

Abstract
The biological functions of coiled coils generally depend on efficient folding and perfect pairing of their α-helices. Dynamic changes in the helical registry that lead to staggered helices have only been proposed for a few special systems and not found in generic coiled coils. Here, we report our observations of multiple staggered helical structures of two canonical coiled coils. The partially folded structures are formed predominantly by coiled coil misfolding and occasionally by helix sliding. Using high-resolution optical tweezers, we characterized their energies and transition kinetics at a single-molecule level. The staggered states occur less than 2% of the time and about 0.1% of the time at zero force. We conclude that dynamic changes in helical registry may be a general property of coiled coils. Our findings should have broad and unique implications in functions and dysfunctions of proteins containing coiled coils.
Keywords: leucine zipper, protein misfolding
Coiled coils widely mediate protein-protein interactions and form rigid structures such as scaffolds, spacers, and levers (1) that are often involved in generating (2), transducing (3), or sensing forces (4) in cells. The biological functions of coiled coils critically depend upon their affinity, specificity, and dynamics of helix pairing. Although the structures and dynamics of coiled coils have been extensively studied, it is unclear whether coiled coils containing staggered helices can form through protein misfolding or helix sliding (5–8). A further understanding of these alternative structures and their mechanisms of formation is needed to better understand the functions and dysfunctions of coiled coils (3).
Because helices in coiled coils have a characteristic seven amino acid repeat (abcdefg)n, their functional folding and assembly generally requires to pair hydrophobic residues in the a and d positions in the dimerization interface and other residues near the interface. However, due to the periodic α-helical structure, alternative structures can form in which one helix shifts its registry, often by one heptad repeat, relative to the other. Although the resultant staggered helical structures were first proposed when the high-resolution crystal structure of the coiled coil GCN4 leucine zipper domain was obtained (5), these alternative conformations have only been observed in a few coiled coils with special sequence patterns deviating from the canonical heptad repeat (3, 7, 8). The difficulty in finding these different conformations is due to functional conformations of most coiled coils being much more stable than staggered conformations. The differential stability can be achieved by specific pairing of polar residues or complementary packing of nonpolar residues in the dimerization interface and by forming interhelical salt bridges between residues near the interface (1, 5, 9). In the GCN4 leucine zipper, substitution of a single asparagine buried in the dimerization interface for a nonpolar residue converts the two-stranded coiled coil to a mixture of two- and three-stranded coiled coils (10). Thus, staggered conformations have not been observed in this model or in other related systems with a canonical heptad repeat.
Increasing evidence shows that helices in coiled coils can dynamically shift their registry. Such helix sliding or rotation has important biological functions, especially for relaying conformational changes in distal domains or across membranes (6). One prominent example is the antiparallel, two-stranded coiled coil contained in the stalk domain of dynein, which connects its ATPase domain and the microtubule-binding domain (3, 7). This 15-nm long coiled coil serves as a shaft to transduce the force generated by the ATPase domain and coordinates interactions of the ATPase domain with ATP and the microtubule-binding domain with microtubules. Helix sliding has also been proposed for other systems, including the leucine zipper of Nek2 kinase (8), the HAMP domain (11), and the tetramerization domain of Mnt repressor (12). However, the kinetics of helix staggering and sliding has not been well characterized, and their molecular mechanisms are unclear. Moreover, it is not known if helix staggering and sliding is limited to specific sequences or if it is a general property intrinsic to coiled coils.
To characterize the mechanisms of helix staggering and sliding and their roles in coiled coil folding and misfolding, we used high-resolution optical tweezers to investigate two of the strongest coiled coils that have canonical sequence compositions: a variant of the GCN4 leucine zipper (pIL) (9), in which three valine residues and one asparagine residue in the a positions in the wild type have been replaced by the most stable isoleucine (13), and a heterodimer (pER) composed of oppositely charged glutamic acid and arginine residues at the e and g positions, respectively (14). Both parallel coiled coils have extraordinary stabilities, with melting temperatures above 100 °C. However, the folding energies and kinetics of both coiled coils have not been characterized. The use of strong coiled coils in our experiments facilitated detection of staggered conformations (15). Our findings suggest that helix staggering and sliding, albeit to various degrees for different coiled coil, is a general feature of coiled coil dynamics intrinsic to its distinctive periodic structure. Our work demonstrates the unique advantages of high-resolution optical tweezers in protein misfolding studies due to their great spatiotemporal resolution and measurement ranges, which extends earlier similar applications (16–19). Observations of misfolding in small protein domains that have been long considered efficient folders suggest the yin and yang of protein folding and misfolding.
Results
pIL Can Misfold or Slide to Three Partially Folded States with Staggered Helices.
To determine its folding energy and kinetics, we pulled a single pIL coiled coil using the same experimental setup and procedure as previously developed for the wild-type GCN4 protein (SI Appendix, Fig. S1) (20). In this setup, the two polypeptides in the coiled coil are crosslinked at the C-termini and pulled from the N-termini through a DNA handle using dual-trap optical tweezers. A representative force-extension curve shows fast extension changes (approximately 10 nm) in a force region centered at approximately 12 pN (Fig. 1A), indicating rapid protein unfolding and refolding. Below or above this force region, the coiled coil remained in folded or unfolded states, respectively. The corresponding force-extension curves are consistent with the prediction of a worm-like chain model (red line). Moreover, the whole extension curve is reproducible regardless of pulling or relaxing the molecule, which suggests that the transition is reversible. The reversibility and the size of the extension change confirm that pIL is a dimer, which can be explained in that our polypeptide constructs are designed to preferentially crosslink dimers not oligomers. Unfolding any higher-order oligomers of the polypeptides would lead to an irreversible force-extension curve due to dissociation of the polypeptide(s) not covalently linked to the protein-DNA tether and/or longer extension changes. Thus, the experiment allowed us to focus on the folding and unfolding kinetics of a single coiled coil and to avoid oligomerization pathways that often compete with high-energy folding states of a coiled coil (10).
Fig. 1.

Formation of staggered coiled coil states through protein misfolding or helix sliding. (A) Force-extension curve (black) of a single pIL molecule. The curve was obtained by separating the two optical traps at a speed of 20 nm/s. A similar curve was measured by relaxing the molecule (not shown). The regions below or above the transition region can be fit (red line) by the work-like chain models for both DNA and polypeptide (SI Appendix), yielding a polypeptide contour length increase of 25.8( ± 0.8) nm due to coiled coil unfolding. (B) Extension-time traces of a single pIL at indicated pulling forces. Data were mean-filtered using a 2.2 ms time window. Red dots indicate the partially folded and staggered states. (C) Probability density distributions of extensions corresponding to the traces in (B) (symbols) and their double-Gaussian-function fit (lines). (D) Close-up view of the extension traces showing the misfolded states (red dots) and intermediate state (cyan dots). Data were mean-filtered using a 1 ms time window. The red lines are the corresponding fit based on the hidden-Markov model. (E) Histogram distribution of the average extensions for the staggered states relative to the folded state. The scaled relative average extensions of the staggered states measured at all trap separations are included. The distribution can be fitted by a triple-Gaussian function, resolving three misfolded states and their relative positions. The total five states involved in pIL transition are numbered as States 1–5 in (C) and (E), with the staggered states numbered in black for traces in D.
To scan the transition region, we increased the separation between two optical traps (or the pulling force) in a stepwise manner (typically a 2-nm step). At each trap separation, the extension and force changes due to spontaneous protein unfolding and refolding were recorded at 10 kHz for a time period of 10–1,000 s. A typical experiment on a single molecule lasted 900–3,000 s. Parts of the time-dependent extension recording are shown for three different trap separations or pulling forces (Fig. 1B). The molecule mainly transited between two average extensions differing by approximately 10 nm as can be seen in the corresponding extension histogram distributions (Fig. 1C). Protein unfolding was accompanied by a sudden force decrease (Fig. 1A). Thus, the pulling force is shown as the arithmetic mean of the average forces of the folded and unfolded states (see SI Appendix), all of which linearly increase with the trap separation in the tested force range of 10–14 pN (20).
Although pIL folded and unfolded in an apparent two-state manner (Fig. 1C), the protein occasionally resided at intermediate extensions, indicating the existence of partially folded states (Fig. 1 B and D, SI Appendix, Fig. S2). Extensive measurements show that these states did not transit between each other and that > 90% of them only transited with the unfolded state, suggesting that these partially folded states are mainly misfolded coiled coil states. Only < 10% of the partially folded states transited with the folded coiled coil state. This observation suggests that the partially folded states may also infrequently serve as intermediate states for folding and unfolding of the coiled coil in alternative indirect pathways.
Interestingly, the partially folded states do not have identical average extensions (Fig. 1D, SI Appendix, Fig. S2), indicating the presence of substates. To determine these substates, we analyzed the extension trace using a five-state hidden Markov model (HMM) (20, 21) and obtained the histogram distribution of the average extensions for three partially folded states (Fig. 1E, see also SI Appendix). The distribution exhibits three peaks and can be fit by a sum of three Gaussian functions. Thus, our high-resolution measurements reveal at least three partially folded states (States 2, 3, and 4) and their positions relative to the folded state. Our histogram distribution shows the total occurrence frequency of the misfolded states, instead of their populations in time. The populations also depend on the lifetimes of the different states (Fig. 2C). A detailed characterization of a total of five states involved in pIL transitions is shown in Fig. 2.
Fig. 2.

Energetics and kinetics of pIL folding and misfolding. (A) Force-dependent probabilities or populations of the five states involved in pIL transitions (symbols) and their best-fits based upon a theoretical model (20) (lines). The states are numbered according to their average extensions in an ascending order. (B) Rates of transition from State 1, 2, or 3 to State 5 (n-5) or from State 5 to States 1–4 (5-n). The pulling force always enhances unfolding and reduces refolding. The unfolding rate of State 4 is shown in SI Appendix, Fig. S4. (C) Average lifetimes of the five states. Results measured from two representative single molecules were shown here.
The Partially Folded States Contain Staggered Helices.
To derive the structures of the partially folded states, we calculated the force-extension curve for each state involved in pIL transitions (SI Appendix, Fig. S3). By fitting the curve with a worm-like chain model, we obtained the contour length or the number of amino acids of the unfolded polypeptide portion of the protein in each state. Thus, we could infer the structures of the protein in various folding stages. The folded state has an average contour length of 26.5 (± 0.9, SD) nm less than the fully unfolded state, indicating folding of 66 (± 2) amino acids (22) in the presence of the pulling force. Assuming in-register helix paring, the folded state detected in our experiment contains three (± 1) frayed amino acids at the N-terminal end of each polypeptide (20, 23).
In contrast to this folded structure, the three partially folded states or States 2, 3, and 4 (Fig. 1E) have 6.5 (± 0.8), 13 (± 1), and 20 (± 3) more amino acids unfolded, respectively. Interestingly, these numbers are close to one, two, and three heptad repeats. Thus, the partially folded states are consistent with a series of alternative coiled coil states in which one alpha helix is shifted relative to the other by one to three heptad repeats (Fig. 3).
Fig. 3.

A quantitative model for folding, misfolding, and unfolding of pIL at zero force. The amino acids in the folded and unfolded regions are aligned vertically and horizontally, respectively. Amino acids are highlighted in blue and red at the a and d positions in the coiled coil region and in cyan in the linker regions, respectively. The misfolded states (States 2–4) have staggered helices with their register successively shifted by one heptad repeat (marked by the red oval). Each heptad shift towards State 4 adds 7 extra amino acids in both the unfolded region and the loop region. The borders of transition states are indicated by the dashed lines with their corresponding contour lengths listed in SI Appendix, Table S1. As an example, the transition state to unfold state 1 is shown in SI Appendix, Fig. S8, state II. The free energy (E), probability (p), and lifetime (τ) of each state are labeled in blue. The minor helix sliding transitions are observed between the folded state and all three staggered states.
Energetics and Kinetics of the Five States (F > 0).
To determine the transition kinetics among the five states identified above, we analyzed the extension trace at each trap separation based on the 5-state hidden-Markov model. The model is further simplified by neglecting the direct transitions within the three staggered states and between the staggered and the folded states, because the frequencies of these transitions are minimal compared to those of other transitions. The HMM analyses yielded the probabilities and average lifetimes of the five states and the transition rates among them (Fig. 2). Consistent with the apparent two-state transitions observed from the extension traces, the folded and unfolded states comprise more than 98% of the protein population over the time course of the entire tested force range (10.5–14 pN). Less than 2% of the population is occupied by the three misfolded states. The populations of these misfolded states decrease as the numbers of heptad shift increase, reaching less than 0.1% for the least populated misfolded state. We found that the populations of the folded and unfolded states are modulated by force in a sigmoidal manner, whereas those of the misfolded states slightly increase and then decrease with force. Thus, the single-molecule manipulation approach used here helps to resolve the rare misfolded states by stabilizing these states by force, compared with the corresponding populations at zero force in Fig. 3.
The measured protein transition rates range from 10-2 to 3 × 102 s-1 (Fig. 2B). As expected from the effect of a force on a chemical reaction, the rate of each transition pathway increases with increasing force as the protein unfolds and decreases as the protein refolds (24). Consistent with this theory, the logarithm of the rate is approximately and linearly dependent on the force in the force range tested, with a slope proportional to the contour length difference between the initial state and the corresponding transition state (20). Based on the transition rates, we calculated the lifetimes of all five states, which range from 4 ms to 2 s (Fig. 2C). The lifetimes of all the folded and misfolded states decrease approximately exponentially with an increase in force, whereas the unfolded state increases exponentially. For a Markov process that underlies most of the chemical reactions, the lifetime of a state is the inverse of the sum of the rates at which the molecule transits away from that state. Thus, the lifetime of a state generally is not a single exponential function of force. However, because only one transition pathway dominates the pathways exiting each state (Fig. 2B), the lifetime appears linear when the logarithm of lifetime is plotted against force (Fig. 2C). Our findings indicate that compared with the GCN4 coiled coil (20), pIL generally has longer lifetimes in its states at higher force ranges. Both features facilitate the detection of three misfolded states with small extension differences of approximately 1 nm using optical tweezers (15, 25) (Fig. 1E).
A Model of Helix Staggering and Sliding in pIL Folding and Misfolding (F = 0).
The force-dependent state populations and transition rates can be extrapolated to a zero force to reveal the intrinsic energetics and kinetics of protein transitions in solution (20, 26). Such an extrapolation requires a model that accounts for the effects of force and other experimental conditions (such as the compliance of the DNA handle attached to the protein) on protein transitions (SI Appendix). By simultaneously fitting the measured populations, rates, state forces, and extension changes, we obtained the folding and activation energies, transition rates, and locations of the transition states of pIL at zero force (Fig. 3, SI Appendix, Table S1).
The fully zippered coiled coil has a folding free energy of -24 ( ± 1) kBT relative to its unfolded state. This energy is equivalent to 0.73 kBT per pair of amino acids in the coiled coil region. Thus, we can estimate the folding energies to be -19 kBT, -14 kBT, and -9 kBT for the misfolded states with one, two, and three unpaired heptad repeats, respectively. These estimates are similar to their corresponding, measured energies of -17 kBT, -13 kBT, and -9 kBT, respectively (SI Appendix, Table S1). This energetic consistency corroborates the staggered coiled coil conformations for the partially folded states derived from extension measurements. The comparison also implies that the C-terminal loop regions do not significantly and differentially destabilize the coiled coil in the folded or partially folded states, consistent with previous observations of DNA hairpins with different loop sizes (23). Based on the measured free energies, we calculated populations of the misfolded states with one, two, and three heptad shifts as 1.1 × 10-3, 1.5 × 10-5, and 3.4 × 10-7, respectively.
Our data are consistent with pIL’s barrierless, down-hill folding to both the folded state and the misfolded state 2 (27, 28). The speed of down-hill folding is limited by diffusion of the polypeptide chain (29) and is approximately 1 × 106 s-1 for proteins comparable to pIL in size (20, 30, 31). In contrast to the folded state and misfolded state 2, there is a small (1 ± 3 kBT) and a large (11 ± 3 kBT) energy barrier for pIL to fold into misfolded states 3 and 4, with corresponding folding rates of 2.9 × 105 s-1 and 23 s-1, respectively. These barriers may be contributed by closing the large C-terminal loops (23). Overall, the small pIL protein can rapidly fold and misfold in comparison to larger proteins. Despite their low occurrence frequencies, the misfolded states have relatively long lifetimes of 17 to 485 s, indicating large energy barriers between the misfolded states and the folded state. These barriers can be accounted for by the coiled coils with shifted registers.
Our model of pIL folding highlights the kinetic partition mechanism of protein folding (32, 33). Despite its extremely high rate directly folding into the registered coiled coil state (from State 5 to State 1), overall pIL folds inefficiently, taking a few hundred seconds to complete (SI Appendix, Fig. S5). The misfolded states 2 and 3 form as quickly as the correctly folded state within a few microseconds, trapping > 50% of pIL population in the misfolded states. Further productive folding has to rely on spontaneous unfolding of the misfolded states, which takes more than 10 s due to their high stability and long lifetime. Therefore, pIL represents a large number of proteins that show multiple time scales in their folding processes and exemplifies the intriguing interplay between folding stability, kinetics, and specificity.
The Highly Charged Coiled Coil pER also Misfolds.
In contrast to pIL, which contains an exclusively hydrophobic dimerization interface with probably three interhelical salt bridges (5, 9), pER contains a single asparagine residue at the a position and as many as eight interhelical salt bridges formed by E and R pairs (14). Whereas hydrophilic residues in the dimerization interface destabilize the coiled coil, the interhelical salt bridges enhance the affinity of the coiled coil (9). Nevertheless, both contribute to the specificity of dimerization. Thus, pER is an ideal system for testing the interplay between affinity and specificity of coiled coil association.
To test whether this more canonical coiled coil also misfolds, we obtained reversible folding and unfolding transitions of pER at high spatiotemporal resolution (Fig. 4 A and B). Similar to pIL, pER showed apparent two-state transition kinetics. Specifically, the transition occurred mainly between a folded state and an unfolded state at a high force ranging from 10 pN to 12.5 pN. Analyses based on a 2-state HMM yielded an equilibrium force of 11.4 ( ± 0.8) pN (Fig. 4C) and a folding energy of -21 ( ± 2) kBT for pER. These equilibrium force and folding energy are slightly lower than those of pIL (12.4 pN, -24 kBT), but significantly higher than those of the GCN4 leucine zipper (7.5 pN, -13 kBT) (20). Surprisingly, we also found that pER occasionally misfolds and slides to several partially folded states (Fig. 4B), which are consistent with the staggered coiled coil states. These findings suggest that the strong affinity between the two helices tends to override their dimerization specificity. Importantly, most salt bridges can be maintained in the staggered conformations. However, throughout the tested force range the partially folded states account for less than 1% of the total protein population (Fig. 4C), preventing us from characterizing these states in more detail. Thus, we only quantified pER’s folding and unfolding kinetics, neglecting the small population of the partially folded states (Fig. 4D).
Fig. 4.

Misfolding of pER into staggered states. (A) Extension-time trace filtered using a time window of 2.2 ms. (B) Close-up view of the misfolded state in the region indicated in (A). Data is shown at 1 kHz. (C) Force-dependent probabilities of the protein being in folded (black), misfolded (red), and unfolded states (blue). (D) Force-dependent transition rates of folding (black) and unfolding (blue). The probabilities and rates were fitted to a two-state model, yielding the best-fit lines shown in (C and D). For simplicity, the misfolded states were neglected in the model due to its low probability.
Despite these similarities, pER shows interesting differences compared to pIL. The average extension change of pER (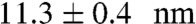 ) at the equilibrium force is greater than that of pIL (10.1 ± 0.3 nm) (SI Appendix, Fig. S6). This result is consistent in that pER is longer than pIL. In addition, pER folds with an activation energy barrier of 3 ( ± 1) kBT, in contrast to the down-hill folding for pIL.
) at the equilibrium force is greater than that of pIL (10.1 ± 0.3 nm) (SI Appendix, Fig. S6). This result is consistent in that pER is longer than pIL. In addition, pER folds with an activation energy barrier of 3 ( ± 1) kBT, in contrast to the down-hill folding for pIL.
Negligible Effects of the Bead Surface and DNA Handle on Protein Transitions.
In our experimental setup, beads were attached directly to one end of a protein molecule and indirectly to the other end using a 2.6 kb DNA spacer, similar to the setup used for the study of the GCN4 leucine zipper (20). The transition kinetics observed for both pIL and pER are homogeneous, which implies minimal nonspecific interactions of the proteins with the bead surfaces and the DNA handle. However, because the misfolded states we observed are new and rare, we further examined the possible nonspecific interactions of pIL or pER with the bead surface and DNA handle that might disturb the transition kinetics of the protein.
To test the effect of surface immobilization, we repeated our single-molecule experiment by isolating the proteins from the bead surface using two DNA handles (26) (SI Appendix, Fig. S7). For both proteins, the transition kinetics were similar to those obtained using one DNA handle, indicating that the bead surface does not appreciably perturb protein transitions in our experiments. Importantly, both proteins show partially folded states indicative of helix staggering and sliding. Moreover, the folding energy and transition kinetics at zero force derived from both pulling setups are consistent. Based on the force-extension curves obtained through pulling individual polypeptides in both pIL and pER, we did not detect any evidence of appreciable interactions between the DNA handle and the polypeptides. This result is further supported by gel-mobility shift assay using a short DNA fragment and pER, which shows no detectable protein-DNA interactions. Overall, these findings suggest that the states with intermediate extensions observed in our experiments are the partially folded, staggered states and not artifacts caused by nonspecific interactions of the tethered proteins with the bead surface and the DNA handle.
Discussion
The Biological Implications of Coiled Coil Staggering, Sliding, and Misfolding.
Using high-resolution optical tweezers, we observed several partially folded states for two strong dimeric coiled coils. We also determined their transition kinetics with the fully folded and unfolded states of these coiled coils. The force-dependent transition kinetics allowed us to determine the structures and energies of the partially folded states as well as their folding and unfolding kinetics at zero force. These structural, thermodynamic, and kinetic analyses suggest that the partially folded structures are a series of staggered helical conformations in which one α-helix is successively shifted by one heptad repeat relative to the other α-helix. These findings confirm the presence of staggered coiled coil conformations previously predicted based upon the periodic structure of the α-helix in the coiled coil (5).
We also show that these staggered coiled coils generally cannot exchange with the fully folded coiled coils, thus representing misfolded structures. Yet, the staggered coiled coils occasionally serve as folding and unfolding intermediates, likely through a helix sliding mechanism observed in other coiled coils (3, 7, 8). Such sliding may take place via polypeptide reptation in the coiled coil without its global unfolding (SI Appendix, Fig. S8). Two-stranded coiled coils are widely believed to fold efficiently without misfolding. To our knowledge, our findings represent a unique example of misfolding and helix sliding of the generic coiled coils.
Most coiled coils mediate protein dimerization and dictate its strength and specificity. Changes in the helical register of coiled coils normally impair their biological functions. Formation of the staggered coiled coil not only reduces the affinity and specificity of protein dimerization, but also alters the overall structures of the resultant protein complexes. Both effects of the staggered coil may compromise protein function. Furthermore, because the staggered states efficiently form but rarely relax to the fully folded states, the coiled coils must unfold and then refold and repeat association, reducing the overall association rate (SI Appendix, Fig. S5). The functional association becomes especially slow for those strong coiled coils like the two proteins studied in this work. In these cases, very stable staggered states with lifetimes that exceed the lifetime of the folded GCN4 leucine zipper (20) can form at rates comparable to the fully folded states. Thus, there seems to be a fundamental limitation for coiled coils to simultaneously meet the demands of high affinity, specificity, and rate for functional association. Additional factors, especially protein chaperones may play important roles in these strong coiled coils.
An important example of such coiled coils is the SNARE proteins, the molecular machines that drive membrane fusion (2). SNAREs generate forces to draw membranes together for fusion and contribute to the specificity of the fusion through ordered protein folding and assembly into an extraordinarily stable four-helix bundle. Particularly, neuronal SNAREs are assembled and disassembled rapidly and repeatedly. SNARE misfolding events impede neuronal communication and are believed to cause various mental disorders and diseases (34). It has been shown in vitro that SNARE proteins can assemble into many nonfunctional structures, leading to reduced fusion rate and specificity (35). Therefore, functional SNARE assembly in vivo is guarded by many chaperone proteins, such as synucleins and Hsc70, and modulated by numerous regulatory proteins (34). Although their molecular mechanisms are unclear, the misfolding of SNARE proteins may be vital to understanding the roles of these diverse protein-protein interactions and their links to various diseases.
The staggered coiled coil states have also been found in many proteins as products of helix sliding, such as the dynein stalk domain (3, 7). Helix sliding is a minor reaction pathway compared with coiled coil misfolding for the two model proteins in our experiments. However, sliding can be greatly enhanced by a shearing force along the helical axis of the coiled coil. This axial force can be induced by motor proteins or by signaling molecules. The sliding reaction can also be facilitated by special sequences in the coiled coil that destabilize the energy barrier for helix sliding. Therefore, helix sliding in coiled coils is a unique mechanism for long-range communication between distant protein domains, especially for signal transduction across membranes (6, 11).
A Minimum of Two-Dimensional Energy Landscape for Protein Folding and Misfolding.
Previous high-resolution measurements from optical tweezers have allowed the full folding energy landscapes of macromolecules to be determined for the first time (28, 36). Because these energy landscapes are deconvoluted from the measured extensions, the derived energy landscapes are intrinsically one-dimensional. Although a 1D energy landscape can describe a sequential folding process, it cannot be used to rationalize the coexistence of both protein folding and misfolding observed in our experiments. In 1D energy landscapes, proteins would be trapped in the misfolded states and would not completely fold. Thus, a higher dimension of energy landscapes is needed to rationalize the misfolding process (37). This conclusion is consistent with the previous observation that a 1D energy landscape is not sufficient to describe the folding kinetics of the three-helix bundle protein αD3 (38). The model used in our data analysis does not rely on the assumption of a 1D energy landscape (SI Appendix). Yet the model allows us to determine several characteristic points of the energy landscape. Further theoretical development in combination with high-resolution measurements may lead to a complete description of protein folding and misfolding in a two-dimensional energy landscape.
Conclusions.
We show that coiled coils are more dynamic than previously thought using high-resolution optical tweezers. Even in strong coiled coils with canonical heptad sequences, helix staggering occurs through either protein misfolding or helix sliding. We suggest that this new dynamic is a general feature of coiled coils that is important in our understanding of the functions and dysfunctions of coiled coils.
Methods
The polypeptides were chemically synthesized and purified to > 95% by HPLC. The sequences of the polypeptides are listed as follows:
pIL-B: Biotin-GGGG R MKQLEDK IEELLSK IYHLENE IARLKKL IGER GGC
pIL-C: Ac-CGGG R MKQLEDK IEELLSK IYHLENE IARLKKL IGER GGC
pER-B: Biotin-GGGG LEIE AAFLEQE NTALETE VAELEQE VQRLENI VSQYETR YGPL GGC
pER-C: Ac-CGGG LEIR AAFLRRR NTALRTR VAELRQR VQRLRNI VSQYETR YGPL GGC
The polypeptides pIL-B and pIL-C were used to form the coiled coil pIL. pER-B and pER-C were used for pER. The sequences in the coiled coil region are listed in blocks of heptad repeat (abcdefg)n, and those at both ends of each polypeptide (in italic) are the linker sequences added to facilitate pulling and crosslinking. The acetylated N-terminal end is indicated by ‘Ac’. The pIL complexes were formed by mixing both polypeptides (pIL-B and pIL-C) in 7 M guanidinium chloride and then gel-filtered to the PBS buffer. The complex was crosslinked to the DNA handle and tethered on bead surfaces as was previously described (20). Tweezer instrument and methods of data analysis are described in SI Appendix.
Supplementary Material
Acknowledgments.
We thank Prof. Arthur Horwich for reading the manuscript and Greg Gundersen for drawing graphs. Support for this work was provided by the National Institute of Health (R01GM093341) to Y.Z.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1116784109/-/DCSupplemental.
References
- 1.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carter AP, Cho C, Jin L, Vale RD. Crystal structure of the dynein motor domain. Science. 2011;331:1159–1165. doi: 10.1126/science.1202393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.del Rio A, et al. Stretching single Talin rod molecules activates Vinculin binding. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oshea EK, Klemm JD, Kim PS, Alber T. X-ray structure of the GCN4 leucine zipper, a 2-stranded, parallel coiled coil. Science. 1991;254:539–544. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 6.Matthews EE, Zoonens M, Engelman DM. Dynamic helix interactions in transmembrane signaling. Cell. 2006;127:447–450. doi: 10.1016/j.cell.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 7.Kon T, et al. Helix sliding in the stalk coiled coil of dynein couples ATPase and microtubule binding. Nat Struct Mol Biol. 2009;16:325–333. doi: 10.1038/nsmb.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Croasdale R, et al. An undecided coiled coil: the leucine zipper of Nek2 kinase exhibits atypical conformational exchange dynamics. J Biol Chem. 2011;286:27537–27547. doi: 10.1074/jbc.M110.196972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harbury PB, Zhang T, Kim PS, Alber T. A switch between 2-stranded, 3-stranded and 4-stranded coiled coils in GCN4 leucine-zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez L, Brown RA, Richardson D, Alber T. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat Struct Biol. 1996;3:1002–1010. doi: 10.1038/nsb1296-1002. [DOI] [PubMed] [Google Scholar]
- 11.Hulko M, et al. The HAMP domain structure implies helix rotation in transmembrane signaling. Cell. 2006;126:929–940. doi: 10.1016/j.cell.2006.06.058. [DOI] [PubMed] [Google Scholar]
- 12.Nooren IM, Kaptein R, Sauer RT, Boelens R. The tetramerization domain of the Mnt repressor consists of two right-handed coiled coils. Nat Struct Biol. 1999;6:755–759. doi: 10.1038/11531. [DOI] [PubMed] [Google Scholar]
- 13.Acharya A, Rishi V, Vinson C. Stability of 100 homo and heterotypic coiled-coil a-a’ pairs for ten amino acids (A, L, I, V, N, K, S, T, E, and R) Biochemistry-Us. 2006;45:11324–11332. doi: 10.1021/bi060822u. [DOI] [PubMed] [Google Scholar]
- 14.Moll JR, Ruvinov SB, Pastan I, Vinson C. Designed heterodimerizing leucine zippers with a range of pIs and stabilities up to 10-15 M. Protein Sci. 2001;10:649–655. doi: 10.1110/ps.39401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffitt JR, Chemla YR, Izhaky D, Bustamante C. Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc Natl Acad Sci USA. 2006;103:9006–9011. doi: 10.1073/pnas.0603342103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oberhauser AF, Marszalek PE, Carrion-Vazquez M, Fernandez JM. Single protein misfolding events captured by atomic force microscopy. Nat Struct Biol. 1999;6:1025–1028. doi: 10.1038/14907. [DOI] [PubMed] [Google Scholar]
- 17.Bechtluft P, et al. Direct observation of chaperone-induced changes in a protein folding pathway. Science. 2007;318:1458–1461. doi: 10.1126/science.1144972. [DOI] [PubMed] [Google Scholar]
- 18.Dong JJ, Castro CE, Boyce MC, Lang MJ, Lindquist S. Optical trapping with high forces reveals unexpected behaviors of prion fibrils. Nat Struct Mol Biol. 2010;17:1422–1430. doi: 10.1038/nsmb.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreon ACM, Deniz AA. Protein folding at single-molecule resolution. Biochim Biophys Acta. 2011;1814:1021–1029. doi: 10.1016/j.bbapap.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Sirinakis G, Zhang YL. Highly anisotropic stability and folding kinetics of a single coiled coil protein under mechanical tension. J Am Chem Soc. 2011;133:12749–12757. doi: 10.1021/ja204005r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin F, Auerbach A, Sachs F. A direct optimization approach to hidden Markov modeling for single channel kinetics. Biophys J. 2000;79:1915–1927. doi: 10.1016/S0006-3495(00)76441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ainavarapu RK, et al. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys J. 2007;92:225–233. doi: 10.1529/biophysj.106.091561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodside MT, et al. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc Natl Acad Sci USA. 2006;103:6190–6195. doi: 10.1073/pnas.0511048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bustamante C, Chemla YR, Forde NR, Izhaky D. Mechanical processes in biochemistry. Annu Rev Biochem. 2004;73:705–748. doi: 10.1146/annurev.biochem.72.121801.161542. [DOI] [PubMed] [Google Scholar]
- 25.Sirinakis G, et al. The RSC chromatin remodeling ATPase translocates DNA with high force and small step size. EMBO J. 2011;30:2364–2372. doi: 10.1038/emboj.2011.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–2060. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 27.Cho SS, Weinkam P, Wolynes PG. Origins of barriers and barrierless folding in BBL. Proc Natl Acad Sci USA. 2008;105:118–123. doi: 10.1073/pnas.0709376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gebhardt JCM, Bornschlogla T, Rief M. Full distance-resolved folding energy landscape of one single protein molecule. Proc Natl Acad Sci USA. 2010;107:2013–2018. doi: 10.1073/pnas.0909854107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang WY, Gruebele M. Folding at the speed limit. Nature. 2003;423:193–197. doi: 10.1038/nature01609. [DOI] [PubMed] [Google Scholar]
- 30.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Mukherjee S, Chowdhury P, Bunagan MR, Gai F. Folding kinetics of a naturally occurring helical peptide: implication of the folding speed limit of helical proteins. J Phys Chem B. 2008;112:9146–9150. doi: 10.1021/jp801721p. [DOI] [PubMed] [Google Scholar]
- 32.Guo ZY, Thirumalai D. Kinetics of protein-folding—nucleation mechanism, time scales, and pathways. Biopolymers. 1995;36:83–102. [Google Scholar]
- 33.Peng Q, Li H. Atomic force microscopy reveals parallel mechanical unfolding pathways of T4 lysozyme: evidence for a kinetic partitioning mechanism. Proc Natl Acad Sci USA. 2008;105:1885–1890. doi: 10.1073/pnas.0706775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burre J, et al. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunger AT. Structure and function of SNARE and SNARE-interacting proteins. Q Rev Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004051. [DOI] [PubMed] [Google Scholar]
- 36.Woodside MT, et al. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001–1004. doi: 10.1126/science.1133601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stigler J, Ziegler F, Gieseke A, Gebhardt JCM, Rief M. The complex folding network of single calmodulin molecules. Science. 2011;334:512–516. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 38.Liu F, et al. A one-dimensional free energy surface does not account for two-probe folding kinetics of protein α3D. J Chem Phys. 2009;130:061101. doi: 10.1063/1.3077008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.