronuclear (1H, 15N, 13C) multidimensional NMR experiments. Excluding the N-terminal methionine, the previously reported backbone and side-chain resonance assignments were nearly complete (> 97%) and have been deposited in the BMRB database under accession number 16177 (45). 13Cα, 13Cβ, 1Hα chemical shifts, short range and sequential 1H-1H nOes, and amides protected from hydrogen-deuterium (H/D) exchange were used to identify secondary structural elements of E73 (Supplementary Figure S1). Each E73 protomer is comprised of an extended β-strand spanning residues 8 to 16 followed by three α-helices designated α1, α2, and α3 that span residues 18–31, 35–53, and 61–69, respectively.
We identified an initial set of 15 interprotomer 1H-1H-nOes consistent with formation of an antiparallel β-sheet and 8 interprotomer 1H-1H-nOes orienting helix α2 close to helix α2′. Using these and other intraprotomer restraints, torsion angle dynamics and simulated annealing calculations using CNS v1.2 and ARIA v2.0 were conducted to determine an initial 3D structure of dimeric E73 in solution. Subsequent rounds of manual and automated nOe assignments using ARIA yielded a final set of nOes (see Table 1) that were used to determine a final ensemble of 20 lowest energy structures with CNS (Figure 2). This ensemble indicated that the E73 dimer adopts a well-ordered conformation that spans residues 10–69, with disordered C- and N-termini. RMSDs for the final set of E73 structures relative to the mean structure was 0.50 Å for backbone atoms of residues 10–15, 18–31, 35–53, and 56–69, which correspond to the well-defined secondary structural elements of E73, and 1.02 Å for all heavy atoms (Table 1). The N- and C-termini of the protein are disordered, as evidenced by the lack of short and long range 1H-1H-nOes and amide hydrogen-deuterium (H/D) exchange (Supplementary Figure S1). 15N NMR relaxation experiments (see below) also indicated that NH bond vectors of amides in these segments of the protein undergo large amplitude ps-ns timescale motions, indicating that the N- and C-termini of E73 are also very flexible.
Figure 2.

Family of accepted E73 structures. The wall-eye stereoview is shown for the overlay of the backbone heavy atoms (N, Ca, C′) of residues 10–69 for the 20 lowest energy structures and was generated with the PYMOL software. The overlay of Cα backbone traces is colored in grey for one of the E73 protomers, and in purple for the second protomer.
As suggested by the initial bioinformatics work and secondary structure assignments from the chemical shift index analysis, the structure of the E73 protomer is comprised of an N-terminal β-strand, followed by the three α-helices α1, α2 and α3. Collectively, the β-strand and helices α1 and α2 form a recognizable ribbon-helix-helix (RHH) substructure, with the β-strand of each E73 protomer interacting together to form a short antiparallel β-sheet (Figure 3). The β-strand and first alpha helix are connected by a sharp turn centered on Asp17. The solvent exposed α1-α2 loop is similarly comprised of charged and polar amino acid residues. Most noteworthy, the amino acid sequence in this loop deviates from the conserved G-X-S/T/N motif found in the loop connecting the α1 and α2 helices in the canonical RHH motif (84). Hydrophobic side chains within helices α1 and α2 (e.g. Leu31, Ile38, Val43 and Trp46) engage in numerous van der Waals contacts within the protein and are responsible for forming a large portion of the E73 hydrophobic core.
Figure 3.

Ribbon representation of a representative conformer from the ensemble of 20 low energy NMR structures of E73. The two respective chains of the E73 homodimer are colored gray and purple. The secondary structure elements are labeled β (beta sheet; residues 11 to 16), α1 (α–helix 1;– residues 18 to 32), α2 (α–helix 2; residues 35 to 53) and– α3 (α-helix 3;– residues 57 to 71). The prime symbol denotes the second protomer chain.
This RHH substructure is augmented by a notable and intriguing embellishment, the addition of a third helix (α3) that is tightly integrated within the RHH homodimer. Specifically, helix α2 is followed by a sharp turn (α2-α3) that positions the α3 helix to run back toward the N-terminus of α1 (Figure 3). Within the context of the homodimer, this sharp α2-α3 turn serves to fold the α3 helix over the α2′ helix of the symmetry related subunit. At the junction of the α1 and α3 helices, we find a constellation of hydrophobic residues that include Phe16, Phe69, Phe70 and Ile7′. These residues appear to position the C-terminal end of the α3 helix toward the N-terminal ends of α1 and β′ in the symmetry related subunit. The net result is that the α1, α2, and α3 helices from one subunit completely encircle the α2′ helix in the neighboring subunit (Figure 3). Similarly, the α1′, α2′ and α3′ helices of the symmetry related subunit also encircle α2. This gives a tightly intertwined dimer, one that cannot dissociate without significant rearrangement of the α3 helices.
We also note that the addition of α3 to the standard RHH fold creates a structural cleft distal to the antiparallel β-sheet of E73’s RHH domain (Figure 4). The tight integration of this third helix within the structural domain and the resulting cleft are features that, to our knowledge, are unique to E73 and have not been observed thus far in structural studies of the RHH fold (see below). The α2-α3 turn creates the upper ridges of this shallow, V-shaped cleft, while the bottom of the cleft is formed by the intertwining of the α3 and α2′ helices (Figures 3 and 4). The α2-α3 loop is largely solvent exposed with the exception of Leu54 whose side-chain forms van der Waals contacts with Leu58 of helix α3′ and with Leu31 of helix α1′.
Figure 4.

Three distinct orientations of the electrostatic potential surface of E73 (blue = positive charge, red = negative charge) calculated as described in the text. The top figures display the different orientations of the ribbon diagram structure of E73 to highlight the orientations of the electrostatic surfaces with respect to the secondary structural elements of the protein. The color scheme for the two protomers is identical to that of Figure 3. (A) Electrostatic potential surface of E73 highlighting the positively charged nature of the top cleft of E73 (arrow); (B) View of electrostatic potential surface of E73 rotated by 90° relative to the orientation shown in (A), with the DNA binding surface of E73 pointing downward and the protein cleft oriented at the top (arrow) and distal to the β-sheet; (C) Molecular surface of E73 rotated by 90° relative to (B) and highlighting the positively charged nature of the protein region engaged in DNA binding.
Structural Homology to Ribbon-Helix-Helix Proteins
DALI (66) and SSM (67) were used to search for structural homologs to E73. Significant structural similarity was limited to ~ 45 residues within the β strand and helices α1 and α2, which constitute the well-known RHH fold described above. However, considering the limited sequence identity, the E73 RHH substructure is quite similar to other bacterial and viral RHH domains. Similar RHH folds identified by the SSM search included PutA [PDB ID 2RBF, 1.52 Å RMSD for 44 aligned residues with 16% sequence identity (85)], CopG [PDB ID 1EA4, 1.55 Å RMSD for 44 aligned residues with 18% sequence identity (86)], the Arc repressor [PDB ID 1MYK, 1.56 Å RMSD for 44 aligned residues with 14% sequence identity (87)], and SvtR from Sulfolobus islandicus Rod-shaped Virus (SIRV1), another hyperthermophilic crenarchaeal virus [PDB ID 2KEL, 1.71 Å for 45 aligned residues sharing 27% sequence identity, (88)]. As an illustration of the structural similarities of E73 RHH domain to that of other RHH proteins, a structural overlay of the RHH motif of E73 with PutA and SvtR is shown in Figure 5. However, none of the RHH proteins that are close homologs of E73 show structural similarity beyond the core RHH motif.
Figure 5.

Structural similarity between E73 and other RHH-containing proteins. (A) Overlay of the ribbon-helix-helix (RHH) domain of E73 (purple) with that of DNA-bound PutA (cyan, PDB ID:2RBF); and in (B) with the RHH structure of the hyperthermophilic protein SvtR (green, PDB ID:2KEL). All structures are shown in their ribbon representations. The 3rd helix of E73 has been omitted in this figure for clarity and to emphasize the close structural similarities of the RHH motifs.
Further scrutiny of the potential structural homologs returned by the DALI and SSM servers did identify more distant proteins with an RHH substructure followed by an additional α-helix. But none of these proteins incorporates the third helix in a manner similar to that seen in the E73 RH3 fold (Figure 6). Specific examples of proteins with a third helix include MetJ [PDB ID 1MJM (89)], the antitoxin FitA [PDB ID 2BSQ (90)] and the Mnt repressor (PDB ID 1MNT, (91)). MetJ consists of a larger domain that incorporates the RHH substructure, with a third helix that follows the RHH motif. However, the helix does not pack against the RHH substructure, and clearly lacks structural equivalence with the third helix of the RH3 fold (see Fig 6A and 6D). For the smaller FitA in the FitA/FitB toxin/antitoxin complex, the RHH domain is followed by a 3rd helix that extends away from the RHH domain to mediate interactions with FitB (Fig 6C). In the absence of FitB, the residues in this third helix are disordered. While in MntR, an additional poorly ordered helix follows the RHH substructure, but runs perpendicular to the α2′ helix, rather than crossing over it to complete encirclement of the α2′ helix (Fig 6B).
Figure 6.

Comparison of the structure of E73 (A) with that of other RHH proteins containing a 3rd helix including (B) MntR (PDB ID:1MNT); (C) FitA (PDB ID: 2BSQ); and (D) MetJ (PDB ID: 1MJM). All structures are shown as ribbon representations with one protomer colored in grey and the other colored in purple (E73), green (MntR), orange (FitA), or blue (MetJ). The 3rd helix of the colored protomers is shown in red for all 4 proteins to highlight that the orientation of the 3rd helix in E73 relative to its RHH motif is unique to E73 and that all three homologs have very distinct 3rd helix topology.
The DNA Binding Site and Superpositional Docking
Least squares superposition was used to dock E73 on the structure of the PutA DNA operator complex to produce a model E73/DNA complex (Figure 7A). This docking model orients a positively charged surface at the E73/DNA interface, and places the anti-parallel β sheet within the DNA major groove where it is poised to make a number of base specific interactions, as well as more generic interactions with the ribose-phosphate backbone (Figure 7A). This superposition suggests potential base specific interactions mediated by Lys11, Thr13 or Ala15, while interactions with the ribose-phosphate backbone are likely to be mediated by basic residues in the N-terminus of E73 including Lys5, Lys6, Lys9, Lys10 and perhaps Lys11. In addition, the main-chain amides at the N-terminal end of helix α2 that do not participate in hydrogen bonding and the α2 helix dipole are also suggested to interact with the negatively charged phosphate in the DNA backbone. Two side chains at the N-terminal end of α2 may also be involved. Specifically, Arg33 and Thr36 might each interact with the ribose-phosphate backbone. Alternatively, Thr36 could also make a base specific interaction. In addition, the docking exercise also places the C-terminus of the α3 helix near the ribose-phosphate backbone, and additional interactions between E73 and DNA could be mediated by basic residues in α3, such as Lys73. Overall, with the exception of α3, this structural arrangement is quite similar to that observed for the PutA and Arc repressor-DNA complexes (85, 92). This further suggests that basic residues within the disordered N-terminus of E73 will interact with the ribose-phosphate backbone.
Figure 7.

(A) Ribbon representation of the E73 homodimer docked by superposition onto Put A DNA (PDB ID:2RBF). The two protomers are colored in purple and grey, DNA is colored cyan. Similar to PutA, the β-sheet interacts with the DNA major groove, and the N-terminus of helix α2 appears to interact with the ribose. (B) EMSA demonstrating the loss of non-specific viral DNA binding affinity by the E73 K11E variant. The E73-DNA intreaction was followed by electromobility shift assay. Increasing concentrations of wild type E73 (lanes 1–5) and the E11K variant (lanes 6–10) were incubated with 182 ng of SSVRH DNA (SSV-RH oligonucleotides 2,900–3,900). Protein (monomer) concentrations were 0 μM (lanes 1, 6), 5 μM (lane 2, 7), 10 μM (lane 3, 8), 15 μM (lane 4, 9) and 20 μM (lane 5, 10). In contrast to wild type E73, the K11E variant failed to shift DNA at any of the concentrations examined (1–20 μM).
E73 Binds dsDNA
A series of overlapping 1000 bp fragments covering the entire SSV-RH genome was generated by PCR and used in an initial screen for potential E73 binding sites in the SSVRH genome. Each 1000 bp fragment was incubated with increasing amounts of E73 (0–20 μM monomer, concentrations at which E73 is a stable dimer as assessed by CD and the thermal denaturation experiments) and analyzed by agarose gel electrophoresis. In general, 1μM E73 shifted each of the 19 PCR amplified products. But as E73 concentrations fell below 1μM (monomer), the DNA band shift was uniformly lost for each for each of the 19 fragments, suggesting the shifts at higher concentrations were due to non-specific interactions. This was confirmed with control experiments using unrelated plasmid DNA that also gave approximate 1 μM dissociation constant. Thus, while we were unable to identify specific E73 binding sites within the SSV-RH genome, this work does confirm the ability of E73 to recognize dsDNA in a non-specific manner with relatively high affinity.
To verify the predicted E73 DNA binding surface, we then characterized the E73 K11E variant in which Lys 11 has been replaced by a glutamic acid residue (Figure 7B) (see Supplemental Figure S6). In comparison to wild-type E73 (lanes 2–5), the EMSA data show that the K11E variant (lanes 7–10) has lost the ability to interact with dsDNA (up to a monomer concentration of 20 μM). The lessened affinity is not due to a structural change, since the K11E variant retains similar biophysical properties and, with the exception of the NMR resonance of K11, exhibits an identical 2D-1H-15N-HSQC spectrum to that of wild type E73.
E73 Backbone Amide Dynamics
15N NMR relaxation parameters (15N-T1, 15N-T2 and 15N-[1H]-nOe) were measured for 58 of the 70 assigned non-proline residue amides of E73. Data for the remaining 12 residues were omitted from the analysis due to overlapped, very weak, or unobservable 15N/1H NMR resonances. The 15N NMR data (see Supplementary Figures S2–S4 and Supplementary Tables S1–S2) were analyzed in terms of spectral density functions and the extended Lipari and Szabo ModelFree formalism, which characterize the internal dynamics of the protein on the ns-ps and μs-ms time scale. As discussed in the Supplementary Information Section, backbone amides in the well-ordered segment of the protein (residues 16–69) display relatively uniform relaxation profiles that are typical of well-folded secondary structural elements of a protein with limited internal dynamics, with a few residues deviating slightly from these trends. More importantly, the 15N NMR relaxation profiles of E73 are very similar to what has been observed for other crenarchaeal viral proteins from hyperthermophiles including the RHH proteins ORF56 (47) and SvtR (88) from Sulfolobus islandicus. This is in contrast to what has been observed for the anti-toxin ParD protein whose N-terminal RHH domain, although structured in solution, is more flexible on a ps-ns timescale than E73, ORF56, or StvtR (93). ParD is also less thermostable than E73 (Tm of 64°C) (94) supporting the notion that restricted internal dynamics is an important factor in the thermostability of hyperthermophilic crenarchaeal viral proteins like E73.
DISCUSSION
Ribbon-Helix-Helix (RHH) containing proteins represent a large and diverse family of proteins found throughout all domains of life, but are particularly common in bacteria, archaea and their viruses where they typically serve as transcription factors (84). For example, the RHH domain is found within larger multidomain proteins such as NikR, the nickel-dependent regulator of nickel uptake in E. coli (95), and PutA, the proline utilization flavoprotein (85). In addition to their RHH domains, these proteins contain associated ligand-binding or catalytic domains. The RHH domain is also found as a standalone module. Examples here include CopG, a transcriptional repressor that in turn represses plasmid replication (96), the Arc repressor, whose DNA binding function initiates the switch between the lytic and lysogenic states in Salmonella bacteriophage P22 (92), and SvtR, a putative transcriptional repressor from the archeal rod shaped virus SIRV1 (88). Structural homology analysis, however, (67, 97, 98) does not identify any RHH containing protein with a third helix structurally equivalent to the α3 helix in E73. The structure of E73 is thus the first observation of the “RH3” fold, in which a third helix (α3) is utilized to complete the encirclement of α2′ in the neighboring RHH subunit.
Interestingly, the tightly intertwined E73 homodimer would appear incapable of dissociating into monomers without significant rearrangement of the α3 helix. At the same time, the 1H/2H exchange and NMR 15N-NMR relaxation data indicate that α3 is well ordered on the surface of the E73 dimer, and is not particularly flexible (Supplemental Figures S2–S4). Combined, these data and the intertwined nature of E73 structure suggest that the α3 helix plays an important role in stabilizing an intrinsic E73 homodimer. In addition, because the α3 helix increases the volume of the hydrophobic core, and because oligomerization is also a well recognized contributor to thermostability (99), α3 is also likely to make a significant contribution to the overall thermostability of E73 (Fig. 1). Thus, α3 clearly plays an important structural role.
However, as evidenced by SvtR (88), RHH domains are clearly thermostable in the absence of α3-like helices. Thus, functional roles for the α3 helix should also be considered. Specifically, within the context of the E73 homodimer, the α2-α3 turn and the N-terminal end of the 3rd helix combine to form a two-fold symmetric cleft that is distal to the canonical DNA binding surface of the RHH fold (Figure 4). This positively charged cleft is clearly the largest pocket or cavity on the protein surface, and for this reason should be considered as a potential ligand binding site (100, 101). We cannot say, however, whether this potential ligand-binding surface is highly conserved, as the unusually high level of sequence conservation among the E73 orthologs does not truly allow the discrimination conserved surface features. If this is a ligand-binding site, its positively charged surface suggests it would recognize a ligand with complementary negative charge. It remains to be determined whether this might be a viral or host protein, nucleic acid, or a regulatory small molecule.
Using an electromobility shift assay (EMSA), we have screened the entire SSV-RH genome for potential E73 binding sites using a series of overlapping 1,000 bp fragments (88). While E73 is capable of shifting each of these fragments, it is unable to discriminate among them, binding each of them with approximately 1 μM dissociation constants. This suggests a non-specific binding interaction with viral DNA, perhaps due to electrostatic interactions of the five lysine residues at the extreme N-terminus (i.e. K5, K6, K9, K10, K11) with the negatively charged ribose phosphate backbone. A structural based alignment of the amino acid sequence of E73 compared to the amino acid sequence of other RHH proteins also highlights the unusual cluster of lysine residues at the extreme N-terminus of E73 that are missing in other RHH homologs (see Supplementary Figure S5). It is interesting to note that replacement of Lys to a Glu at residue position 11 abrogates non-specific DNA binding providing supporting evidence for a functional role of the N-terminal lysine residues of E73 for DNA binding.
The observation of non-specific interactions with viral DNA leads us to postulate that instead of regulating viral DNA expression, the true DNA targets of E73 may instead reside within the host genome and that E73 may regulate expression of Sulfolobus host genes. This conclusion is consistent with the rich abundance of putative transcription factors found in fuselloviral genomes (11, 17, 27). However, a simple gene candidate approach is impractical for screening the larger host genome, and strategies that evaluate the whole genome such as ChIP-on-Chip or ChIP-Seq will probably be required to address this issue. We also note that each of the stand alone RHH containing proteins showing greatest structural similarity to E73, i.e. CopG, Arc Repressor, and SvtR, serve as transcriptional repressors. In light of the minimal structural features present in E73, it is likely the E73 will also serve as a repressor, targeting the host genome.
This scenario would also be consistent with a recent microarray study that observed tight temporal control of SSV1 gene expression and viral production following UV irradiation (24). The process begins with expression of the immediate early gene B49, followed by expression of the T5 and T6 early transcripts. SSV1 E51, an E73 ortholog is found in transcript T5, and is thus expressed early in the viral life cycle. Were it to serve as general repressor of viral transcription, this would be at odds with the continued temporal expression of the viral genome and viral production. It is also at odds with a CopG-like function for E73 and its homologs, suggesting that direct functional analogies to CopG, which represses expression of the RepB nickase needed to initiate rolling circle replication, should be avoided (6).
In conclusion, our structural and functional analysis of SSVRH E73 has revealed a unique adaptation on the RHH fold that we designate RH3. The addition of the third helix results in a tightly intertwined RHH homodimer, which in the case of E73 shows significant thermostability, and a notable cleft distal to the canonical DNA binding site. Structural similarity to CopG, the Arc repressor, and SvtR suggests that E73 is most likely to act as a transcriptional repressor. However, the inability of E73 to discriminate a high affinity binding site in the SSVRH genome suggests that rather than regulating transcription of the viral genome, it will instead target elements of the host genome. Further, in conjunction with the microarray study of Frols et al. on SSV1 (24), it appears this will occur early in the viral life cycle.
Supplementary Material
Acknowledgments
We acknowledge Montana State University Research Experience for Undergraduate (REU) program (NSF-0852043), which supported REU students Guthrie Lewis and Kelly Mildenberger. We are grateful to Guthrie and Kelly for help with expression and purification of 15N-labeled and 15N/13C labeled protein.
ABBREVIATIONS
- APBS
Adaptive Poisson-Boltzman Solver
- ARIA
Ambiguous Restraints for Iterative Assignment
- BMRB
BioMagResBank database
- CD
Circular Dichroism
- CPMG
Carr-Purcell-Meiboom-Gill multipulse sequence
- DIPSI
decoupling in the presence of scalar interactions
- dsDNA
double-stranded DNA
- EDTA
ethylenediaminetetraacetic acid
- EMSA
electromobility shift assay
- GdnHCl
guanidine hydrochloride
- HSQC
heteronuclear single quantum coherence spectroscopy
- IPTG
isopropyl β-thio-galactoside
- NMR
nuclear magnetic resonance spectroscopy
- NOESY
nuclear Overhauser enhancement spectroscopy
- nOe
nuclear Overhauser effect
- ORF
Open Reading Frame
- PCR
polymerase chain reaction
- PDB
Protein Data Bank
- RHH
Ribbon-Helix-Helix
- RMSD
root-mean-squared deviation
- SSM
Secondary Structure Matching
- SSV-RH
Sulfolobus spindle shaped virus - Ragged Hills
- TBE buffer
Tris/Borate/EDTA buffer
- WALTZ
Wideband Alternative-phase Low-power Technique for Zero residual splitting
Footnotes
This work has been supported by NSF grant # MCB-0920312. The NMR experiments were conducted at Montana State University on a Bruker DRX600 NMR spectrometer purchased in part with funds from the NIH shared instrumentation grant program (SIG grant # 1-S10RR13878-01). The Montana State University Research Experience for Undergraduate (REU) program (NSF-0852043) provided support for Taylor Willi.
Supporting information available:
Six Supplementary Figures (Figures S1–S6) and four Supplementary Tables (Tables S0–S3) are included as additional information. The supplementary figures include: a summary of amide H/D exchange data, patterns of sequential and short range NOEs, and 13Cα/β and 1Hα chemical shifts differences from random coil values for SSV-RH E73 (Figure S1); Plots of 15N NMR relaxation profiles and dynamics parameters resulting from reduced spectral density function and Model Free analysis of the 15N NMR relaxation data of E73 (Figures S2–S4); a structure-based amino acid sequence alignment of E73 highlighting structurally equivalent residues among RHH homologs (Figure S5); and a structural representation of E73 residues that may be involved in DNA binding (Figure S6). The three Supplementary Tables provide a list of experimental parameters used in the structure calculations of E73 (Table S0); 15N-NMR relaxation parameters 15N-T1, 15N-T2, 15N-[1H]-nOe (Table S1); Reduced spectral density functions Jeff(0), J(ωN) and J(0.87ωH) (Table S2); and motional parameters (S2, τe, Rex, and model selection) computed for E73 (Table S2). In addition to the Supplementary Figures and Tables, a small section of text describing the 15N NMR relaxation analysis and internal dynamics results is provided.
“This material is available free of charge via the Internet at http://pubs.acs.org”
References
- 1.Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005;13:278–284. doi: 10.1016/j.tim.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Suttle CA. Marine viruses--major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–812. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 3.Pina M, Bize A, Forterre P, Prangishvili D. The archeoviruses. FEMS Microbiol Rev. 2011;35:1035–1054. doi: 10.1111/j.1574-6976.2011.00280.x. [DOI] [PubMed] [Google Scholar]
- 4.Forterre P. Three RNA cells for ribosomal lineages and three DNA viruses to replicate their genomes: a hypothesis for the origin of cellular domain. Proc Nat’l Acad Sci U S A. 2006;103:3669–3674. doi: 10.1073/pnas.0510333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell PJ. Viral eukaryogenesis: was the ancestor of the nucleus a complex DNA virus? J Mol Evol. 2001;53:251–256. doi: 10.1007/s002390010215. [DOI] [PubMed] [Google Scholar]
- 6.Ortmann AC, Wiedenheft B, Douglas T, Young M. Hot crenarchaeal viruses reveal deep evolutionary connections. Nat Rev Microbiol. 2006;4:520–528. doi: 10.1038/nrmicro1444. [DOI] [PubMed] [Google Scholar]
- 7.Prangishvili D, Forterre P, Garrett RA. Viruses of the Archaea: a unifying view. Nat Rev Microbiol. 2006;4:837–848. doi: 10.1038/nrmicro1527. [DOI] [PubMed] [Google Scholar]
- 8.Haring M, Peng X, Brugger K, Rachel R, Stetter KO, Garrett RA, Prangishvili D. Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae. Virology. 2004;323:233–242. doi: 10.1016/j.virol.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Bath C, Cukalac T, Porter K, Dyall-Smith ML. His1 and His2 are distantly related, spindle-shaped haloviruses belonging to the novel virus group, Salterprovirus. Virology. 2006;350:228–239. doi: 10.1016/j.virol.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 10.Bath C, Dyall-Smith ML. His1, an archaeal virus of the Fuselloviridae family that infects Haloarcula hispanica. J Virol. 1998;72:9392–9395. doi: 10.1128/jvi.72.11.9392-9395.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence CM, Menon S, Eilers BJ, Bothner B, Khayat R, Douglas T, Young MJ. Structural and functional studies of archaeal viruses. J Biol Chem. 2009;284:12599–12603. doi: 10.1074/jbc.R800078200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng X, Blum H, She Q, Mallok S, Brugger K, Garrett RA, Zillig W, Prangishvili D. Sequences and replication of genomes of the archaeal Rudiviruses SIRV1 and SIRV2: Relationships to the archaeal Lipothrixvirus SIFV and some eukaryal viruses. Virology. 2001;291:226–234. doi: 10.1006/viro.2001.1190. [DOI] [PubMed] [Google Scholar]
- 13.Redder P, Peng X, Brugger K, Shah SA, Roesch F, Greve B, She Q, Schleper C, Forterre P, Garrett RA, Prangishvili D. Four newly isolated fuselloviruses from extreme geothermal environments reveal unusual morphologies and a possible interviral recombination mechanism. Environ Microbiol. 2009;11:2849–2862. doi: 10.1111/j.1462-2920.2009.02009.x. [DOI] [PubMed] [Google Scholar]
- 14.Wiedenheft B, Stedman K, Roberto F, Willits D, Gleske AK, Zoeller L, Snyder J, Douglas T, Young M. Comparative Genomic Analysis of Hyperthermophilic Archaeal Fuselloviridae Viruses. J Virol. 2004;78:1954–1961. doi: 10.1128/JVI.78.4.1954-1961.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palm P, Schleper C, Grampp B, Yeats S, McWilliam P, Reiter WD, Zillig W. Complete nucleotide sequence of the virus SSV1 of the archaebacterium Sulfolobus shibatae. Virology. 1991;185:242–250. doi: 10.1016/0042-6822(91)90771-3. [DOI] [PubMed] [Google Scholar]
- 16.Reiter WD, Palm P, Henschen A, Lottspeich F, Zillig W, Grampp B. Identification and characterization of the genes encoding three structural proteins of the Sulfolobus virus-like particle SSV1. Mol Genet Genom. 1987;206:144–153. [Google Scholar]
- 17.Menon SK, Maaty WS, Corn GJ, Kwok SC, Eilers BJ, Kraft P, Gillitzer E, Young MJ, Bothner B, Lawrence CM. Cysteine usage in Sulfolobus spindle-shaped virus 1 and extension to hyperthermophilic viruses in general. Virology. 2008;376:270–278. doi: 10.1016/j.virol.2008.03.026. [DOI] [PubMed] [Google Scholar]
- 18.Argos P, Landy A, Abremski K, Egan JB, Haggard-Ljungquist E, Hoess RH, Kahn ML, Kalionis B, Narayana SV, Pierson LS, 3rd, et al. The integrase family of site-specific recombinases: regional similarities and global diversity. EMBO J. 1986;5:433–440. doi: 10.1002/j.1460-2075.1986.tb04229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muskhelishvili G, Palm P, Zillig W. SSV1-encoded site-specific recombination system in Sulfolobus shibatae. Mol Gen Genet. 1993;237:334–342. doi: 10.1007/BF00279436. [DOI] [PubMed] [Google Scholar]
- 20.Clore AJ, Stedman KM. The SSV1 viral integrase is not essential. Virology. 2007;361:103–111. doi: 10.1016/j.virol.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Letzelter C, Duguet M, Serre MC. Mutational analysis of the archaeal tyrosine recombinase SSV1 integrase suggests a mechanism of DNA cleavage in trans. J Biol Chem. 2004;279:28936–28944. doi: 10.1074/jbc.M403971200. [DOI] [PubMed] [Google Scholar]
- 22.Serre MC, Letzelter C, Garel JR, Duguet M. Cleavage properties of an archaeal site-specific recombinase, the SSV1 integrase. J Biol Chem. 2002;277:16758–16767. doi: 10.1074/jbc.M200707200. [DOI] [PubMed] [Google Scholar]
- 23.Koonin EV. Archaebacterial virus SSV1 encodes a putative DnaA-like protein. Nucl Acids Res. 1992;20:1143. doi: 10.1093/nar/20.5.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frols S, Gordon PM, Panlilio MA, Schleper C, Sensen CW. Elucidating the transcription cycle of the UV-inducible hyperthermophilic archaeal virus SSV1 by DNA microarrays. Virology. 2007;365:48–59. doi: 10.1016/j.virol.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 25.Prangishvili D, Garrett RA, Koonin EV. Evolutionary genomics of archaeal viruses: Unique viral genomes in the third domain of life. Virus Res. 2006;117:52–67. doi: 10.1016/j.virusres.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Moult J, Melamud E. From fold to function. Curr Opin Struct Biol. 2000;10:384–389. doi: 10.1016/s0959-440x(00)00101-9. [DOI] [PubMed] [Google Scholar]
- 27.Kraft P, Oeckinghaus A, Kummel D, Gauss GH, Gilmore J, Wiedenheft B, Young M, Lawrence CM. Crystal Structure of F-93 from Sulfolobus Spindle-Shaped Virus 1, a Winged-Helix DNA Binding Protein. J Virol. 2004;78:11544–11550. doi: 10.1128/JVI.78.21.11544-11550.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kraft P, Kummel D, Oeckinghaus A, Gauss GH, Wiedenheft B, Young M, Lawrence CM. Structure of D-63 from Sulfolobus Spindle-Shaped Virus 1: Surface Properties of the Dimeric Four-Helix Bundle Suggest an Adaptor Protein Function. J Virol. 2004;78:7438–7442. doi: 10.1128/JVI.78.14.7438-7442.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larson ET, Eilers B, Menon S, Reiter D, Ortmann A, Young MJ, Lawrence CM. A winged-helix protein from Sulfolobus turreted icosahedral virus points toward stabilizing disulfide bonds in the intracellular proteins of a hyperthermophilic virus. Virology. 2007;38:249–261. doi: 10.1016/j.virol.2007.06.040. [DOI] [PubMed] [Google Scholar]
- 30.Larson ET, Eilers BJ, Reiter D, Ortmann AC, Young MJ, Lawrence CM. A new DNA binding protein highly conserved in diverse crenarchaeal viruses. Virology. 2007;363:387–396. doi: 10.1016/j.virol.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 31.Larson ET, Reiter D, Young M, Lawrence CM. Structure of A197 from Sulfolobus turreted icosahedral virus: a crenarchaeal viral glycosyltransferase exhibiting the GTA fold. J Virol. 2006;80:7636–7644. doi: 10.1128/JVI.00567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khayat R, Tang L, Larson ET, Lawrence CM, Young M, Johnson JE. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc Nat’l Acad Sci U S A. 2005;102:18944–18949. doi: 10.1073/pnas.0506383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lintner NG, Frankel KA, Tsutakawa SE, Alsbury DL, Copie V, Young MJ, Tainer JA, Lawrence CM. The structure of the CRISPR-associated protein Csa3 provides insight into the regulation of the CRISPR/Cas system. J Mol Biol. 2011;405:939–955. doi: 10.1016/j.jmb.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menon SK, Eilers BJ, Young MJ, Lawrence CM. The Crystal Structure of D212 from Sulfolobus Spindle-shaped Virus Ragged Hills reveals a new member of the PD-(D/E)XK nuclease superfamily. J Virol. 2010;84:5890–5897. doi: 10.1128/JVI.01663-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lintner NG, Kerou M, Brumfield SK, Graham S, Liu H, Naismith JH, Sdano M, Peng N, She Q, Copie V, Young MJ, White MF, Lawrence CM. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE) J Biol Chem. 2011;286:21643–21656. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stedman KM, She Q, Phan H, Arnold HP, Holz I, Garrett RA, Zillig W. Relationships between fuselloviruses infecting the extremely thermophilic archaeon Sulfolobus: SSV1 and SSV2. Res Microbiol. 2003;154:295–302. doi: 10.1016/S0923-2508(03)00074-3. [DOI] [PubMed] [Google Scholar]
- 37.Marchler-Bauer ABS. CD-Search: protein domain annotations on the fly. Nucl Acids Res. 2004;32:W327–331. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 40.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucl acids Res. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dlakic M. HHsvm: fast and accurate classification of profile-profile matches identified by HHsearch. Bioinformatics. 2009;25:3071–3076. doi: 10.1093/bioinformatics/btp555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Golovanov AP, Barilla D, Golovanova M, Hayes F, Lian LY. ParG, a protein required for active partition of bacterial plasmids, has a dimeric ribbon-helix-helix structure. Mol Microbiol. 2003;50:1141–1153. doi: 10.1046/j.1365-2958.2003.03750.x. [DOI] [PubMed] [Google Scholar]
- 43.Madl T, Van Melderen L, Mine N, Respondek M, Oberer M, Keller W, Khatai L, Zangger K. Structural Basis for Nucleic Acid and Toxin Recognition of the Bacterial Antitoxin CcdA. J Mol Biol. 2006;364:170–185. doi: 10.1016/j.jmb.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 44.Larson JD, Jenkins JL, Schuermann JP, Zhou Y, Becker DF, Tanner JJ. Crystal structures of the DNA binding domain of Eschericia coli proline utilization A flavoprotein and analysis of the role of Lys 9 in DNA recognition. Protein Sci. 2006;15:2630–2641. doi: 10.1110/ps.062425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schlenker C, Menon S, Lawrence CM, Copié V. 1H, 13C, 15N backbone and side chain NMR resonance assignments for E73 from Sulfolobus spindle-shaped virus ragged hills, a hyperthermophilic crenarchaeal virus from Yellowstone National Park. Biomol NMR Assign. 2009;3:219–222. doi: 10.1007/s12104-009-9179-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 47.Weininger U, Zeeb M, Neumann P, Lowm C, Stubbs MT, Lipps G, Balbach J. Structure-based stability analysis of an extremely stable dimeric DNA binding protein from Sulfolobus islandicus. Biochemistry. 2009;48:10030–10037. doi: 10.1021/bi900760n. [DOI] [PubMed] [Google Scholar]
- 48.Zeeb M, Lipps G, Lilie H, Balbach J. Folding and association of an extremely stable dimeric protein from Sulfolobus islandicus. J Mol Biol. 2004;336:227–240. doi: 10.1016/j.jmb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 49.Chao H, Houston ME, Jr, Grother S, Kay CM, O’connor-McCourt M, Irvin RT, Hodges RS. Kinetic study on the formation of a de novo designed heterodimeric coiled-coil: use of surface plasmon resonance to monitor the association and dissociation of polypeptide chains. Biochemistry. 1996;35:12175–12185. doi: 10.1021/bi9530604. [DOI] [PubMed] [Google Scholar]
- 50.Kay LE, Ikura M, Tschudin R, Bax A. Three-dimensional triple-resonance NMR spectroscopy of isotopically enriched proteins. J Mag Res. 1990;89:496–514. doi: 10.1016/j.jmr.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with {alpha} and proton and nitrogen resonances with alpha and beta carbon resonances in proteins. J Mag Res. 1993;101:201–205. [Google Scholar]
- 52.Grzesiek S, Bax A. An Efficient Experiment for Sequential Backbone Assignment of Medium-Sized Isotopically Enriched Proteins. J Mag Res. 1992;99:201–207. [Google Scholar]
- 53.Grzesiek S, Anglister J, Bax A. Correlation of backbone amide and aliphatic side-chain resonances in 13C/15N-enriched proteins by isotropic mixing of 13C magnetization. J Mag Res Ser B. 1993;101:114–119. [Google Scholar]
- 54.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 55.Vuister GW, Bax A. Resolution enhancement and spectral editing of uniformy [13]C-enriched proteins by homonuclear braodband [13]C decoupling. J Mag Res. 1992;98:428–435. [Google Scholar]
- 56.Bax A, Clore MG, Gronenborn AM. 1H-1H correlation via isotropic mixing of 13C magnetization, a new three-dimensional approach for assigning 1H and 13C spectra of 13C enriched proteins. J Mag Res. 1990;88:425–431. [Google Scholar]
- 57.Brunger AT, Adams PD, Clore MG, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system.CNS: A new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:901–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 58.Habeck M, Rieping W, Linge JP, Nilges M. NOE assignment with ARIA 2.0: the nuts and bolts. Meth Mol Biol. 2004;278:379–402. doi: 10.1385/1-59259-809-9:379. [DOI] [PubMed] [Google Scholar]
- 59.Cornilescu G, Delaglio F, Bax Ad. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 60.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 61.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luthy R, Bowie JU, Eisenberg D. Assessment of protein models with three-dimensional profiles. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 63.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl acids Res. 2007;35(suppl 2):W407–W410. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeLano WL. The PyMOL Molecular Graphics System. 2002 htttp:// http://www.pymol.org.
- 66.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Cryst Sect D. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 68.Kabsch W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Cryst Sect A. 1978;34:827–828. [Google Scholar]
- 69.Kleywegt GJ. LSQMAN. Uppsala University; Uppsala, SWEDEN: [Google Scholar]
- 70.Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- 71.Grzesiek S, Bax A. The importance of not saturating H2O in protein NMR: application to sensitivity enhancement and NOE measurements. J Am Chem Soc. 1993;115:12593–12594. [Google Scholar]
- 72.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 73.Goel A, Tripet BP, Tyler RC, Nebert LD, Copie V. Backbone amide dynamics of apo-L75F-TrpR, a temperature sensitive mutant of the tryptophan repressor protein (TrpR): comparison with the 15N NMR relaxation profiles of wild type and A77V mutant TrpR apo-repressors. Biochemistry. 2010;49:8006–8019. doi: 10.1021/bi100508u. [DOI] [PubMed] [Google Scholar]
- 74.Tripet BP, Goel A, Copié V. Internal dynamics of the tryptophan repressor (TrpR) and two functionally distinct TrpR variants, L75F-TrpR and A77V-TrpR, in their L-trp bound forms. Biochemistry. 2011;50:5140–5153. doi: 10.1021/bi200389k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carr HY, Purcell EM. Effects of Diffusion on Free Precession in Nuclear Magnetic Resonance Experiments. Phys Rev. 1954;94:630. [Google Scholar]
- 76.Meiboom S, Gill D. Modified Spin-Echo Method for Measuring Nuclear Relaxation Times. Rev Sci Inst. 1958;29:688–691. [Google Scholar]
- 77.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax Ad. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 78.Goddard TD, Kneller DG. SPARKY. Vol. 3. University of California; San Fransisco USA: [Google Scholar]
- 79.Bracken C, Carr PA, Cavanagh J, Palmer AG. Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: implications for the entropy of association with DNA. J Mol Biol. 1999;285:2133–2146. doi: 10.1006/jmbi.1998.2429. [DOI] [PubMed] [Google Scholar]
- 80.Cole R, Loria JP. FAST-model free: a program for rapid automated analysis of solution NMR spin-relaxation data. J Biomol NMR. 2003;26:203–213. doi: 10.1023/a:1023808801134. [DOI] [PubMed] [Google Scholar]
- 81.Palmer A. ModelFree Version 4.0. 1998 http://cpmcent.columbia.edu/dept/gsas/biochem/labs/palmer.
- 82.Lipari G, Szabo A. Model-Free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 83.Lipari G, Szabo A. Model-Free approach to the interpretation of nuclear magnetic-resonance relaxation in macromolecules. 2. Analysis of experimental results. J Am Chem Soc. 1982;104:4559–4570. [Google Scholar]
- 84.Schreiter ER, Drennan CL. Ribbon-helix-helix transcription factors: variations on a theme. Nat Rev Microbiol. 2007;5:710–720. doi: 10.1038/nrmicro1717. [DOI] [PubMed] [Google Scholar]
- 85.Zhou Y, Larson JD, Bottoms CA, Arturo EC, Henzl MT, Jenkins JL, Nix JC, Becker DF, Tanner JJ. Structural Basis of the Transcriptional Regulation of the Proline Utilization Regulon by Multifunctional PutA. J Mol Biol. 2008;381:174–188. doi: 10.1016/j.jmb.2008.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costa M, Sola M, del Solar G, Eritja R, Hernandez-Arriaga AM, Espinosa M, Gomis-Ruth FX, Coll M. Plasmid Transcriptional Repressor CopG Oligomerises to Render Helical Superstructures Unbound and in Complexes with Oligonucleotides. J Mol Biol. 2001;310:403–417. doi: 10.1006/jmbi.2001.4760. [DOI] [PubMed] [Google Scholar]
- 87.Schildbach JF, Milla ME, Jeffrey PD, Raumann BE, Sauer RT. Crystal structure, folding, and operator binding of the hyperstable Arc repressor mutant PL8. Biochemistry. 1995;34:1405–1412. doi: 10.1021/bi00004a035. [DOI] [PubMed] [Google Scholar]
- 88.Guilliere F, Peixeiro N, Kessler A, Raynal B, Desnoues N, Keller J, Delepierre M, Prangishvili D, Sezonov G, Guijarro JI. Structure, function, and targets of the transcriptional regulator SvtR from the hyperthermophilic archaeal virus SIRV1. J Biol Chem. 2009;284:22222–22237. doi: 10.1074/jbc.M109.029850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garvie CW, Phillips SE. Direct and indirect readout in mutant Met repressor-operator complexes. Structure. 2000;8:905–914. doi: 10.1016/s0969-2126(00)00182-9. [DOI] [PubMed] [Google Scholar]
- 90.Mattison K, Wilbur JS, So M, Brennan RG. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J Biol Chem. 2006;281:37942–37951. doi: 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 91.Burgering MJM, Boelens R, Gilbert DE, Breg JN, Knight KL, Sauer RT, Kaptein R. Solution Structure of Dimeric Mnt Repressor (1–76) Biochemistry. 1994;33:15036–15045. doi: 10.1021/bi00254a012. [DOI] [PubMed] [Google Scholar]
- 92.Breg JN, van Opheusden JHJ, Burgering MJM, Boelens R, Kaptein R. Structure of Arc represser in solution: evidence for a family of [beta]-sheet DMA-binding proteins. Nature. 1990;346:586–589. doi: 10.1038/346586a0. [DOI] [PubMed] [Google Scholar]
- 93.Oberer M, Zangger K, Gruber K, Keller W. The solution structure of ParD, the antidote of the ParDE toxin-antitoxin module, provides the structural basis for DNA and toxin binding. Protein Sci. 2007;16:1676–1688. doi: 10.1110/ps.062680707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oberer M, Lindner H, Glatter O, Krafky C, Keller W. Thermodynamic properties and DNA binding of the ParD protein from the broad host-range plasmid RK2/RP4 killing system. J Biol Chem. 1999;380:1413–1420. doi: 10.1515/BC.1999.181. [DOI] [PubMed] [Google Scholar]
- 95.Schreiter ER, Wang SC, Zamble DB, Drennan CL. NikR - operator complex structure and the mechanism of repressor activation by metal ions. Proc Natl Acad Sci USA. 2006;103:13676–13681. doi: 10.1073/pnas.0606247103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rasooly A, Rasooly RS. How rolling circle plasmids control their copy number. Trends Microbiol. 1997;5:440–446. doi: 10.1016/S0966-842X(97)01143-8. [DOI] [PubMed] [Google Scholar]
- 97.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 98.Gibrat JF, Madej T, Bryant SH. Surprising similarities in structure comparison. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 99.Vieille C, Zeikus GJ. Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Laskowski RA, Luscombe NM, Swindells MB, Thornton JM. Protein clefts in molecular recognition and function. Protein Sci. 1996;5:2438–2452. doi: 10.1002/pro.5560051206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.