

Abstract
Domain swapping creates protein oligomers by exchange of structural units between identical monomers. At present, no unifying molecular mechanism of domain swapping has emerged. Here we used the protein Cyanovirin-N and 19F-NMR to investigate the process of domain swapping. CV-N is an HIV inactivating protein that can exist as a monomer or a domain-swapped dimer. We measured thermodynamic and kinetic parameters of the conversion process and determined the size of the energy barrier between the two species. The barrier is very large and of similar magnitude to that for equilibrium unfolding of the protein. Therefore, for CV-N, overall unfolding of the polypeptide is required for domain swapping.
INTRODUCTION
Under physiological conditions most proteins exhibit a unique, narrowly distributed ensemble of conformations, broadly termed the native state. Within this native state ensemble, relatively low kinetic barriers separate the individual, very similar conformational sub-states.1 Under specific circumstances, proteins may sample multiple sub-states, and such structural plasticity is exploited in molecular switches. For example, proteins that bind different substrates often employ alternative binding modes that optimize the inter-molecular interactions, which are facilitated by their conformational adaptability. Likewise, oligomerization may occur in different geometries, depending on the environmental conditions. Among thousands of homo-oligomers, a special type of oligomerization involves ‘domain swapping’.2 In domain-swapped structures one monomeric subunit exchanges one or more identical structural elements (domains, subdomains or secondary structure elements) with another monomer. The three-dimensional structure of the pseudo-monomer within the domain-swapped multimer is identical to its corresponding monomer structure, except for the ‘hinge’ region that links the exchanged units.2
Currently, more than 100 domain-swapped structures are deposited in the Protein Data Bank (PDB).3 The analysis of their chain lengths, structural class or amino acid composition does not reveal any special properties, suggesting that almost any protein may be capable of undergoing domain swapping, and that domain swapping is a specialized form of oligomer assembly.4 Furthermore, domain swapping cannot be solely an in vitro artifact, given that some domain-swapped structures constitute biologically important species5,6 or cause disease-related aggregation.7,8 Therefore, understanding the mechanism of domain swapping is desirable.
Despite considerable efforts by several experimental and computational groups, a general explanation for how proteins exchange domains still remains elusive; each protein seemingly behaves in a distinctive and individual fashion.4,9-18 What seems to emerge as a common theme is that domain swapping is closely associated with the unfolding/folding process of proteins. Comparing the closed conformation of the monomeric polypeptide chain with the open conformation of the same chain in the domain-swapped structure does not immediately suggest a pathway by which all intra-molecular interactions can be replaced by inter-molecular ones. Hydrophobic contacts, hydrogen-bonding, electrostatic interactions, and even disulfide bridges can be exchanged, and only the loop region in the monomer adopts a different conformation from the hinge in the domain-swapped dimer.4,19 Therefore, starting with a folded monomer structure, the expectation would be that breaking and re-establishing interactions in conjunction with backbone conformational changes in the hinge-loop may require considerable energy. We call this energy the activation energy for 3D domain swapping starting from folded monomers.2,4 Folding from the unfolded polypeptide chain can result in either the closed monomer or the domain-swapped dimer, with partitioning between the two products determined by their free energy difference.
Here, we experimentally investigated domain swapping by NMR using the fluorine nucleus as the NMR-active probe. Fluorine has several favorable properties: it is the smallest atom that can be substituted for a hydrogen in a molecule; it possesses a nuclear spin of 1/2, 100% natural abundance, and a high gyromagnetic ratio (0.94 of that of a proton).20 In addition, the 19F lone pair electrons can participate in non-bonded interactions with the local environment, rendering 19F chemical shifts extremely sensitive to even very small changes in van der Waals contacts, electrostatic fields, and hydrogen bonding in proteins.21 These advantages render fluorine labeling extremely attractive for NMR studies of complex systems. Although not plentiful, applications of 19F-NMR have been previously used to monitor conformational changes in proteins and to evaluate kinetic parameters associated with conformational transitions.22-26
The system that we selected for our studies is Cyanovirin-N (CV-N),27 a well-characterized protein with domain swapping abilities.28,29 Using 19F-NMR, we investigated the thermodynamics and kinetics of the conversion process between monomeric form and domain-swapped dimer for the wild type (wt) CV-N and its variants (Figure 1). Our results permit us to assess the energy landscape for interconversion between monomer and domain-swapped dimer, including the enthalpy barrier height between the two states.
Figure 1.
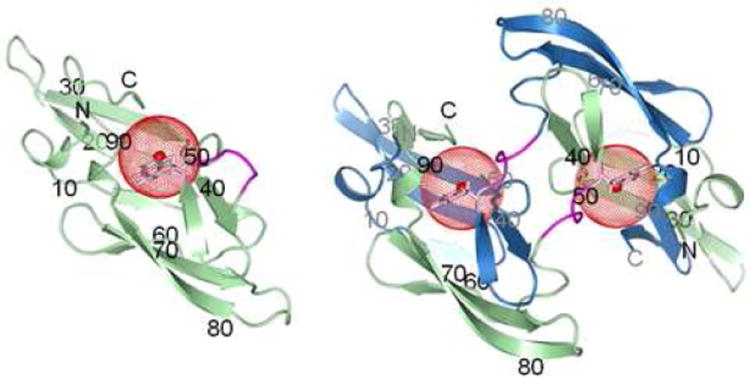
Structures of wt CV-N monomer (left, PDB ID: 2EZM)27 and domain-swapped dimer (right, PDB ID: 3EZM).29 Ribbon diagrams are shown with chains A and B colored in green and blue, respectively, and the hinge-loop in magenta. The side chain of W49 is shown in stick representation (pink) with a red sphere of radius 5 Å drawn around the fluorine atom at position 5 of the tryptophan ring. Amino acid sequence positions are labeled for every 10th residue, in black for chain A and in gray for chain B.
EXPERIMENTS AND METHODS
Sample Preparation
The genes for mutant variants (CV-NP51G, CV-NΔQ50) of wt CV-N were prepared using the QuikChange Site-directed Mutagenesis kit (Stratagene Corp., La Jolla, CA). The presence of the desired mutations was confirmed by sequencing. All proteins were expressed using the pET26b(+) (Novagen Inc., Madison, WI) vector in Escherichia coli BL-21 (DE3). Cultures were grown at 37 °C in modified minimal medium, and 5-19F-DL-tryptophan (Sigma-Aldrich Corp., St. Louis, MO) was added to the medium at a final concentration of 500 mg/L 15 minutes prior to induction with 0.5 mM IPTG. Cells were harvested 3 hours after induction by centrifugation and suspended in ice-cold PBS buffer (40 ml/1 L culture) for opening by sonication. Insoluble material was removed by centrifugation. The soluble protein present in the supernatant was fractionated by anion-exchange chromatography on a Q HP column (GE Healthcare, Piscataway, NJ) using a linear gradient of NaCl (0-1000 mM) for elution. Additional purification was achieved by gel filtration on Superdex 75 (HiLoad 2.6 × 60 cm, GE Healthcare, Piscataway, NJ), equilibrated in 20 mM sodium phosphate buffer (pH 6.0). Fractions with different quaternary states were collected: monomeric wt CV-N, monomeric CV-NP51G, and dimeric CV-NΔQ50. A sample of domain-swapped dimeric wt CV-N was obtained by incubating an ~ 10 mM monomeric sample at 39 °C for a week.30 Dimeric domain-swapped CV-NP51G was obtained by unfolding ~ 4 mM monomer in 8 M GdnHCl overnight, followed by extensive dialysis against 20 mM sodium phosphate buffer (pH 6.0) at 4 °C overnight for refolding. The domain-swapped dimer species was separated from the monomer species on a Superdex 75 gel filtration column equilibrated in 20 mM sodium phosphate, pH 6.0, containing 0.02% sodium azide, 2 mM DTT at 4 °C. The extent of fluorine labeling (> 95%), purity and identity of all proteins were assessed and verified by mass spectrometry and SDS-PAGE. All samples were prepared in 20 mM sodium phosphate buffer, pH 6.0, and kept at 4 °C until used. D2O was added to a final concentration of 8% to all NMR samples.
Differential Scanning Calorimetry (DSC)
20 mM sodium phosphate buffer (pH 6.0) was degassed overnight, and samples at a protein concentration of 1 mg/mL were dialyzed against the degassed buffer for at least 12 hours. DSC measurements were carried out using a VPDSC instrument (MicroCal Inc., Northampton, MA) at a heating scan rate of 1 °C per minute from 20 °C to 100 °C. Data were analyzed using the Microcal Origin 7.0 software (MicroCal Inc., Northampton, MA).
NMR Spectroscopy
Experiments were performed on Bruker Avance 600 or 900 MHz NMR spectrometers equipped with TCI triple-resonance, z-axis gradient cryoprobes (Bruker, Billerica, MA). External 2,2-dimethyl-2-silapentene-5-sulfonate (DSS) solution (1mM) was used for 1H chemical shift referencing.31 19F-NMR spectra were obtained on a Bruker Avance 600 spectrometer equipped with a Bruker CP TXO triple-resonance, X-nuclei observe, z-axis gradient cryoprobe (Bruker, Billerica, MA). External trifluoroacetic acid (TFA) solution (10 mM) was used for 19F chemical shift referencing.25,32 The temperature was calibrated using 100% ethylene glycol.33
Data Analysis
Conversion between CV-N monomer and CV-N domain-swapped dimer on an accessible timescale occurs only at elevated temperatures.30 The conversion was followed by NMR. The fractions of polypeptide chains in the monomeric and dimeric states, fM and fD, were determined from the relative signal integrals of their associated resonances, either 19F- or 1H-spectra. Integration of the peak areas (volumes) was carried out in Topspin (Bruker, Billerica, MA). The absolute concentrations of CV-N monomer [M] and CV-N dimer [D] were calculated based on their respective initial concentrations, CM and CD, before incubation at elevated temperatures as:
| (1) |
These equations are derived using the following properties: (i) each dimer contains two polypeptide chains, while each monomer contains only one; (ii) the total number of polypeptide chains (participating in either monomers or dimers) is conserved, i.e., [M] + 2[D] = constant. For domain swapping, both conversions and occur simultaneously. According to classical chemical kinetics theory,34 the order of a reaction and the rate constant k for a reaction can be obtained by monitoring the change in the concentration of the reactant during the time course of the reaction and fitting the data by appropriate models. The reaction is observed in our case to obey a first-order reaction kinetics, such that the integrated rate law reads:
| (2) |
where [A] is the instantaneous concentration of the reactant (monomer or dimer) and ka is the effective rate constant (ka = k1 + k-1). Additionally, the relative resonance integrals ratio fM/fD at equilibrium is governed by the ratio of k1/k-1, allowing for the extraction of k1 and k-1 values.
The temperature dependence of the reaction rate constant k permits us to calculate the Gibbs free energy of activation ΔG‡ at any given temperature using the Eyring equation:
| (3) |
which leads to:
| (4) |
using ΔG‡ = ΔH‡ - T ΔS‡, with the gas constant R = 1.986 cal/(mol·K), the Boltzmann factor kB = 1.38 × 10-23 J/K, and the Planck’s constant h = 6.63 × 10-34 J·s. Plotting ln(k/T) vs. 1/T yields a straight line with slope equal to −ΔH‡/R.
The equilibrium constant Keq and the Gibbs free energy change ΔGD-M for the conversion reaction are given by:
| (5) |
| (6) |
RESULTS AND DISCUSSION
CV-N System
CV-N is a 101 amino acid cyanobacterial lectin that was originally isolated from an aqueous extract of Nostoc ellipsosporum.27 CV-N exhibits potent anti-HIV activity and is being developed as a general virucidal agent against HIV and other enveloped viruses.27 The original solution structure found the protein to be monomeric27 while in the subsequently solved X-ray structures domain-swapped dimers were observed28,29 (Figure 1). Manipulating experimental conditions, both quaternary states can be generated for CV-N, and the CV-N system has been used extensively for biophysical, structural, and functional studies.27-30,35-40 The monomer structure exhibits a compact, bilobal fold with C2 pseudosymmetry. Each domain comprises a triple-stranded β-sheet with a β-hairpin packed on top. A helical linker is located in the middle of the sequence. In the domain-swapped dimer structure, this linker acts as a hinge to open the monomers which pair up to form a dimer exhibiting essentially the same interactions as present in the monomer, but now inter-molecular. Residues in the hinge region (Q50-N53) provide important determinants for domain swapping. For instance, changing the single proline at position 51 to glycine results in substantial stabilization of the mutant, compared to the wild type, for both the monomer and the domain-swapped dimer.30 The S52P mutant yields predominantly dimeric protein,30 and the deletion mutant, ΔQ50, exists solely as a domain-swapped dimer.35
CV-N contains only one tryptophan (W49) in its sequence, and the side chain sits at the junction between the pseudo-symmetric halves, close to the pseudo two-fold axis, occupying a pivotal region during domain swapping. We therefore introduced 5-19F-tryptophan into CV-N (Figure 1), for exploring the mechanism of domain swapping by 19F-NMR. Incorporation of a single or a few 5-19F-tryptophan residues into proteins has been shown previously to cause no discernible effects on global and local structure or thermodynamic stability of 19F labeled proteins.21,24,25
19F Spectroscopy
Since there is only one tryptophan in CV-N sequence, a single 19F resonance is expected in the 1D 19F spectrum. If, on the other hand, more than one species of the same protein exists, multiple resonances corresponding to the number of the species will be observed. Given the extreme sensitivity of the 19F chemical shift to conformational and electronic influences, combined with its large chemical shift range, little overlap in the 19F spectra of F-labeled proteins ensues.24 In addition, the temperature dependence of the 19F chemical shift is small in the present case, with chemical shift differences of 0.12 ppm and 0.28 ppm observed for free 5-19F-tryptophan and monomeric CV-NP51G, respectively, between 278 and 323 K. In addition, essentially identical linewidths were observed for free 5-19F-tryptophan over the temperature range 278-323 K, indicating that the rotational correlation time does not appreciably vary within this temperature range (Figure S1). For the CV-N monomer and the domain-swapped dimer, however, increases in linewidths were noted in the 19F resonance when the temperature was reduced, reflecting the slower overall tumbling of the protein at lower temperature. This effect was more pronounced for dimer, due its larger size (Figure S1).
Figure 2 displays the 19F spectra of 5-19F-tryptophan labeled CV-N at 298 K. and pertinent spectral parameters are listed in Table 1. Interestingly, the single amino acid change from proline to glycine at position 51 did not significantly affect the chemical shift and linewidth of the 19F resonance of the 5-19F-tryptophan labeled CV-N monomer species. However, a significant difference was observed for the CV-NP51G dimer, with the 19F resonance substantially upfield shifted, compared to wt CV-N monomer, wt CV-N dimer, and CV-NP51G monomer. In addition, the linewidth for the wt CV-N dimer (71.83 Hz) was noticeably larger than that of the CV-NP51G dimer (56.42 Hz). This is consistent with the fact that the wild type sequence contains a proline residue, and prolines are known for imparting reduced motional freedom to polypeptide backbones caused by their fixed dihedral angle ϕ. Since W49 is adjacent to the hinge-loop region, these observations suggest that the influence of millisecond backbone motion of the proline containing wt CV-N hinge is removed in the CV-NP51G variant. Since the 19F resonance of 5-19F- tryptophan labeled wt CV-N monomer and domain-swapped dimer species are partially overlapping, we used the well separated Nε1 proton resonances of the tryptophan side chain of the monomer and the domain-swapped dimer30 for monitoring the conversion time course for wt CV-N.
Figure 2.
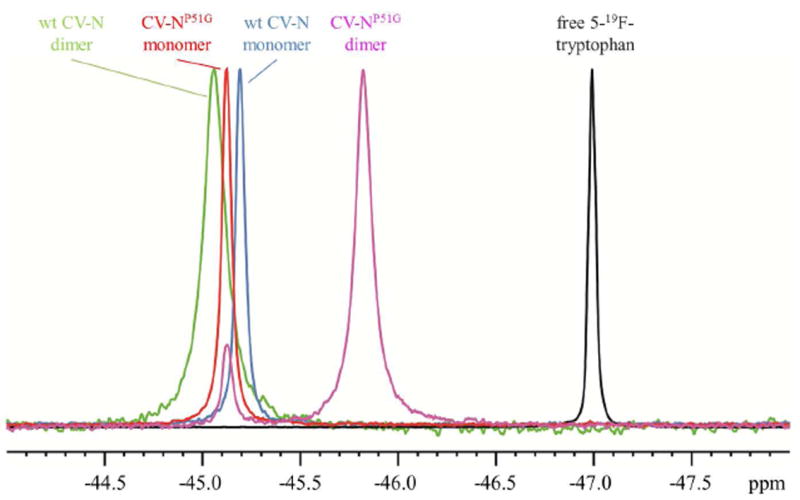
19F-NMR spectra of 5-19F-tryptophan labeled CV-N samples and free 5-19F-tryptophan at 298 K.
Table 1.
19F-NMR Parameters of 5-19F-Tryptophan Labeled CV-N Samples at 298 K
| free 5-19F-tryptophan | wt CV-N | CV-NP51G | |||
|---|---|---|---|---|---|
| M | D | M | D | ||
| resonance frequency (ppm) | -46.99 | -45.19 | -45.06 | -45.12 | -45.82 |
| linewidth at half-height (Hz) | 23.69 | 31.60 | 71.83 | 32.79 | 56.42 |
Kinetics of the Conversion between Domain-Swapped Dimer and Monomer
For CV-NP51G, the monomer and domain-swapped dimer 19F resonances are well separated and conversion between the two species can be followed readily using 1D spectra (Figure 3). The predominantly dimeric sample was incubated at 330.5 K for increasing amounts of time, and 19F spectra were recorded at 298 K, where the conversion process is slowed sufficiently to not interfere with accurate determination of the relative integrals/amounts. The data provided in Figure 3 clearly show that after ~ 4 hours of incubation at 330.5 K, ~ 50% of the swapped-dimer species had converted into monomer. Spectra were also recorded for the CV-NP51G dimer conversion at other temperatures, as well as for the wt CV-N conversion process. The excellent spectral quality allowed to fit the data using eq 2 and permitted us to extract rate constants, for example: k1 of 3.3 × 10-5 s-1 for the reaction D → 2M at 330.5K.
Figure 3.
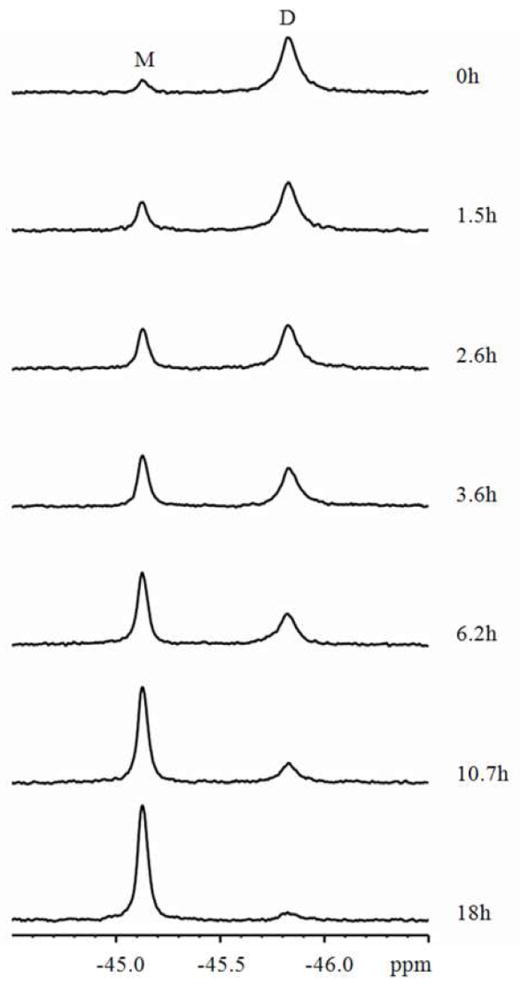
19F-NMR spectra recorded at 298 K following the conversion process from domain-swapped dimer to monomer of 5-19F-tryptophan labeled CV-NP51G at 330.5 K. The length of incubation at 330.5 K is indicated at the right side of each spectrum. NMR spectra were recorded at 298 K to prevent any conversion during the time of the NMR measurement.
The same analysis was repeated for a series of temperatures. The time-courses for the conversion of the wt CV-N swapped dimer at different temperatures are displayed in Figure 4A. For each temperature, the resonance integrals decreased exponentially. Not surprisingly, faster rates were observed at higher temperatures. Using the experimentally determined temperature dependence of the rate constant k, the activation enthalpy ΔH‡D-M, entropy ΔS‡D-M and Gibbs free energy ΔG‡D-M for the conversion from domain-swapped dimer to monomer was calculated using eqs 3 and 4 (Figure 4A inset).
Figure 4.
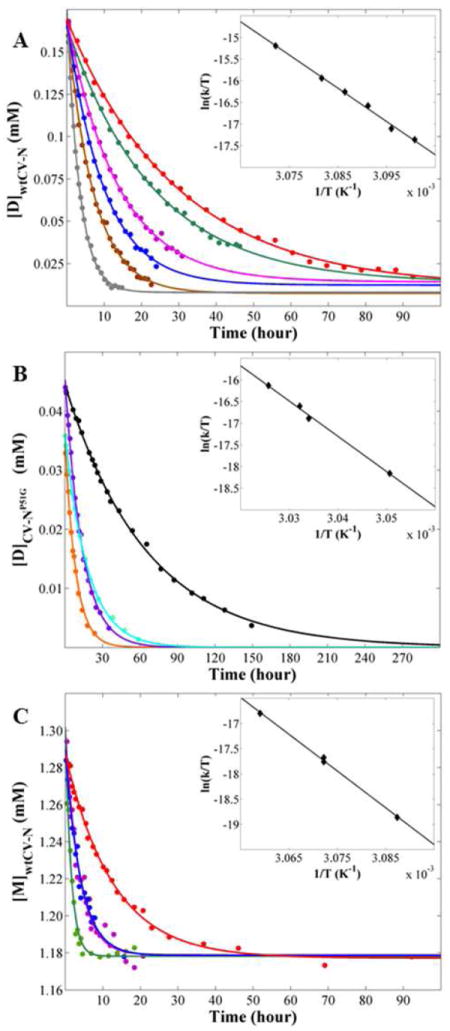
Time dependence of the conversion reactions for wt CV-N and CV-NP51G at different temperatures. Each point represents the concentration of the domain-swapped dimer (or monomer) species at a particular point in time as measured by the relative peak integrals of the dimer and monomer resonances. The inset shows the temperature dependence of reaction rate constant. The data fits a straight line whose slope ( ) and intercept ( ) yield the activation enthalpy ΔH‡ and entropy ΔS‡, respectively, using eq 4. (A) The conversion from wt CV-N domain-swapped dimer to monomer. The incubation temperatures are: 322.5 K, red; 323 K, green; 323.5 K, magenta; 324 K, blue; 324.5 K, brown; and 325.5, gray. (B) The conversion from CV-NP51G domain-swapped dimer to monomer. The incubation temperatures are: 327.8 K, black; 329.6 K, cyan; 329.8 K, purple; and 330.5 K, orange. (C) The conversion for wt CV-N monomer to domain-swapped dimer. The incubation temperatures are: 323.9 K, red; 325.5 K (1), blue; 325.5 K (2), magenta; and 326.9 K, green.
The series of gray data points in Figure 4A represents the conversion at 325.5 K, the fastest reaction for wt CV-N domain-swapped dimer (k1 = 8.2 × 10-5 s-1). At a very similar temperature, 327.8 K, conversion for the CV-NP51G domain-swapped dimer was the slowest reaction in the series (k1 = 4.3 × 10-6 s-1, black data points in Figure 4B), and required more than six days to reach the equilibrium. Therefore, the accessible temperature windows for the conversion reaction for wt CV-N and CV-NP51G are distinctly different and non-overlapping: at 327.8 K, the conversion for wt CV-N is too fast, while the conversion for CV-NP51G at 325.5 K is too slow. As a consequence, temperature dependent ΔG‡D-M values could only be extracted for different sets of temperatures (Table 2). Given that smaller activation energies are seen with increasing temperatures, it is safe to assume that the ΔG‡D-M for the wt CV-N domain-swapped dimer conversion at 327.8 K should be lower than 25.2 kcal/mol, the measured ΔG‡D-M for the wt CV-N domain-swapped dimer conversion at 325.5 K. Comparison of this value with the ΔG‡D-M for CV-NP51G (27.3 kcal/mol at 327.8 K) reveals that less energy is required for the wt CV-N conversion than for the CV-NP51G dimer at the same temperature. This is consistent with the experimentally observed faster equilibration during the conversion of wt CV-N dimer into monomer.
Table 2.
Energetics of Domain Swapping and Protein Unfolding of WT CV-N and its Variants
| Kinetic parameters for domain swapping measured by NMR | Thermodynamic parameters for unfolding measured by DSC | |||||
|---|---|---|---|---|---|---|
| wt CV-N (M → D) | wt CV-N (D → M) | CV-NP51G (D → M) | CV-NP51G (M → U) | CV-NP51G (D → U) | CV-NΔQ50 (D → U) | |
| ΔH‡ or ΔH kcal/mol | 145 ± 22 (323.9-326.9K) | 153 ± 15 (322.5-325.5K) | 162 ± 32 (327.8-330.5K) | 130 ± 1 | 171 ± 4 | 142 ± 1 |
| ΔS‡ cal/(mol·K) | 363 ± 66 (323.9-326.9K) | 391 ± 45 (322.5-325.5K) | 410 ± 97 (327.8-330.5K) | - | - | - |
| ΔG‡ kcal/mol | 26.8 ± 0.1 (325.5K) | 25.2 ± 0.1 (325.5K) | 27.3 ± 0.1 (327.8K) | - | - | - |
| k1 or k-1 × 10-6 s -1 | 6.6 ± 0.3 (325.5K) | 82.0 ± 2.6 (325.5K) | 4.3 ± 0.5 (327.8K) | - | - | - |
Since equivalent experiments were carried out for wt CV-N and CV-NP51G, we can directly compare the activation barriers for conversion. The ΔH‡ values are listed in Table 2. Interestingly, these ΔH‡ values are very similar in magnitude to the unfolding enthalpy changes, ΔH, observed by DSC. Since both wt CV-N and CV-NP51G comprise monomeric and dimeric species that can undergo interconversions, we used a unique mutant, CV-NΔQ50, that exists only as an unfolded monomer or a folded domain-swapped dimer for the control DSC experiment. The ΔHD-U value for CV-NΔQ50 unfolding was 142 kcal/mol; this value is of the same order of magnitude as the activation enthalpy ΔH‡D-M for the conversion from domain-swapped dimer to monomer for wt CV-N (153 kcal/mol) and CV-NP51G (162 kcal/mol) extracted for the NMR kinetic study. This surprising result implies that the monomer/swapped dimer conversion proceeds via complete unfolding of the protein, rather than partially un/folded states.
We also followed the reverse reaction for wt CV-N, namely conversion from monomer to domain-swapped dimer (Figure 4C). At 325.5 K, the reaction was carried out twice to evaluate and confirm the reliability of the experimental data. Both datasets agree extremely well (magenta and blue symbols) and can be fit to the same curve. In addition, the extracted ΔH‡M-D value for the conversion of the wt CV-N monomer to the domain-swapped dimer (145 kcal/mol) agrees well with the DSC result (130 kcal/mol) and the derived value (125 kcal/mol) for the CV-NP51G monomer to domain-swapped dimer conversion. This is very gratifying and again implies that complete unfolding is involved in the conversion process.
Both conversion reactions (monomer to dimer and dimer to monomer) exhibit exponential time dependence, suggesting that both are first order reactions. This observation appears to be at odds with the assumption that a molecular reaction of the type M + M → D might be a second order reaction. Although puzzling at first, the observed first order kinetics is in perfect agreement with the fact that complete unfolding occurs in the conversion reaction. The observations are indeed consistent with the presence of the rate-limiting steps of M → U and D → 2U for conversion of monomer to domain-swapped dimer and conversion from domain-swapped dimer to monomer, respectively. Each conversion process consists of two steps, with the unfolded state (U) as the intermediate.
Our current system is particularly suitable to investigate the kinetics given our excellent fluorine labeling efficiency. However, even if incomplete labeling were the case, resulting in sample heterogeneity,25 it should be possible to follow the first order reaction and determine the reaction rate constant. Kinetic parameters (but not thermodynamic ones) are extracted from the temperature dependence of the reaction rate, and thus do not depend on the concentration. Therefore, only the labeled fraction of the protein is contributing to the data and correct kinetic information is obtained.
In addition to the Gibbs free energy barrier ΔG‡ and the activation enthalpy ΔH‡ discussed above, the average entropy change ΔS‡ can also be extracted using eq 4. The entropy change ΔS‡D-M was 391 cal/(mol·K) for the wt CV-N domain-swapped dimer to monomer conversion, ~ 30 cal/(mol.K) larger than ΔS‡M-D, the value extracted for the wt CV-N monomer to dimer conversion of 363 cal/(mol·K). Given that in the conversion reaction one dimer molecule converts into two unfolded single-chain molecules, the total number of molecules in the system increases while the number of polypeptide chains remains the same. Therefore, the system becomes more disordered and its entropy change is larger than for unfolding of a single folded to an unfolded chain, for which no increase in the number of molecules occurs. The slight increase in entropy for the CV-NP51G domain-swapped dimer conversion compared to the wt CV-N dimer of 410 cal/(mol·K) can be explained by the increased flexibility in the linker introduced by the P51G mutation.
Equilibrium Properties
The data presented in Figure 4 also allows for the extraction of the monomer-dimer equilibrium constant, Keq, since the final flat part of each curve at long conversion times yields the equilibrium concentration. For the conversion starting from the wt CV-N domain-swapped dimer all reactions reached a similar equilibrium concentration of 11.2 ± 2.8 μM. Taking the reaction D → 2M into account, we then extracted an average equilibrium constant Keq of 15.3 mM, which leads to a Gibbs free energy ΔGD-M of 2.4 ± 0.3 kcal/mol at 293 K based on eq 6. Neglecting a possible, small temperature dependence in Keq, for the temperature interval from 322.5 K to 325.5K, ΔGD-M can be equated with the difference between thermal unfolding of the wt CV-N domain-swapped dimer, and twice the value for the unfolding of the wt CV-N monomer.
Although the mechanism(s) for unfolding by chaotrops, such as urea and guanidine hydrochloride (GdnHCl) may be different from thermal unfolding, it is expected that the free energy difference between monomer and dimer for the two unfolding reactions is similar. In particular, it is reasonable to assume that the free energy difference between reactants and products of the unfolding reaction is mainly determined by their intrinsic interaction difference. Previously reported unfolding free energies for wt CV-N monomer and the obligate domain-swapped dimer form are ΔGwtM-U = 4.2 ± 0.2 kcal/mol and ΔGΔQ50D-U = 10.6 ± 0.5 kcal/mol, respectively,30,41 yielding a chemical reaction energy of about 2.2 (10.6 − 2 × 4.2) kcal/mol. Since the previous chemical unfolding and the current thermal conversion/unfolding were performed for identical buffer conditions and temperature (293 K), it is satisfying to observe the excellent agreement between these values.
The conversion of the CV-NP51G domain-swapped dimer into monomer (Figure 4B) yields a final equilibrium concentration of dimer around zero, given the experimental precision. (A very small amount of dimer (< 5%) cannot reliably be distinguished from the noise in the spectra.) In order to derive a lower limit Keq value we used the last/smallest available concentration as the approximate equilibrium concentration and obtained a value of Keq = 2.9 ± 0.9 mM.
For both, wt CV-N and CV-NP51G, the interconversion ΔGD-M is very small, in excellent agreement with the fact all interactions within the monomeric and swapped-dimeric structures are extremely similar; only the hinge-loop conformation is different. Therefore, any measurable free energy difference has to be associated with the hinge-loop that can either introduce or relieve strain in the monomer-dimer interconversion.
The Energy Landscape of Domain Swapping
The available thermodynamic and kinetic parameters (Table 2) permit an estimation of the overall energy landscape for domain swapping of CV-NP51G (black profile). This is depicted in Figure 5, with the unfolding enthalpies for the monomer ΔHM-U and domain-swapped dimer ΔHD-U of CV-NP51G obtained from DSC measurements and the activation enthalpy ΔH‡D-M (at 327.8-330.5K) for the CV-NP51G dimer to monomer conversion extracted from the 19F-NMR study. The activation enthalpy ΔH‡M-D for the CV-NP51G monomer to dimer conversion can also be estimated (ΔH‡M-D − ΔH‡*D-M) should be equal to their unfolding enthalpy difference (ΔHM-U − ΔH* D-U). The asterisks indicate that half the dimer values from Table 2 have to be used for the normalization, to ascertain that an identical number of polypeptide chains is taken into account. A similar treatment yields the gray profile for wt CV-N. The wt CV-N ΔH‡M-D and ΔH‡D-M values were extracted from the NMR study and ΔHD-U for unfolding of the CV-NΔQ50 domain-swapped dimer was determined by DSC. As can be easily appreciated, the activation barrier for domain swapping is comparable in magnitude to the unfolding barrier for both wt CV-N and CV-NP51G. In addition, as observed previously,30 the single amino acid change in P51G mutant stabilizes both monomer and domain-swapped dimer of this variant.
Figure 5.
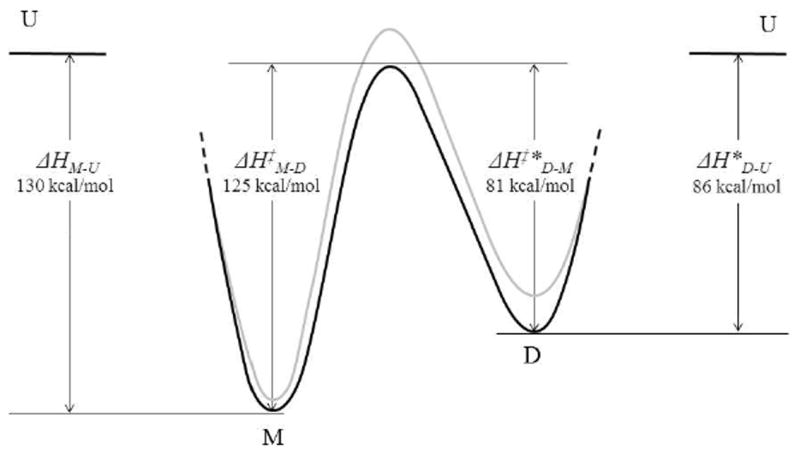
Schematic enthalpy diagram for domain swapping of CV-NP51G (black) and wt CV-N (gray).
The thermodynamic and kinetic parameters of domain swapping have attracted interest by the protein folding community for a long time.14,15,42 For example, a kinetic study on Stefin A dimerization reported an activation enthalpy of 99 kcal/mol,15 consistent with the large activation enthalpy barrier for domain swapping of CV-N determined here. In addition, for p13suc1 it had been proposed that domain swapping could occur via the unfolded state.11 Alternatively, the existence of partially folded monomers that have been suggested for some other proteins2,10,18 imply that complete unfolding maybe not necessarily always be a prerequisite for domain swapping of every protein.
CONCLUSION
We carried out an extensive investigation of the thermodynamic and kinetic behavior for domain swapping of wt CV-N and CV-NP51G, primarily using 19F-NMR. Both proteins can exist at room temperature either as monomers or domain-swapped dimers in solution, and interconversion between these quaternary states is slow at room temperature or below. Here, we determined that the kinetic barrier between the monomer and domain-swapped dimer is significant (of the order of ~100 ± 20 kcal/mol) and of similar magnitude to that for equilibrium unfolding. This is suggestive that, at least for CV-N, complete unfolding is required for domain swapping.
Supplementary Material
Acknowledgments
We gratefully acknowledge M. Delk for NMR technical support and Jozef Hritz for useful discussions. This work was supported by National Institutes of Health Grants 5R01GM086238 (IB) and GM080642 (AMG).
Footnotes
Supporting Information. A figure showing the line-widths of 19F resonances for different samples as a function of temperature. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. Science. 1991;254:1598. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MJ, Schlunegger MP, Eisenberg D. Protein Sci. 1995;4:2455. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu L, Gronenborn AM. Comprehensive Biophysics. Chapter 3.9 Elsevier; 2011. [Google Scholar]
- 5.Cafaro V, De LC, Piccoli R, Bracale A, Mastronicola MR, Di DA, D’Alessio G. FEBS Lett. 1995;359:31. doi: 10.1016/0014-5793(94)01450-f. [DOI] [PubMed] [Google Scholar]
- 6.Czjzek M, Letoffe S, Wandersman C, Delepierre M, Lecroisey A, Izadi-Pruneyre N. J Mol Biol. 2007;365:1176. doi: 10.1016/j.jmb.2006.10.063. [DOI] [PubMed] [Google Scholar]
- 7.Sanders A, Jeremy CC, Higgins LD, Giannini S, Conroy MJ, Hounslow AM, Waltho JP, Staniforth RA. J Mol Biol. 2004;336:165. doi: 10.1016/j.jmb.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Yamasaki M, Li W, Johnson DJ, Huntington JA. Nature. 2008;455:1255. doi: 10.1038/nature07394. [DOI] [PubMed] [Google Scholar]
- 9.Stroud JC, Wu Y, Bates DL, Han A, Nowick K, Paabo S, Tong H, Chen L. Structure. 2006;14:159. doi: 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Staniforth RA, Giannini S, Higgins LD, Conroy MJ, Hounslow AM, Jerala R, Craven CJ, Waltho JP. EMBO J. 2001;20:4774. doi: 10.1093/emboj/20.17.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rousseau F, Schymkowitz JWH, Wilkinson HR, Itzhaki LS. Proc Natl Acad Sci U S A. 2001;98:5596. doi: 10.1073/pnas.101542098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ogihara NL, Ghirlanda G, Bryson JW, Gingery M, DeGrado WF, Eisenberg D. Proc Natl Acad Sci U S A. 2001;98:1404. doi: 10.1073/pnas.98.4.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray AJ, Head JG, Barker JJ, Brady RL. Nat Struct Biol. 1998;5:778. doi: 10.1038/1816. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Alonso JP, Bruix M, Font J, Ribo M, Vilanova M, Rico M, Gotte G, Libonati M, Gonzalez C, Laurents DV. J Biol Chem. 2006;281:9400. doi: 10.1074/jbc.M510491200. [DOI] [PubMed] [Google Scholar]
- 15.Jerala R, Zerovnik E. J Mol Biol. 1999;291:1079. doi: 10.1006/jmbi.1999.3045. [DOI] [PubMed] [Google Scholar]
- 16.Ding F, Prutzman KC, Campbell SL, Dokholyan NV. Structure. 2006;14:5. doi: 10.1016/j.str.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Chen YW, Stott K, Perutz MF. Proc Natl Acad Sci U S A. 1999;96:1257. doi: 10.1073/pnas.96.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byeon IJ, Louis JM, Gronenborn AM. J Mol Biol. 2003;333:141. doi: 10.1016/s0022-2836(03)00928-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Eisenberg D. Protein Sci. 2002;11:1285. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolbier WR. Guide to fluorine NMR for organic chemists. Wiley; New Jersey: 2009. [Google Scholar]
- 21.Campos-Olivas R, Aziz R, Helms GL, Evans JN, Gronenborn AM. FEBS Lett. 2002;517:55. doi: 10.1016/s0014-5793(02)02577-2. [DOI] [PubMed] [Google Scholar]
- 22.Abbott GL, Blouse GE, Perron MJ, Shore JD, Luck LA, Szabo AG. Biochemistry. 2004;43:1507. doi: 10.1021/bi035618a. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed AH, Loh AP, Jane DE, Oswald RE. J Biol Chem. 2007;282:12773. doi: 10.1074/jbc.M610077200. [DOI] [PubMed] [Google Scholar]
- 24.Danielson MA, Falke JJ. Annu Rev Biophys Biomol Struct. 1996;25:163. doi: 10.1146/annurev.bb.25.060196.001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuler B, Kremer W, Kalbitzer HR, Jaenicke R. Biochemistry. 2002;41:11670. doi: 10.1021/bi026293l. [DOI] [PubMed] [Google Scholar]
- 26.Toptygin D, Gronenborn AM, Brand L. J Phys Chem B. 2006;110:26292. doi: 10.1021/jp064528n. [DOI] [PubMed] [Google Scholar]
- 27.Bewley CA, Gustafson KR, Boyd MR, Covell DG, Bax A, Clore GM, Gronenborn AM. Nat Struct Biol. 1998;5:571. doi: 10.1038/828. [DOI] [PubMed] [Google Scholar]
- 28.Botos I, O’Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. J Biol Chem. 2002;277:34336. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 29.Yang F, Bewley CA, Louis JM, Gustafson KR, Boyd MR, Gronenborn AM, Clore GM, Wlodawer A. J Mol Biol. 1999;288:403. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- 30.Barrientos LG, Louis JM, Botos I, Mori T, Han Z, O’Keefe BR, Boyd MR, Wlodawer A, Gronenborn AM. Structure. 2002;10:673. doi: 10.1016/s0969-2126(02)00758-x. [DOI] [PubMed] [Google Scholar]
- 31.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes BD. J Biomol NMR. 1995;6:135. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 32.Maurer T, Kalbitzer HR. J Magn Reson B. 1996;113:177. doi: 10.1006/jmrb.1996.0172. [DOI] [PubMed] [Google Scholar]
- 33.Van Geet AL. Anal Chem. 1968;40:2227. [Google Scholar]
- 34.Atkins P, De Paula J. Atkins Physical Chemistry. Oxford University Press; 2006. [Google Scholar]
- 35.Kelley BS, Chang LC, Bewley CA. J Am Chem Soc. 2002;124:3210. doi: 10.1021/ja025537m. [DOI] [PubMed] [Google Scholar]
- 36.Matei E, Zheng A, Furey W, Rose J, Aiken C, Gronenborn AM. J Biol Chem. 2010;285:13057. doi: 10.1074/jbc.M109.094938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fromme R, Katiliene Z, Giomarelli B, Bogani F, Mc MJ, Mori T, Fromme P, Ghirlanda G. Biochemistry. 2007;46:9199. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 38.Zweckstetter M, Bax A. J Am Chem Soc. 2000;122:3791. [Google Scholar]
- 39.Rohl CA, Baker D. J Am Chem Soc. 2002;124:2723. doi: 10.1021/ja016880e. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka H, Chiba H, Inokoshi J, Kuno A, Sugai T, Takahashi A, Ito Y, Tsunoda M, Suzuki K, Takenaka A, Sekiguchi T, Umeyama H, Hirabayashi J, Omura S. Proc Natl Acad Sci U S A. 2009;106:15633. doi: 10.1073/pnas.0907572106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barrientos LG, Lasala F, Delgado R, Sanchez A, Gronenborn AM. Structure. 2004;12:1799. doi: 10.1016/j.str.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 42.Zerovnik E, Lohner K, Jerala R, Laggner P, Turk V. Eur J Biochem. 1992;210:217. doi: 10.1111/j.1432-1033.1992.tb17411.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.