

Abstract
With the rapid growth of publicly available high-throughput transcriptomic data, there is increasing recognition that large sets of such data can be mined to better understand disease states and mechanisms. Prior gene expression analyses, both large and small, have been dichotomous in nature, in which phenotypes are compared using clearly defined controls. Such approaches may require arbitrary decisions about what are considered “normal” phenotypes, and what each phenotype should be compared to. Instead, we adopt a holistic approach in which we characterize phenotypes in the context of a myriad of tissues and diseases. We introduce scalable methods that associate expression patterns to phenotypes in order both to assign phenotype labels to new expression samples and to select phenotypically meaningful gene signatures. By using a nonparametric statistical approach, we identify signatures that are more precise than those from existing approaches and accurately reveal biological processes that are hidden in case vs. control studies. Employing a comprehensive perspective on expression, we show how metastasized tumor samples localize in the vicinity of the primary site counterparts and are overenriched for those phenotype labels. We find that our approach provides insights into the biological processes that underlie differences between tissues and diseases beyond those identified by traditional differential expression analyses. Finally, we provide an online resource (http://concordia.csail.mit.edu) for mapping users’ gene expression samples onto the expression landscape of tissue and disease.
Keywords: large-scale analysis, personalized medicine, phenotype classification
Although gene expression microarrays have been a standard, widely utilized biological assay for many years, we still lack a comprehensive understanding of the transcriptional relationships between various tissues and disease states. Even with the hundreds of thousands of expression array datasets available through public repositories such as National Center for Biotechnology Information’s (NCBI’s) Gene Expression Omnibus (1) (GEO), the lack of standardized nomenclature and annotation methods has made large-scale, multiphenotype analyses difficult. Thus, expression analyses have typically used the decade-old approach of comparing expression levels across two states (e.g., case vs. control) or a limited number of phenotype classes (2–4). Even recent large-scale gene expression investigations, whether they have attempted to elucidate phenotypic signals (5–7) or applied those signals for downstream analyses such as drug repurposing (8, 9), involve comparisons between two states or classes.
Comparative analyses, where transcriptional differences are directly measured between two phenotypes, inherently impose subjective decisions about what constitutes an appropriate control population. Importantly, such analyses are fundamentally limited in scope and cannot differentiate between biological processes that are unique to a particular phenotype or part of a larger process that is common to multiple phenotypes (e.g., a generic “cancer pathway”). Moreover, the results of such comparative analyses can be limited in generalizability as they make assumptions about the phenotypes being compared (10). Alternatively, in a data-rich environment, we can take a holistic view of gene expression analyses.
In this paper we introduce scalable and robust statistical approaches that leverage the full expression space of a large diverse set of tissue and disease phenotypes to accurately perform and glean biological insights from both sample- and gene-centric analyses. By viewing a given phenotype in the context of this comprehensive transcriptomic landscape, we circumvent the need for predefined control groups and presupposed relationships between phenotypes (Fig. 1A). We devise, implement and validate the accuracy of an enrichment statistic that provides detailed phenotypic information for new samples when they are mapped onto and compared with the transcriptomic landscape (http://concordia.csail.mit.edu).
Fig. 1.

Comprehensive view of gene expression. (A) A comprehensive perspective on expression analysis enables the elucidation of biological signals that are thematically coherent but provide an alternative view to traditional dichotomous approaches. For example, the gene-signature for “breast cancer” is enriched for breast-specific development and carbohydrate and lipid metabolism in our comprehensive approach, as opposed to being dominated by a more general “cancer” signal. (B) The gene expression landscape, as represented by the first two principal components of the expression values of 20,252 genes from 3,030 microarray samples separates into three distinct clusters: blood, brain, and soft tissue. The shading of the regions corresponds to the amount of data located in that particular region of the landscape such that the darker the color, the more data exists at that location. Interestingly, the area where the soft tissue intersects the blood tissue corresponds to bone marrow samples, and where it intersects the brain tissue, mostly corresponds to spinal cord tissue samples. (C) There is a clear separation of reproductive and gastrointestinal tissue samples in the soft-tissue cluster.
Our perspective on interpreting gene expression space helps uncover phenotype-specific marker genes beyond those discovered by traditional dichotomous views of gene expression. We introduce a method based on a finite impulse response filter (11) used in signal processing to reveal, for instance, marker genes involved in carbohydrate and lipid metabolism as key processes in breast cancer. Such findings are in contrast to those of traditional over- and underexpression based analyses, which focus on generic cancer processes not specific to breast cancer such as cell cycle and cell adhesion (12). Capitalizing on the hierarchical nature of the phenotypic labels associated with our samples, we also demonstrate that genes previously linked to specific types of carcinomas may actually be part of a broader “carcinoma” process. Finally, we illustrate how metastasized tumor samples are transcriptomically more proximal to other cancer samples from their respective primary sites, as opposed to cancerous tissue from the metastasis sites from which the samples were resected.
Results
Making Sense of the Transcriptomic Landscape.
As an initial step towards a holistic approach to gene expression analysis, we must make sense of the substructure of the global transcriptomic landscape. We first constructed a curated gene expression database of 3,030 diverse samples (from 192 series) obtained from NCBI’s GEO (1). These samples were annotated with their phenotypes (tissue of origin, disease state, etc.) using the anatomical and disease concepts in a custom subset of the Unified Medical Language System (13) (UMLS) concept ontology via both natural language processing and manual validation (Methods).
Although visualizing the full transcriptomic landscape encompassing all genes is not feasible, the first two principal components (PCs) of the expression level of 20,252 genes across the database provide a representation of the phenotypic relationships that captures roughly 20% of the variance in the data (Methods). Although others have suggested that the primary factors driving the organization of the global transcriptomic landscape can largely be attributed to hematopoietic and malignant programming (14), we alternatively see the cell and tissue-specific signatures of blood, brain, and soft tissue are dominant (Fig. 1B). Furthermore, these PCs recapitulate the phenotypic relationships captured in a tissue network (Supporting Information) derived from a de novo tissue correlation analysis (Methods). Indeed, when analyzing the tissue-specific characteristics of these clusters, we observe the overexpression of fibrillar and epithelial genes such as COL3A1, COL6A3, KRT19, KRT14, and CADH1 in the soft-tissue cluster and neural genes such as GFAP, APLP1, GRIA2, PLP1, and SLC1A2 in the brain cluster. Gene ontology (GO) enrichment analysis of the top 250 tissue-specific genes for each cluster further points to overenrichment for terms related to each of the three tissue types (Supporting Information). Several reports have stated that data from different datasets are not comparable as the dataset signal is dominant (10, 15); however, we find that the tissue signal is dominant in this macroscopic view (Supporting Information).
By additionally performing principal component analysis on soft-tissue samples (all noncancerous samples that are also not blood or brain), it becomes apparent that phenotypic grouping occurs on multiple levels of phenotypic granularity. Not only are individual tissue samples in confined regions, they are also organized by functionality. Tissues sensitive to reproductive hormones (ovary, uterus, myometrium, endometrium, prostate, penis, and breast) group together to form a distinct subregion in the smooth landscape (Fig. 1C). Juxtaposed to them are primarily gastrointestinal tract samples from tissues such as colon, stomach, intestine, liver, and esophagus.
Concordia: Phenotypic Concept Enrichment.
Although correlation analyses and the visual representation of the transcriptomic landscape provide insight into the broad relationships between various phenotypes, our ability to harness these expression signals to map previously unseen samples into a database of expression samples is compelling. Beginning with our customized UMLS concept annotation of the 3,030 samples, we restricted the set of UMLS concepts to the 1,489 anatomy and disease concepts that mapped to at least three expression samples (Supporting Information). We developed a sample-centric method based on the Kolmogorov–Smirnov statistic to label new samples with UMLS concepts that are overrepresented in their local expression neighborhoods (Methods). No hard boundaries are drawn when a new input sample is labeled, but rather the concepts pertinent to the transcriptomic neighborhood for the input sample are reported. Importantly, as it is often difficult to define an appropriate control, this approach has the advantage that it does not require case-control type input but, rather, just a single microarray sample. To illustrate its function, we provide a web resource, Concordia (http://concordia.csail.mit.edu), that allows users to submit their own microarray samples performed on the popular Affymetrix HG-U133 Plus 2.0 array and obtain their overenriched tissue and disease concepts.
We performed leave-one-sample-out cross-validation to validate the accuracy of our method for assigning an unknown sample to the correct phenotype. The receiver operating characteristic (ROC) curve was computed for each of the 1,489 UMLS concepts, and the standard measure of area under the curve (AUC) that summarizes both the true-positive and false-positive rates was used as a measure of accuracy. We see an average accuracy of 92.8% after restricting the set of UMLS concepts to the 1,209 that have samples from two or more expression series in GEO to ensure that a diverse set of data is used. Even when we restrict the concepts to the 450 that have at least 50 samples originating from at least five different data series, the average accuracy is approximately 89.8%. Table 1 contains the performance of a selection of UMLS concepts, along with the number of samples and series that were associated with that concept. Unsurprisingly, “broader” concepts have poorer performance compared to the more specific concepts, as the former encompass a much more diverse expression signal. The performance values are in Supporting Information, and the ROC curves are available on the web site. Note that many of these concepts are similar and have samples in common; consequently, many of the concepts have similarly high (low) AUC values.
Table 1.
Concordia cross-validation performance on selected UMLS concepts

We see a significant increase in accuracy as more data is added to the underlying database. For example, when half of the samples associated with each concept are removed, the global performance is a mere 44%, compared to the aforementioned 93% (Supporting Information). This implies that the phenotypic signal becomes stronger and the power of this type of macroscopic analysis increases with the amount of underlying data. As our approach employs a nonparametric enrichment statistic that only requires the concept annotation of the samples in the original gene expression database, it can be updated in real-time without having to “retrain” the database. A system such as this could thus be deployed in a research or clinical setting where new samples are continually being added and analyzed, with minimal alteration of normal protocols.
Primed with the 3,030 labeled samples, we applied Concordia to 15,904 other GEO samples performed on the Affymetrix HG-U133 Plus 2.0 array. These enrichment scores represent the expression patterns as characterized by the 1,489 anatomy and disease-related concepts and can be used as an additional source of biological information when performing future large-scale gene expression analyses (Supporting Information).
Phenotypic-Specific Marker Genes.
We developed a method to identify marker genes that characterize a specific phenotype in the context of broad transcriptomic landscapes, and not in the context of dichotomous classes. Instead of defining a marker gene as one that is over- or underexpressed in a case vs. control study using methods akin to t-tests, we define a marker gene as a gene that has a “localized” expression signature for a phenotype; i.e., how grouped together all of the samples are corresponding to that phenotype for that gene. If all of the samples for a phenotype have a very similar expression level (all high, all low, etc.), the gene may be considered a marker gene for that phenotype. We employ a finite impulse response filter (FIRF) (11) on each gene’s expression values across the entire database of 3,030 diverse expression samples to quantify the degree of expression level localization for a given phenotype. To generate the set of genes most relevant to a phenotype, we use the marker gene localization scores to rank all genes and then we identify the cutoff for the number of genes to include by balancing the set’s ability to accurately classify samples of its own phenotype while minimizing the presence of non-phenotype-specific signal (Methods). Not only does this method sidestep the requirement of defining appropriate “control” phenotype(s), it also facilitates the identification of thematically coherent gene signatures that reveal very different aspects of biology from traditional ones.
As an example, we derived the breast cancer gene set from a landscape of 673 samples representing 17 different cancerous tissues. The 74 genes that comprise this set are functionally enriched for processes related to breast-specific development, and carbohydrate and lipid metabolism (Supporting Information). These pathways, revealed through gene expression, are consistent with independent clinical and genetic data suggesting an important role for carbohydrate and lipid metabolism in breast cancer. For example, women with type 2 diabetes may have higher susceptibility to breast cancer (16). Three genes specifically implicated in this analysis, ENPP1, ADIPOQ, and PPARA, are of particular interest. ADIPOQ is expressed in adipose tissue exclusively. Variants in the ADIPOQ gene and protein levels are implicated in prostate cancer (17) and breast cancer (18). Similarly, ENPP1 levels have been correlated to progression-free survival in tamoxifen-treated patients with breast cancer (19). PPARA is one of a family of nuclear transcription factors that has been found to stimulate both adipocyte (fat cell) differentiation and fatty acid oxidation (20). Moreover, the PPARA signaling pathway has been implicated in breast cancer progression (21), and in a case-control study a polymorphism of PPARA was identified to be associated with a twofold increase in breast cancer (22).
Notably missing from this list of enriched pathways are processes commonly associated with cancer, such as cell–cycle and cell–adhesion (12). We can recreate this conventional perspective by selecting the set of candidate marker genes using a traditional permutation t-test-based method (Methods). This reveals enrichment for processes that are associated with cancer in general, but not specific to breast cancer, such as “cellular response to tumor necrosis factor,” “induction of apoptosis,” and other tumor-related processes (Supporting Information). Furthermore, according to the permutation t-test method, PPARA is less significant than nearly 17% of the other genes (ADIPOQ is in the top 2% and ENPP1 is in the top 0.5%). In comparison, using the FIRF, the tumor-necrosis-related genes, such as RIPK1, TRADD, and TNFRSF25, do not appear until, respectively, 18%, 54%, and 97% of the other more breast-cancer-specific genes appear first.
To ascertain the “cancer” gene set using our FIRF-based method, however, we expanded the landscape of data to include not only 17 cancers but also 2,187 samples across 30 noncancerous tissue types. By comparing all cancers against all noncancers, we unsurprisingly then find that the most significant genes are functionally enriched for processes that are typically associated with tumors: “cell division,” “cell cycle,” and “DNA repair,” to name but a few. Taken together, landscape-based gene signature discovery can recapitulate canonical cancer pathways but also can identify a complementary set of gene signatures with distinct biological implications.
Specificity of Marker Genes.
It has been suggested that the so-called “incidentalome” of incidental findings is a threat that has yet to be addressed in either biological or clinical settings (23). The consequences of noncomprehensive views of biomarkers, such as prostate-specific antigen, continue to cause needless harm and costs (24). By performing analyses in the context of a large database of biological samples, however, we see that many genes are not specific to a single disease.
To illustrate this, we took the 459 carcinoma samples in our database and computed the “carcinoma” marker gene localization scores by comparing them to the 270 other tumor samples. As the UMLS concepts are in a structured ontology, we computed the marker gene scores for the 13 concepts subordinate to “carcinoma” (e.g., “adenocarcinoma,” “adenosquamous carcinoma”) for which we had at least three expression samples. From the list of genes sorted by their carcinoma marker gene score p-value, we removed all genes that had a better p-value in any of the 13 subordinate concepts. This yielded a list of 5,805 genes that had better p-values at the more general concept “carcinoma” than at any of the more specific subordinate carcinoma types. Functional enrichment analyses of the top 10, 20, 50, 100, and 150 genes in this list reveals processes such as “regulation of cell adhesion,” “response to growth factors,” and other morphogenesis and development terms. Furthermore, within the sorted list of carcinoma genes, we see genes previously implicated in carcinomas such as COL1A1 (25, 26) and ELF3 (27) in the top five. As such, these genes that have previously been implicated in particular types of carcinomas may instead be part of a larger carcinoma process, rather than specific to breast or colorectal cancer.
This sort of quantification of phenotype specificity is of course relevant to the diagnostic accuracy of putative biomarkers and for developing suitably broad-spectrum or targeted therapeutics. As such, we computed the gene–phenotype expression localization scores for all 20,252 genes and 1,489 concepts (Supporting Information).
Tissue-Specific Signal of Tumor Metastases.
The clinical problem of distinguishing whether a cancerous lesion represents a primary tumor, or a metastasis from a distant malignancy, presents a test case for our ability to localize a sample to the appropriate phenotypic group within the transcriptomic landscape. By combining the aforementioned sample-and gene-centric methods, we are able to map new tumor metastasis tissue samples onto the expression landscape, providing an unbiased measure of their phenotypic predisposition based on gene expression. It is commonly known by pathologists that tumor metastasis tissue biopsies viewed “under the microscope” resemble the tissue of the primary site rather than that of the tissue in the metastasized location. Nevertheless, the proper identification of the primary site of a metastasis can be critical in determining the appropriate clinical treatment plan (28). Indeed, we find that metastatic tissue samples localize in the vicinity of their tissue of origin in the transcriptomic landscape (Fig. 2), even without the use of specially tuned primary site detection methods (28, 29).
Fig. 2.

Sample- and gene-centric expression analyses show that metastasized samples more closely resemble their primary sites than their biopsy site. Legend applies to both plots. (A) Breast tumors that metastasized to the lung, brain, and bone (GSE14107) still appear to be more closely related to other breast samples (red shaded region) than to their metastasis sites when placed in the transcriptomic landscape of 3,030 other expression samples. (B) Recomputing the PCs using only the 164 genes of the breast gene set, as opposed to all 20,252 genes, recapitulates the proximity of the metastasized breast cancer samples to breast tissue samples (orange shaded region), and shows that they lie within the confines of the other breast cancer samples in the database.
For instance, in an analysis of 29 metastasized breast cancer samples resected from lung, brain, and bone (GSE14107), the metastases more closely resemble breast tissue than their biopsy locations (Fig. 2A). Overenriched UMLS concepts from Concordia for the metastasized samples include “white adipose tissue,” “subcutaneous fat,” “subcutaneous tissue,” “lactiferous duct,” “mammary lobe,” and “glandular structure of breast.” When we restrict the analysis to use only the 164 genes in the breast gene set identified using our aforementioned FIRF-based method, we observe that these metastasized breast samples lie within the context of other primary breast cancer samples in the database, which in turn are juxtaposed to normal breast tissue (Fig. 2B). Similarly, 15 of the 17 metastasized colorectal cancer samples that were removed from liver (GSE10961) were all labeled with “rectum and sigmoid colon,” “colonic diseases, functional,” and “colon carcinoma” with a false-positive rate (FPR) below 0.05; the other two samples had FPRs of 0.06 for “colon carcinoma.” The top UMLS concepts for other metastatic samples obtained from GEO are in Supporting Information.
Interestingly, the mislabeled metastases provide an unbiased measure of the degree of overlap between the biological signals of related tissues. This is particularly evident within the soft-tissue cluster (Fig. 1B, Lower, Left), in which the tissue-specific signal can be dwarfed by the larger variances caused by the blood and brain tissue samples. Although the use of supervised learning approaches could mitigate these issues (29), they minimize the significant biological overlap of some of these samples, which may have implications for therapeutic selection (30). For example, due to the proximity of breast and ovarian tissue samples in the global transcriptomic landscape, we had difficulty distinguishing between breast metastases in the ovary and primary ovarian carcinoma (GSE20565).
Discussion
With the ever-growing amounts of transcriptomic data, it has become not only possible but also imperative to embrace the full transcriptomic continuum of tissue and disease. Employing a comprehensive, non case- vs.-control approach and making use of the multidimensional nature of gene expression data, we capture biological processes that are typically overshadowed in traditional analyses. Furthermore, we are able to recapitulate the biologically and medically relevant concepts relating to a new expression sample through Concordia. Indeed, as the power of this macroscopic analysis increases with the amount of data, this approach has the potential to more fully leverage large databases with biological data and to benefit further as more data are added. Although we have presented our sample-and gene-centric methods utilizing medically relevant concepts and gene expression data, the data-driven nature of these methods implies that by changing the scope or domain of the labels and/or the underlying quantitative data, they can be applied to analyses in different contexts with relative ease. For instance, it is not far fetched to imagine using these methods to create a transcriptomic landscape based on RNAseq expression data (31) annotated with concepts from RxNorm, a clinical drug vocabulary.
As suggested by some (32), systematic application of molecular pathology measurements will allow a shifting of the conventionally employed diagnostic classification boundaries to include intermediate pathotypes that cross the boundaries of the conventional medical classifications. These intermediate pathotypes are more closely coupled to the actual underlying pathology, thus revealing not only shared pathology but also opportunities for development of shared treatment (30, 33). It may be the case that the expression signatures of diseases provide clues to a disease network (34) other than what classical medical knowledge dictates, thus providing insights to previously unknown disease relationships.
It has been proposed that the future of personalized medicine, and the proper application of genomic and genetic data, requires an understanding of both who the patient is and the characteristics of the subpopulation to which the patient belongs (35). Clinical applications of our approach, together with other genetic, environmental, and phenotypic information, could more accurately and consistently annotate clinical samples and provide an impartial view of the landscape of clinico-pathological classification. As we employ an enrichment statistic that only requires the usual standard of care in the labeling of samples, this system could be deployed in a clinical setting with minimal alteration of normal procedures. By shifting away from a dichotomous view and employing the global transcriptomic landscape, we hope to address one of the key requirements of personalized medicine and begin to answer one of its fundamental questions, “what other samples am I most similar to so that the most effective treatment can be administered?”
Methods
Normalizing the gene expression samples.
Our database is comprised of 3,030 gene expression samples belonging to 192 series performed on the Affymetrix HG-U133 Plus 2.0 arrays that were obtained from NCBI’s GEO (1). The original CEL files were downloaded from GEO and Microarray Suite (MAS) 5.0 normalized. Subsequently all probe-specific values were converted to gene-specific values using a trimmed mean. For the gene selection procedure, we log-normalized all of the expression values to be between -1 and 1 to ensure a normal distribution. For all of the other analyses, the expression values were additionally rank normalized.
UMLS Annotation.
We follow the lead of Butte, et al. (36) and extracted the title, description, and source fields from each of the 3,030 expression samples and annotated them using the Java implementation of the National Library of Medicine’s (NLM) MetaMap program, MMTx (37). A custom UMLS (13) thesaurus containing concepts from the UMLS, Medical Subject Heading, and Systematized Nomenclature of Medicine ontologies was generated using NLM’s MetaMorphosys program. The automated annotations were manually verified and 672 UMLS concepts were kept. As these concepts only represented the most detailed level of annotation, they were mapped up the ontology such that a sample labeled with a specific concept also received labels corresponding to all of its ancestor concepts. Due to the domain of the data, we filtered the concepts to only those that are descendants of either “disease” or “anatomy,” resulting in 1,489 concepts.
Making Sense of the Transcriptomic Landscape.
The transcriptomic landscape visualization is the first two PCs of the PC projection of the 3,030 centered and scaled gene expression samples. The phenotypic clusters portrayed by shaded regions were created by iteratively using the convex hull function (chull) in the R statistical language package. We performed the hierarchic analysis of the landscape by taking the 1,065 phenotypically normal samples in the soft-tissue cluster and recalculating the PCs. The convex hulls for the gastrointestinal and reproductive clusters were computed in the aforementioned fashion.
The tissue similarity network was generated by computing correlations of a representative sample of a tissue type to all other representatives of the other tissues. The representative was chosen to be the sample that was closest to the centroid in the set of samples for that phenotype. To contend with sampling bias, the correlations were computed 100 times, the centroid for each phenotype having been chosen from a random 75% subset of the samples for that phenotype. The network was then created based on the tissue–tissue relationships with an average correlation greater than 0.8 across all 100 subsampling runs. The colors of the nodes denote the general tissue class (blood, brain, gastrointestinal, reproductive, and other).
Our online resource also provides a visualization of where an input sample lies in the transcriptomic landscape. An input sample’s coordinates are computed by centering and scaling its expression values by constants learned from the database and then applying the loadings from the first two PCs.
Picking Blood, Brain, and Soft Tissue-Specific Genes.
Tissue-specific genes were selected by performing permutation t-tests comparing, for example, the log-normalized expression values for the blood samples for a given gene to the log-normalized expression values of the samples associated with brain and soft tissue. Each permutation run consisted of computing the t statistic for the actual labeling of the samples and comparing it to the t statistics produced when the labels were randomly permuted 200 times while keeping the sample size distribution constant. To counter the potential influence of sampling bias, this entire procedure was performed 100 times, each time using only a random 75% of the data for each tissue type. Genes with a false discovery rate corrected p-value of 0.05 or lower in all 100 runs were deemed significant. As there were genes with identical p-values, the genes were then sorted such that a gene with a larger difference in means between the phenotypes was ordered before those with a smaller difference. GO enrichment was performed on the top 50, 100, and 250 genes for each tissue type using FuncAssociate 2 (38). We report only the GO terms that had a resampling-based p-value less than 0.05.
Computing Phenotype-Specific Gene Signatures.
To determine the level of localization of the expression intensities for a given gene, we employed a FIRF (11). For each gene g, phenotype p pair, we sort all of the expression samples by their expression intensities for g. Using a “sliding window” of size equal to the number of samples corresponding to p, we compute the fraction of samples in that window that are associated with p. The value is 1 if all samples in the window are associated with p and 0 if none of them are. This window is iteratively moved across the sorted list of samples to obtain a value for all positions. The marker gene score for a particular gene–phenotype pair is the maximum value that is achieved in any of the windows. A p-value is computed for each score using a binomial distribution.
To determine the appropriate cutoff for the number of genes to include in the gene set for phenotype p, the genes are first sorted according to their marker gene score from highest to lowest. We then iteratively examine the quality of the top n genes, balancing their positive predictive capability with the amount of additional noise. Starting with the first two highest scoring genes, we iteratively remove each sample s and compute its correlation to all other samples using only those two genes. We generate an ROC curve for s and use the AUC as a summary statistic. The ROC curve is generated by sorting all samples by their correlation to s and incrementing the true-positive count when that sample is associated with p and incrementing the false-positive count when that sample is not associated with p. Once all AUCs are computed for two genes, we add the next highest scoring gene and recompute all AUC values. We define the mean “hit” AUC as the average AUC obtained by all samples associated with p, and the mean “miss” AUC as the average AUC of all samples not associated with p. By taking the ratio of the mean hit AUC and mean miss AUC at each number of genes n, we determine the relevant set of genes as all genes in the sorted list up until the number of genes that maximizes this ratio.
To compare the performance of the FIRF to the traditional over- and underexpression-based analyses relying on differences in the mean expression levels in the phenotypes being studied, we performed a t-test for each gene and computed the empirical p-value based on 1,000 random permutations of the phenotype labels. As many of the p-values were 0 (or the same), we sorted the list of genes by the z score of the actual t statistic as compared to the 1,000 t statistics generated by the random permutations. GO enrichment was then performed using the Bioconductor GOstats (39) library in R.
Enrichment Score Calculation.
We use the database of gene expression samples to assess overenrichment for particular disease- and tissue-specific signals. Given a new expression profile, for each concept represented in the database, we calculate a statistic that measures the strength of association between the sample and concept, as implied by its similarity to the labeled database samples.
The statistic is calculated as follows. First, the database consisting of n curated expression samples {s1,s2,s3,…,sn} is sorted (in decreasing order) according to each observation’s Spearman correlation, ρ, with the new profile. Let s1′,s2′,s3′, ..,sn′ represent the samples ordered according to their correlation coefficients ρs1′,ρs2′,ρs3′,…,ρsn′. For a given concept c in the set C, the set of all UMLS concepts in our database, let Sc be the set of all database samples associated with the concept. That is, Sc = {si|si is associated with c}. We define an ordered list of xi values:
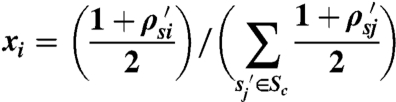 |
when sample si′ is associated with concept c, and
for all other samples that are not associated with concept c. Intuitively, when si is associated with the concept in question, the xi value corresponds to the fraction of total correlation between the new sample and all database samples associated with the concept. All of the xi values for the concept “hits” sum to 1, and all of the xi values for the concept “misses” sum to -1.
Then we compute a running sum of xi across all n database samples and take the maximum value achieved by this running sum as our enrichment score (ES) for the concept in question:
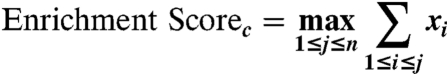 |
This sum across all n samples is zero. The concepts where there is strong positive deviation from 0 are the concepts whose associated samples are more highly correlated with the new profile than those samples that are not associated with the concept.
Quantifying Performance.
To quantify the ability of the method to recover UMLS concepts based on an input expression profile, we generate an ROC curve and calculate the AUC as a summary statistic for each concept represented in the database. To compute the ROC curve for each concept c in the database, we iteratively leave out each sample s and compute s’s enrichment score for c using the remaining database samples. We compute the running true-positive (TP) and false-positive (FP) counts by walking down the list of samples sorted by their enrichment score for c. The TP is incremented if the ith sample in the list is actually labeled with concept c. If the sample is not labeled with concept c, the FP is incremented. The true-positive results (TPRs) and FPRs are obtained by dividing TP and FP, respectively, by the number of known positives and negatives at each position i. By plotting the TPR vs. FPR we obtain the ROC curve. The larger the area under the ROC curve (AUC), the greater the gene expression signal for that concept as the samples with the highest enrichment scores for the concept were truly labeled with that concept.
When using this method to label a new sample, we compute its ES (w.r.t. the entire database) for each concept. We then report the system’s estimated FPR for each concept at the sample’s observed concept-specific enrichment score. These FPR values are derived from the running statistics used to generate the ROC plots: Look up the new sample’s score position in the list of sorted scores and report the FPR at that position (if there is not an exact match, report the next-worst FPR).
Supplementary Material
ACKNOWLEDGMENTS.
The authors would like to thank Alvin Kho, Joseph Loscalzo, and Stanley Shaw for their comments and insightful input and Leslie Gaffney for her input on the figures.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118792109/-/DCSupplemental.
References
- 1.Barrett T, et al. NCBI GEO: Archive for functional genomics data sets—10 years on. NAR. 2010:D1005–D1010. doi: 10.1093/nar/gkq1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian Z, et al. A practical platform for blood biomarker study by using global gene expression profiling of peripheral whole blood. PloS One. 2009;4:e5157. doi: 10.1371/journal.pone.0005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley JT, Tibshirani R, Deshpande T, Butte AJ. Disease signatures are robust across tissues and experiments. Mol Syst Biol. 2009;5:307. doi: 10.1038/msb.2009.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golub TR, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 5.Rhodes DR, et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. NEO. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X, Yu X, Zack DJ, Zhu H, Qian J. TiGER: A database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogasawara O, et al. BodyMap-Xs: Anatomical breakdown of 17 million animal ESTs for cross-species comparison of gene expression. NAR. 2006;34:D628–D631. doi: 10.1093/nar/gkj137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sirota M, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med. 2011;3:96ra77–96ra77. doi: 10.1126/scitranslmed.3001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamb J. The connectivity map: A new tool for biomedical research. Nat Rev Cancer. 2007;7:54–60. doi: 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- 10.Ransohoff DF. Bias as a threat to the validity of cancer molecular-marker research. Nat Rev Cancer. 2005;5:142–149. doi: 10.1038/nrc1550. [DOI] [PubMed] [Google Scholar]
- 11.McClellen JH, Schafer RW, Yoder MA. DSP First: A Multimedia Approach. Englewood Cliffs, NJ: Prentice Hall; 1998. [Google Scholar]
- 12.Rhodes DR, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. NAR. 2004;32:D267–D270. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukk M, et al. A global map of human gene expression. Nature Biotechnol. 2010;28:322. doi: 10.1038/nbt0410-322. 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owzar K, Barry WT, Jung S-H, Sohn I, George SL. Statistical challenges in preprocessing in microarray experiments in cancer. Clin Cancer Res. 2008;14:5959. doi: 10.1158/1078-0432.CCR-07-4532. 5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michels KB, et al. Type 2 diabetes and subsequent incidence of breast cancer in the Nurses’ Health Study. Diabetes Care. 2003;26:1752–1758. doi: 10.2337/diacare.26.6.1752. [DOI] [PubMed] [Google Scholar]
- 17.Dhillon PK, et al. Common polymorphisms in the adiponectin and its receptor genes, adiponectin levels and the risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2011;12:2618–2627. doi: 10.1158/1055-9965.EPI-11-0434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaklamani V, et al. Polymorphisms of ADIPOQ and ADIPOR1 and prostate cancer risk. Metabolism. 2011;60:1234–1243. doi: 10.1016/j.metabol.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umar A, et al. Identification of a putative protein profile associated with tamoxifen therapy resistance in breast cancer. Mol Cell Proteomics. 2009;8:1278–1294. doi: 10.1074/mcp.M800493-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
20.Lee J-Y, et al. Activation of peroxisome proliferator-activated receptor-
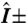 enhances fatty acid oxidation in human adipocytes. Biochem Biophys Res Commun. 2011;407:818–822. doi: 10.1016/j.bbrc.2011.03.106. [DOI] [PubMed] [Google Scholar]
enhances fatty acid oxidation in human adipocytes. Biochem Biophys Res Commun. 2011;407:818–822. doi: 10.1016/j.bbrc.2011.03.106. [DOI] [PubMed] [Google Scholar] - 21.Shi Z, Derow CK, Zhang B. Co-expression module analysis reveals biological processes, genomic gain, and regulatory mechanisms associated with breast cancer progression. BMC Syst Biol. 2010;4:74. doi: 10.1186/1752-0509-4-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golembesky AK, et al. Peroxisome proliferator-activated receptor-alpha (PPARA) genetic polymorphisms and breast cancer risk: a Long Island ancillary study. Carcinogenesis. 2008;29:1944–1949. doi: 10.1093/carcin/bgn154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohane IS, Masys DR, Altman RB. The incidentalome: a threat to genomic medicine. JAMA. 2006;296:212–215. doi: 10.1001/jama.296.2.212. [DOI] [PubMed] [Google Scholar]
- 24.Steenhuysen J. PSA test for prostate cancer not recommended: panel. Reuters. 2011. Oct 7, pp. 1–2.
- 25.Zhao H, et al. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 2006;3:e13. doi: 10.1371/journal.pmed.0030013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons TR, et al. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med. 2011;17:1109–1115. doi: 10.1038/nm.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang J, et al. Over-expression of ERT(ESX/ESE-1/ELF3), an ets-related transcription factor, induces endogenous TGF-beta type II receptor expression and restores the TGF-beta signaling pathway in Hs578t human breast cancer cells. Oncogene. 2000;19:151–154. doi: 10.1038/sj.onc.1203252. [DOI] [PubMed] [Google Scholar]
- 28.Bridgewater J, van Laar R, van’t Veer L. Gene expression profiling may improve diagnosis in patients with carcinoma of unknown primary. Br J Cancer. 2008;98:1425–1430. doi: 10.1038/sj.bjc.6604315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaner ME, et al. Gene expression patterns in ovarian carcinomas. Mol Biol Cell. 2003;14:4376–4386. doi: 10.1091/mbc.E03-05-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dudley JT, Butte AJ. Biomarker and drug discovery for gastroenterology through translational bioinformatics. Gastroenterology. 2010;139:735–741. doi: 10.1053/j.gastro.2010.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loscalzo J, Kohane IS, Barabási A-L. Human disease classification in the postgenomic era: A complex systems approach to human pathobiology. Mol Syst Biol. 2007;3:124. doi: 10.1038/msb4100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- 34.Barabási A-L, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohane IS. The twin questions of personalized medicine: Who are you and whom do you most resemble? Genome Med. 2009;1:4. doi: 10.1186/gm4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Butte AJ, Kohane IS. Creation and implications of a phenome-genome network. Nature Biotech. 2006;24:55–62. doi: 10.1038/nbt1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aronson AR. Effective mapping of biomedical text to the UMLS Metathesaurus: The MetaMap program. Proc AMIA Symp. 2001:17–21. [PMC free article] [PubMed] [Google Scholar]
- 38.Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP. Next generation software for functional trend analysis. Bioinformatics. 2009;25:3043–3044. doi: 10.1093/bioinformatics/btp498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.