

Abstract
Surface charges of proteins have in several cases been found to function as “structural gatekeepers,” which avoid unwanted interactions by negative design, for example, in the control of protein aggregation and binding. The question is then if side-chain charges, due to their desolvation penalties, play a corresponding role in protein folding by avoiding competing, misfolded traps? To find out, we removed all 32 side-chain charges from the 101-residue protein S6 from Thermus thermophilus. The results show that the charge-depleted S6 variant not only retains its native structure and cooperative folding transition, but folds also faster than the wild-type protein. In addition, charge removal unleashes pronounced aggregation on longer timescales. S6 provides thus an example where the bias toward native contacts of a naturally evolved protein sequence is independent of charges, and point at a fundamental difference in the codes for folding and intermolecular interaction: specificity in folding is governed primarily by hydrophobic packing and hydrogen bonding, whereas solubility and binding relies critically on the interplay of side-chain charges.
Keywords: folding cooperativity, protein aggregation, protein charges, protein engineering, protein folding
Protein folding is not only about optimizing the native state, but is also about avoiding misfolded traps (1–5). Such traps would otherwise compete thermodynamically with the native structures and decrease protein stability. Misfolded states that fail to properly bury “sticky” sequence material are also undesired because of their coupling to protein-aggregation disease (6–9). The general idea is that, to avoid misfolding, natural proteins have cooperative folding transitions with strong bias toward native interactions (10–13): they fold as if they were blind to alternative conformations. A clue to how this “Go-like” behavior arises is hinted by the ribosomal protein S6 (14). In essence, the S6 sequence is found to comprise certain “gatekeeper” residues (5) that block competing misfolded states by negative design (15), biasing the folding-energy landscape toward native interactions (5, 12). Mutation of these folding gatekeepers increases the propensity for S6 to misfold prior to the global folding transition in stopped-flow experiments. The phenomenon is most clearly seen in the presence of Na2SO4 where the mutations induce a pronounced retardation of the refolding kinetics and characteristic roll-overs in the refolding limbs of the chevron plots (5). Notably, the chemical identity of the folding gatekeepers of S6 is not uniform but includes the buried V85, the solvent exposed E22, as well as the strain-relieving mutation A35G. The reason for this chemical diversity, as well as the detailed action of the gatekeepers, is yet not known. From a chemical standpoint it is nevertheless expected that the ubiquitous surface charges of globular proteins (16) would play a general role in negative design by their intrinsic desolvation penalties; i.e., misfolding that leads to burial of unmatched charges is strongly disfavored (17). Moreover, the evolutionary freedom of using surface charges to block misfolding is comparatively large as their effect on native-state stability is often small. Consistently, surface charges are naturally employed to prevent unwanted association of folded proteins (18–21) and play a key role in solubility of denatured states (22). This class of side chains, which we term “aggregation gatekeepers” (20), need not have any influence on folding and stability (20) but decorate as a rule β-sheet edges in crystal structures (21). In addition to the folding gatekeepers, S6 contains two pairs of such charged aggregation gatekeepers in β-strand 2: their removal triggers transient coil aggregation, native-state tetramerization, and fibrillation on longer timescales (20).
In this study, we examine at more general level the role of side-chain charges in protein folding and aggregation by removing them completely. S6 is normally rich in charges and carries 16 negatively and 16 positively charged side chains, comprising 32% of its sequence content. By a combination of protein engineering and lowered pH we produced a protein that is altogether noncharged, save the positive N-terminal (S6+1). The results show that complete charge removal, if anything, favors the folding process: S6+1 not only maintains a classically v-shaped chevron plot, but also folds faster than the wild-type protein. On longer timescales, however, the protein starts to aggregate, both with native and denatured S6+1 as starting material. Our conjecture from these data is that the profusion of charges scattered in the sequences of natural proteins are not required for folding per se, but play their major role in solubility, recognition, and biological function.
Results
Design of a Charge-free Protein.
Charge removal was done in two steps (Fig. 1). First, all K and R side chains in wild-type S6+17-17 were mutated to S. The resulting supercharged variant S6+1-17 expressed in soluble form with good yields in Escherichia coli, showing that the bacterial transcription machinery has no problems with handling extreme, uniform charge. Second, the remaining negatively charged D and E moieties were neutralized by protonation at pH 2.3 to obtain the charge-depleted variant S6+1, containing only one positive charge at the N-terminal. S6 is particularly well suited for this approach as the native-state pKA shifts are relatively modest and the protein is fully protonated and still folded at pH 2.3 (Fig. S1). To probe for protonation of the backbone carbonyls, we examined S6+1 stability by transient unfolding down to pH 0.3. The results indicate that S6+1 carries solely its N-terminal charge between pH 1 and pH 3 (Fig. S1). We also attempted to produce S6+1 directly by the global substitutions K and R to S, D to N and E to Q, but this construct failed to express.
Fig. 1.

Charge removal of S6. A. Wild-type S6+17-17 comprises 16 negative (red) and 16 positive (blue) charges on the protein surface. B. Supercharged S6+1-17 was produced by mutation of all K and R to S. C. Charge-depleted S6+1 was finally obtained by transferring S6+1-17 to pH 2.3 where all negative side-chain charges as well as the C-terminal becomes neutralized by protonation. D. Positions of positively- (blue) and negatively (red) charged side chains in the S6 sequence.
Charge-depleted S6 Maintains Native-like Solution Structure.
Wild-type S6+17-17 has a fixed, tertiary-ordered structure in solution, which is indistinguishable from that in crystals (23). In this study, we see that S6 retains this ordered structure, as well as a cooperative folding transition, in its supercharged state S6+1-17. The NMR HSQC spectrum of S6+1-17 at pH 6.3 and 100 mM NaCl reveals well-dispersed cross-peaks similar, but not identical to, those of wild-type S6+17-17 (Fig. 2). The observed difference between the S6+1-17 and S6+17-17 spectra, however, is expected since the proteins have only 84% sequence identity. Backbone assignment was obtained unambiguously for 84 of the 101 residues of S6+1-17. The missing assignments are mainly scattered throughout the protein but leads to short gaps at positions 40–43 and 59–63 in the sequence. We attribute the missing assignments to spectral overlap and exchange broadening, consistent with higher than average dynamic motions in the affected regions of the wild-type S6+17-17 structure (24). Even so, Cα - Cβ secondary-chemical-shift analysis indicate that the secondary-structure elements remain wild-type like at all positions where the assignments of S6+17-17 and S6+1-17 overlap (Fig. S2). To further examine the conservation of secondary structure we plotted the secondary chemical shifts of wild-type S6+17-17 versus those of S6+1-17 (Fig. S2). The results yield a linear correlation of r = 0.95, which closely resembles that between wild-type S6+17-17 and the structurally ordered permutant S654-55 (23, 24) (Fig. S2), and supports the conclusion that K and R removal does not significantly alter the S6 structure. Neutralization of the remaining 17 negative charges at low pH (Table 1) is then unlikely to have any further structural effects: if anything, the complete alleviation of electrostatic repulsion within the S6+1 molecule is expected to be beneficial. Consistently, the charge-depleted S6+1 structure yields highly dispersed HSQC spectra under carefully tuned conditions at pH 1 (Fig. 2), indicating a homogeneous, tertiary-ordered population. However, the disappearance of signal due to aggregation on longer timescale has so far precluded detailed structural assignment of the charge-depleted protein.
Fig. 2.

NMR HSQC spectra of the different charge variants of S6. All spectra show wild-type like dispersion, suggesting that the supercharged S6+1-17 and the charge-depleted S6+1 maintain fixed, three-dimensional structures. Additional NMR evidence for conserved structure of S6+1-17 is presented in Fig. S2, whereas detailed structural analysis of S6+1 has so far been precluded by aggregation on longer timescales (cf. Fig. 5).
Table 1.
Kinetic parameters and protein stabilities
| S6+17-17 | S6+1-17 | S6+1 | |
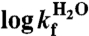 * *
|
2.53 ± 0.03 | 1.48 ± 0.03 | 3.28 ± 0.06 |
| mf*(M-1) | −1.22 ± 0.01 | −0.82 ± 0.04 | −1.10 ± 0.04 |
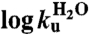 * *
|
−3.51 ± 0.08 | −0.64 ± 0.05 | 0.35 ± 0.04 |
| mu* (M-1) | 0.54 ± 0.02 | 0.43 ± 0.01 | 0.27 ± 0.01 |
| MP † (M) | 3.44 ± 0.03 | 1.71 ± 0.05 | 2.15 ± 0.04 |
| mD-N‡ (M-1) | 1.76 ± 0.02 | 1.24 ± 0.04 | 1.36 ± 0.04 |
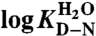 § §
|
6.04 ± 0.09 | 2.12 ± 0.06 | 2.93 ± 0.07 |
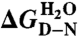 ¶ (kcal/mol) ¶ (kcal/mol) |
8.24 ± 0.12 | 2.89 ± 0.08 | 3.99 ± 0.09 |
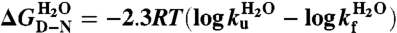
Charges are not Required for Cooperative Folding of S6.
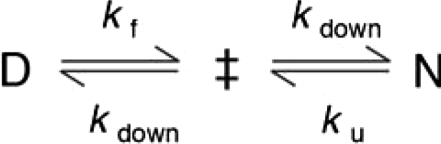
Folding of S6 is a two-state process with two competing pathways, the bias between which can be altered by circular permutation (24, 25) (Fig. S3). Wild-type S6+17-17 employs mainly one of these pathways (26), manifested in a v-shaped chevron plot characteristic of a cooperative transition between the denatured (D) and native (N) states over a single transition-state (‡) (25–27) where kf and ku are the refolding- and unfolding-rate constants, and kdown is the kinetic prefactor; i.e., the rate constant for jumping down the barrier (28). The intersect of log kf and log ku in the S6+17-17 chevron plot gives a transition midpoint (MP) at 3.4 M GdmCl and an extrapolated stability of 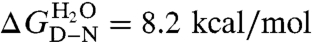 (Fig. 3, Eq. 4, Table 1). Upon mutational removal of all positively charged side chains, the transition midpoint decreases to 1.7 M GdmCl (Fig. 3, Table 1) and the chevron changes yield a global ϕ-value of 0.20; i.e., the mutation to S6+1-17 has the largest impact on log ku [Eq. 5]. This means that global removal of alkaline residues destabilizes N more that than ‡. Considering the high ionic strength under the experimental conditions; i.e., [GdmCl] > 0.4 M, this destabilization is not dominated by long-range electrostatic repulsion but seems rather to arise from mutant-induced contact losses and/or changes in solvent interactions. At low ionic strength with added charge-charge repulsion the destabilization of S6+1-17 becomes even larger, leading to mixed populations of D and N (Fig. S4). Finally, to obtain the folding kinetics of the charge-depleted species S6+1, we adjusted the refolding- and unfolding buffers to pH 2.3, to assure full protonation of the acidic side chains in the reaction chamber of the stopped-flow apparatus. Two notable features are revealed. First, the charge-depleted protein maintains a v-shaped chevron plot and apparent two-state behavior in the absence of side-chain charges. Second, charge depletion increases both
(Fig. 3, Eq. 4, Table 1). Upon mutational removal of all positively charged side chains, the transition midpoint decreases to 1.7 M GdmCl (Fig. 3, Table 1) and the chevron changes yield a global ϕ-value of 0.20; i.e., the mutation to S6+1-17 has the largest impact on log ku [Eq. 5]. This means that global removal of alkaline residues destabilizes N more that than ‡. Considering the high ionic strength under the experimental conditions; i.e., [GdmCl] > 0.4 M, this destabilization is not dominated by long-range electrostatic repulsion but seems rather to arise from mutant-induced contact losses and/or changes in solvent interactions. At low ionic strength with added charge-charge repulsion the destabilization of S6+1-17 becomes even larger, leading to mixed populations of D and N (Fig. S4). Finally, to obtain the folding kinetics of the charge-depleted species S6+1, we adjusted the refolding- and unfolding buffers to pH 2.3, to assure full protonation of the acidic side chains in the reaction chamber of the stopped-flow apparatus. Two notable features are revealed. First, the charge-depleted protein maintains a v-shaped chevron plot and apparent two-state behavior in the absence of side-chain charges. Second, charge depletion increases both 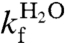 and
and 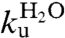 : at the transition midpoint S6+1 folds about 300 times faster than wild-type S6+17-17 (Fig. 3,Table 1). The corresponding data for wild-type S6 at pH 2.3 are shown in Fig. S5. As the acceleration upon charge depletion is not accompanied by a correspondingly large change in protein stability (Fig. 4, Table 1), it seems to arise from either a selective stabilisation of the transition-state ensemble (‡) or an increase of the folding prefactor; i.e., kdown in Scheme 1 (10, 28). The chevron plots of S6+1-17 and S6+1 reveal also slight reductions of mf and mu [Eqs. 2–3] and S6+1-17 displays a small downward curvature at high [GdmCl] (Fig. 3). Similar m-value changes and curvatures are seen upon point mutation of the wild-type and circularly permuted S6 (29), and are generally attributed to transition-state shifts (30) (Fig. 4, Fig. S3) or ground-state fraying (27). In the present study, however, it is also possible that the m-value changes stem from alterations of the protein’s interaction with the solvent/denaturant molecules or changes of the compactness of the denatured ensemble as described below (Fig. 4). Elimination of 33 charges represents, after all, a considerable change of the chemical properties of the polypeptide chain. Even so, it is clear that the charge depletion of S6 has little effect on the principal features of the folding process, if anything the protein seems to fold faster without charges.
: at the transition midpoint S6+1 folds about 300 times faster than wild-type S6+17-17 (Fig. 3,Table 1). The corresponding data for wild-type S6 at pH 2.3 are shown in Fig. S5. As the acceleration upon charge depletion is not accompanied by a correspondingly large change in protein stability (Fig. 4, Table 1), it seems to arise from either a selective stabilisation of the transition-state ensemble (‡) or an increase of the folding prefactor; i.e., kdown in Scheme 1 (10, 28). The chevron plots of S6+1-17 and S6+1 reveal also slight reductions of mf and mu [Eqs. 2–3] and S6+1-17 displays a small downward curvature at high [GdmCl] (Fig. 3). Similar m-value changes and curvatures are seen upon point mutation of the wild-type and circularly permuted S6 (29), and are generally attributed to transition-state shifts (30) (Fig. 4, Fig. S3) or ground-state fraying (27). In the present study, however, it is also possible that the m-value changes stem from alterations of the protein’s interaction with the solvent/denaturant molecules or changes of the compactness of the denatured ensemble as described below (Fig. 4). Elimination of 33 charges represents, after all, a considerable change of the chemical properties of the polypeptide chain. Even so, it is clear that the charge depletion of S6 has little effect on the principal features of the folding process, if anything the protein seems to fold faster without charges.
Fig. 3.

The folding kinetics of the different charge variants of S6 analyzed by stopped-flow mixing. A. The chevron plots of wild-type S6+17-17, supercharged S6+1-17 and charge-depleted S6+1 are all classically v-shaped, showing that the folding transition remains cooperative and does not rely on side-chain charges. Of particular interest is that complete charge removal even speeds-up folding: at the transition midpoint S6+1 folds > 300 times faster than wild-type S6+17-17. B. Na2SO4 titration of the refolding reaction at 0.4 M GdmCl shows that the propensity of the coil to undergo premature collapse in the mixing dead-time slightly increases upon removal of all positively charged side chains. C. At 1.6 M GdmCl, it is seen that charge-depleted S6+1 has the highest collapse propensity of the three proteins and also displays a change of rate-limiting step at high [Na2SO4] (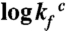 ). The origin of this change could be the population of an alternative, parallel, folding route to the native state according to Scheme 2.
). The origin of this change could be the population of an alternative, parallel, folding route to the native state according to Scheme 2.
Fig. 4.

Folding free-energy profiles of wild-type S6+17-17 (blue), supercharged S6+1-17 (red), and charge-depleted S6+1 (black). Barrier heights were calculated from kf and a prefactor of 106s-1 (28). Charge removal leads to faster folding kinetics and an apparent stabilization of the transition-state ensemble (‡), whereas the native state is destabilized. The positions of D, ‡ and N along the progress coordinate have been scaled according to the m-values in Table 1 and normalized to N.
Scheme 1.
Charge-Depletion Affects Collapse Propensity.
To determine how charge removal affects the misfolding propensity, we measured and compared the refolding kinetics of S6+17-17, S6+1-17, and S6+1 at increasing concentrations of Na2SO4. Titration with stabilizing 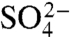 ions has previously been found to induce misfolding of S6, accompanied by a characteristic retardation of kf (5). The rationale behind this experiment is that titration with cosmotropic
ions has previously been found to induce misfolding of S6, accompanied by a characteristic retardation of kf (5). The rationale behind this experiment is that titration with cosmotropic 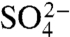 ions gradually increases the contact free energies to a point where misfolding start to retard kf (5); i.e., the frustration in the folding-energy landscape is increased to promote nonnative collapse (12, 31). A minimalist model for such a collapse is shown in Scheme 2, where I∗ is a competing, misfolded state and
ions gradually increases the contact free energies to a point where misfolding start to retard kf (5); i.e., the frustration in the folding-energy landscape is increased to promote nonnative collapse (12, 31). A minimalist model for such a collapse is shown in Scheme 2, where I∗ is a competing, misfolded state and 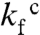 denotes tentatively an alternative folding route to the native state (5). To assure suitable windows for the refolding kinetics the experiments were performed at a background of 0.4 and 1.6 M GdmCl (Fig. 3). At 0.4 M GdmCl, kf of S6+17-17 first increases as ‡ is stabilized relative to D by a “reversed” denaturant effect (Fig. 3); i.e., the
denotes tentatively an alternative folding route to the native state (5). To assure suitable windows for the refolding kinetics the experiments were performed at a background of 0.4 and 1.6 M GdmCl (Fig. 3). At 0.4 M GdmCl, kf of S6+17-17 first increases as ‡ is stabilized relative to D by a “reversed” denaturant effect (Fig. 3); i.e., the 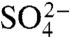 ions favor compact states by being preferentially excluded from the protein’s hydration shell. Then, around 0.2 M Na2SO4, kf starts to decrease as misfolding commence (Fig. 3). In an earlier study, we ascribed this misfolding to premature collapse of the coil in the mixing dead time (5), which can slow down folding by either ground-state stabilization or retardation of the diffusive motions. Consistently, the maximum of kf shifts to higher [Na2SO4] under better solvent conditions at 1.6 M GdmCl (Fig. 3). A similar shift of the kf maximum is observed for the supercharged S6+1-17 (Fig. 3), indicating that the increased negative charge suppresses coil collapse, cf. (32). The very opposite effect is observed for S6+1: complete charge removal increases slightly the misfolding propensity (Fig. 3). Despite this tendency, the role of charges in smoothening the folding funnel seems overall marginal as this increased collapse propensity does not compromise folding in the absence of Na2SO4, (12).
ions favor compact states by being preferentially excluded from the protein’s hydration shell. Then, around 0.2 M Na2SO4, kf starts to decrease as misfolding commence (Fig. 3). In an earlier study, we ascribed this misfolding to premature collapse of the coil in the mixing dead time (5), which can slow down folding by either ground-state stabilization or retardation of the diffusive motions. Consistently, the maximum of kf shifts to higher [Na2SO4] under better solvent conditions at 1.6 M GdmCl (Fig. 3). A similar shift of the kf maximum is observed for the supercharged S6+1-17 (Fig. 3), indicating that the increased negative charge suppresses coil collapse, cf. (32). The very opposite effect is observed for S6+1: complete charge removal increases slightly the misfolding propensity (Fig. 3). Despite this tendency, the role of charges in smoothening the folding funnel seems overall marginal as this increased collapse propensity does not compromise folding in the absence of Na2SO4, (12).
Scheme 2.

The Compact Detour.
From the S6+1 data in Fig. 3, it is evident that log kf does not continue to decrease with increasing [Na2SO4] but levels off and describes a slight positive slope above 0.8 M Na2SO4. A corresponding change of log kf is seen upon lowering the GdmCl concentration at a background of 1.0 M Na2SO4 (Fig. S6). For simplicity, we denote this new phase 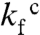 (Scheme 2). The phenomenon has previously been assigned to a change of rate-limiting step: when the free-energy difference between the collapsed state and the normal ‡ reaches a critical value, folding switches to a parallel pathway (5). Since the denaturant and Na2SO4 dependencies of
(Scheme 2). The phenomenon has previously been assigned to a change of rate-limiting step: when the free-energy difference between the collapsed state and the normal ‡ reaches a critical value, folding switches to a parallel pathway (5). Since the denaturant and Na2SO4 dependencies of 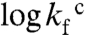 are very much weaker than for log kf, this parallel pathway was suggested to show reconfigurations between collapsed species of similar solvent accessible surface area—a
are very much weaker than for log kf, this parallel pathway was suggested to show reconfigurations between collapsed species of similar solvent accessible surface area—a 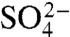 -induced detour through compact regions of the conformational space (5). In this study, we observe further that the change of rate-limiting step is coupled to the emergence of several slower refolding phases (Fig. S7). One possibility is that these phases describe the onset of transient aggregation occurring in parallel with the coil collapse, as observed upon removal of the charged aggregation gatekeepers in β2 (20), or stem from a more complex partitioning of trapped species under high misfolding pressure. In keeping with the recent discussion of downhill folding (33, 34), the complex time course of
-induced detour through compact regions of the conformational space (5). In this study, we observe further that the change of rate-limiting step is coupled to the emergence of several slower refolding phases (Fig. S7). One possibility is that these phases describe the onset of transient aggregation occurring in parallel with the coil collapse, as observed upon removal of the charged aggregation gatekeepers in β2 (20), or stem from a more complex partitioning of trapped species under high misfolding pressure. In keeping with the recent discussion of downhill folding (33, 34), the complex time course of  could also reflect a change to stretched-exponential behavior arising from barrierless reconfigurations in an increasingly rugged landscape. The collapse transition would then break down the delicate imbalance between entropy and contact free energy that shape the barrier, promoting noncooperative downhill folding.
could also reflect a change to stretched-exponential behavior arising from barrierless reconfigurations in an increasingly rugged landscape. The collapse transition would then break down the delicate imbalance between entropy and contact free energy that shape the barrier, promoting noncooperative downhill folding.
Charges are Critical for Solubility on Longer Timescales.
Wild-type S6+17-17 and supercharged S6+1-17 display no tendency to aggregate in this study, not even in the presence of Na2SO4. The proteins lack characteristic aggregation phases on longer time courses in the stopped-flow mixing experiments and the equilibrated starting- and end-material remain soluble for weeks. Nevertheless, the protein begins typically to precipitate at protein concentrations above a few μM upon acidification of S6+1-17 to obtain S6+1 and is easily detected by centrifugation (Fig. 5). Aggregation of charge-depleted S6+1 is also clear to the eye in cuvettes, both in the absence and presence of [GdmCl] (Fig. 5). The latter observation shows that both the folded- and denatured-states of S6 lose solubility without side-chain charges, even at high concentrations of denaturant. Due to the relatively slow time course of the S6+1 aggregation, however, it commences after refolding and unfolding has been completed in stopped-flow experiments: aggregation under typical refolding and unfolding conditions occurs on a timescale that is > 10 times slower than the folding transitions (Fig. 3). Moreover, transient coil aggregation during refolding, which is initiated by truncation of charged aggregation gatekeepers in β2 (20), is expected to have marginal impact on folding data at 1 μM protein concentration (20). Consistently, charge-depleted S6+1 displays an archetypically v-shaped chevron plot without observable distortion from competing aggregation processes (Fig. 3).
Fig. 5.

Charge depletion leads to S6 aggregation. A. The three charge variants of S6 were incubated at 11 μM protein concentration for 48 h and then centrifuged at 17,000 × g for 2 h. SDS-PAGE gels show that wild-type S6+17-17 and supercharged S6+1-17 remain soluble in the supernatant, whereas the charge-depleted S6+1 aggregates go into the pellet. The different densities of the bands are due to weakened interaction between the SYPRO Orange stain and the supercharged S6+1-17. During the electrophoresis at pH 6.8, charge-depleted S6 deprotonates and becomes supercharged S6+1-17. B. Photograph of S6 samples showing aggregation as increased scatter upon flash illumination. Charge-depleted S6+1 aggregates both in its folded and unfolded states at high concentration of denaturant.
Discussion
The data in this study demonstrate that the native structure and folding behavior of S6 does not rely on the presence of side-chain charges: the protein displays a swift and cooperative folding transition both with and without side-chain charges (Figs. 2–3). The result concurs with the earlier conclusion by Loladze and Makhatadze that surface charge-charge interactions are not essential for protein folding, based on thermodynamic analysis of chemically charge-depleted ubiqutin (35). Judging by the accelerated folding kinetics of S6+1 (Fig. 3), it can even be said that charges are a burden to protein folding. The origin of this acceleration, however, is not yet clear. One possibility is that side-chain charges restrict the protein’s reconfigurations or ability to collapse (36) by the way they interact with the solvent. Along this line, elimination of charges could speed up folding by increasing the degree of unspecific hydrophobic contacts in the transition-state ensemble, as observed for the α-spectrin SH3 domain upon Tyr-Phe exchange (37, 38). Although the explanation seems perfectly consistent with the increased collapse propensity of S6+1 in Na2SO4 assays, the window for this acceleration is then rather narrow: as soon as the collapse continues into the glassy regime folding will seize up by increased internal friction, as implicated by the rollover above ≈0.3 M Na2SO4 (Fig. 3). Even so, the overall limited influence of charge on S6 folding is remarkable considering that the protein functions in a thermophilic bacterium where high charge content is generally believed to reflect an optimization to high thermal stability (39–41). The principal role of the S6 charges seems rather to be in solubility. As opposed to folding, protein solubility and intermolecular interactions are found to depend critically on charge. In the case of S6, elimination of edge-strand charges promotes the assembly of nonnative tetramers (20) and, in other systems, the introduction of edge-strand charges have reversely been observed to break up aggregates into monomeric beta-sheet proteins (42). Moreover, the rates of aggregation of unfolded proteins and polypeptides show generally a distinct dependence on charge content (22), and reduction of a protein’s net negative charge by merely one unit is sufficient for triggering fatal neurodegenerative disease (43). These observations go hand-in-hand with the general idea that charges enhance interaction specificity, not primarily by attraction, but because of large penalties for unmatched burial (17, 44, 45). In this context it is interesting to note that the S6 charges do a better job to prevent aggregation than high concentrations of denaturant (Fig. 5). Surveys of structural data banks shows consistently that lysine, which is the most abundant residue on protein surfaces (46), is the most underrepresented amino acid at interfaces between proteins in functional complexes (47, 48). To this end there are also numerous examples where surface charges control protein interactions in a more attractive way; e.g., in protein-protein recognition (49, 50), in binding to membranes (51) and binding of metal ions (52). As a first approximation, we distinguish these charge-controlled interactions, including solubility and spatial organization, as “functional,” since they orchestrate the biological function of the elementary folding units. With this distinction, the ability of S6 to fold without charges raises the question if there is a principal division of amino-acid use in the self-organization of proteins: folding can evidently be driven by hydrophobicity and hydrogen bonding alone, whereas function and intermolecular organization tend to rely critically on the interplay of charges. An advantage of such a separation of “driving forces” could be that, in its pure form, it biases folding and function from being intermingled; i.e., folding and function have chemical space to evolve independently. It is nevertheless inevitable that charges will still have a pronounced influence on their protein scaffolds, be it through favorable ion pairing or as a side effect of conflicting, functional optimization (49). In some cases, charges control even the structural order of entire protein domains. An intriguing example is the “dome-like” active-site envelope of Cu/Zn superoxide dismutase, which is built almost exclusively by polar and charged side chains, pulled together at its center by a single Zn2+ ion (53, 54). The metal ion seems here to substitute for a local hydrophobic core and creates effectively a functional subdomain that structures separately and does not interfere with the folding of the main hydrophobic core (55). This split architecture and folding behavior of superoxide dismutase lends further support to the conjecture of an underlying, chemical bias in codes for folding and function of proteins. A clue to the question “why are proteins charged” (16) could then be: not for folding of the basic structural domains.
Materials
Mutagenisis, Expression, and Purification.
S6+1-17 gene synthesis, codon optimization for overexpression in E. coli, subcloning into a pET-3a vector using 5′ Nde1 and 3′ BamH1 restriction sites, and construct sequencing were performed by GenScript. Transformation into E. coli BL21 (DE3) cells was by standard heat-shock procedures. Expression and purification were as previously described for wild-type S6+17-17 (27), whereas the supercharged S6+1-17 required a modified purification protocol (Supporting Information Data Analysis and Methods. Mutagenesis, expression, and purification of S6+1-17.). Purity was analyzed by Ready Gel SDS-PAGE system (Bio-Rad) and electrospray ionization mass spectrometry and Edman degradation performed by the Protein Analysis Center (Karolinska Institute). Edman degradation showed that S6+1-17 lacks the N-terminal methione present in wild-type S6+17-17.
NMR Spectroscopy.
HSQC NMR data were obtained at 25 °C with protein concentrations ranging from 30 (pH 1) to 500 μM (pH 6.3, 100 mM NaCl), on a Bruker 700 MHz spectrometer (Bruker Avance) equipped with a cryogenically cooled triple resonance probe. Backbone assignment was obtained from a set of standard 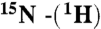 -HSQC, HNCA, HN(CO)CA, HN(CA)CO, HNCO, experiments on a 800 MHz Varian (Varian). Spectra were transformed using NMRPipe and analyzed with the program Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, UCSF).
-HSQC, HNCA, HN(CO)CA, HN(CA)CO, HNCO, experiments on a 800 MHz Varian (Varian). Spectra were transformed using NMRPipe and analyzed with the program Sparky (T. D. Goddard and D. G. Kneller, SPARKY 3, UCSF).
Kinetic Measurements.
Refolding and unfolding kinetics were monitored at 25 °C with PiStar-180 and SX.18-MV stopped-flow fluorimeters (Applied Photophysics). Excitation was at 280 nm and emission was collected with a 320 nm long-pass filter. Protein concentration after mixing was 1 μM. Buffers were: 50 mM Mes (Sigma-Aldrich) at pH 6.3; 50 mM formate at pH 3.5–4.5 (Scharlau), and at pH ≤ 3.0, the concentration of HCl corresponding to the pH. Between pH 1.3 and pH 3.0, NaCl (VWR) was added to achieve a final ionic strength of 50 mM unless otherwise stated. Ultrapure guanidinium hydrochloride (AppliChem) and proanalysi Na2SO4 (Merck) were used in the denaturation experiments.
Two-state Assumption and Fitting of Chevron Data.
Following standard protocols (56), S6 was assumed to display two-state folding between the denatured (D) and native (N) states (27, 56, 57),
| [1] |
where KD-N is the equilibrium constant and kf and ku are the folding- and unfolding- rate constants, respectively, and linear free-energy relations were described by
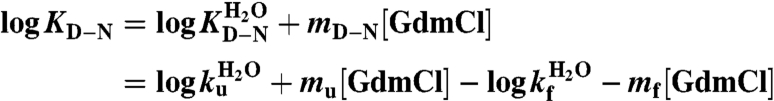 |
[2] |
where mD-N = mu-mf are the m-values, and 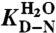 ,
, 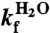 and
and 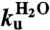 are the extrapolated values of KD-N, kf and ku at 0 M GdmCl. Chevron data was fitted to
are the extrapolated values of KD-N, kf and ku at 0 M GdmCl. Chevron data was fitted to
| [3] |
where kobs is the observed rate constant. Protein stability at 0 M GdmCl was calculated as
| [4] |
and the ϕ-values were derived from the standard equation
| [5] |
where 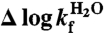 and
and 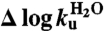 are the changes in rate constants upon mutation at 0 M GdmCl (56). Data analysis was done with Pro-Data Viewer (Applied Photophysics) and Kaleidagraph (Synergy Software).
are the changes in rate constants upon mutation at 0 M GdmCl (56). Data analysis was done with Pro-Data Viewer (Applied Photophysics) and Kaleidagraph (Synergy Software).
Supplementary Material
ACKNOWLEDGMENTS.
We thank Håkan Wennerström for stimulating discussions. Financial support was from the Swedish Research Council (VR 2009–5580), the Knut and Alice Wallenberg Foundation, the Bertil Hållsten Foundation, and Hjärnfonden.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1118640109/-/DCSupplemental.
References
- 1.Anderson TA, Cordes MH, Sauer RT. Sequence determinants of a conformational switch in a protein structure. Proc Natl Acad Sci USA. 2005;102:18344–18349. doi: 10.1073/pnas.0509349102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berezovsky IN, Zeldovich KB, Shakhnovich EI. Positive and negative design in stability and thermal adaptation of natural proteins. PLoS Comp Biol. 2007;3:e52. doi: 10.1371/journal.pcbi.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capaldi AP, Kleanthous C, Radford SE. Im7 folding mechanism: misfolding on a path to the native state. Nat Struct Biol. 2002;9:209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
- 4.Jin W, Kambara O, Sasakawa H, Tamura A, Takada S. De novo design of foldable proteins with smooth folding funnel: automated negative design and experimental verification. Structure. 2003;11:581–590. doi: 10.1016/s0969-2126(03)00075-3. [DOI] [PubMed] [Google Scholar]
- 5.Otzen DE, Oliveberg M. Salt-induced detour through compact regions of the protein folding landscape. Proc Natl Acad Sci USA. 1999;96:11746–11751. doi: 10.1073/pnas.96.21.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colletier JP, et al. Molecular basis for amyloid-{beta} polymorphism. Proc Natl Acad Sci USA. 2011;108:16938–16943. doi: 10.1073/pnas.1112600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jahn TR, Radford SE. Folding versus aggregation: polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sievers SA, et al. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thirumalai D, Klimov DK, Dima RI. Emerging ideas on the molecular basis of protein and peptide aggregation. Curr Opin Struct Biol. 2003;13:146–159. doi: 10.1016/s0959-440x(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 10.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 11.Klimov DK, Thirumalai D. Factors governing the foldability of proteins. Proteins. 1996;26:411–441. doi: 10.1002/(SICI)1097-0134(199612)26:4<411::AID-PROT4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 12.Oliveberg M, Wolynes PG. The experimental survey of protein-folding energy landscapes. Q Rev Biophys. 2005;38:245–288. doi: 10.1017/S0033583506004185. [DOI] [PubMed] [Google Scholar]
- 13.Sali A, Shakhnovich E, Karplus M. How does a protein fold? Nature. 1994;369:248–251. doi: 10.1038/369248a0. [DOI] [PubMed] [Google Scholar]
- 14.Lindahl M, et al. Crystal structure of the ribosomal protein S6 from Thermus thermophilus. EMBO J. 1994;13:1249–1254. doi: 10.2210/pdb1ris/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richardson JS, et al. Looking at proteins: Representations, folding, packing, and design. Biophysical Society National Lecture, 1992. Biophys J. 1992;63:1185–1209. [PMC free article] [PubMed] [Google Scholar]
- 16.Gitlin I, Carbeck JD, Whitesides GM. Why are proteins charged? Networks of charge-charge interactions in proteins measured by charge ladders and capillary electrophoresis. Angew Chemie Int Edit. 2006;45:3022–3060. doi: 10.1002/anie.200502530. [DOI] [PubMed] [Google Scholar]
- 17.Tanford C. Protein denaturation C. Theoretical models for the mechanism of denaturation. Adv Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 18.Doye JP, Louis AA, Vendruscolo M. Inhibition of protein crystallization by evolutionary negative design. Phys Biol. 2004;1:P9–13. doi: 10.1088/1478-3967/1/1/P02. [DOI] [PubMed] [Google Scholar]
- 19.Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: A new approach to understanding sickle cell disease. Proc Natl Acad Sci USA. 1974;71:4864–4868. doi: 10.1073/pnas.71.12.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otzen DE, Kristensen O, Oliveberg M. Designed protein tetramer zipped together with a hydrophobic Alzheimer homology: a structural clue to amyloid assembly. Proc Natl Acad Sci USA. 2000;97:9907–9912. doi: 10.1073/pnas.160086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richardson JS, Richardson DC. Natural beta -sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci USA. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 23.Ohman A, Oman T, Oliveberg M. Solution structures and backbone dynamics of the ribosomal protein S6 and its permutant P(54–55) Protein Sci. 2010;19:183–189. doi: 10.1002/pro.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haglund E, et al. The HD-exchange motions of ribosomal protein S6 are insensitive to reversal of the protein-folding pathway. Proc Natl Acad Sci USA. 2009;106:21619–21624. doi: 10.1073/pnas.0907665106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindberg MO, Oliveberg M. Malleability of protein folding pathways: A simple reason for complex behaviour. Curr Opin Struct Biol. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Haglund E, Lindberg MO, Oliveberg M. Changes of protein folding pathways by circular permutation. Overlapping nuclei promote global cooperativity. J Biol Chem. 2008;283:27904–27915. doi: 10.1074/jbc.M801776200. [DOI] [PubMed] [Google Scholar]
- 27.Otzen DE, Kristensen O, Proctor M, Oliveberg M. Structural changes in the transition state of protein folding: alternative interpretations of curved chevron plots. Biochemistry. 1999;38:6499–6511. doi: 10.1021/bi982819j. [DOI] [PubMed] [Google Scholar]
- 28.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 29.Lindberg MO, Haglund E, Hubner IA, Shakhnovich EI, Oliveberg M. Identification of the minimal protein-folding nucleus through loop-entropy perturbations. Proc Natl Acad Sci USA. 2006;103:4083–4088. doi: 10.1073/pnas.0508863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveberg M. Alternative explanations for multi-state kinetics in protein folding: transient aggregation and changing transition-state ensambles. Accounts Chem Res. 1998;31:765–772. [Google Scholar]
- 31.Camacho CJ, Thirumalai D. Denaturants can accelerate folding rates in a class of globular proteins. Protein Sci. 1996;5:1826–1832. doi: 10.1002/pro.5560050908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weinkam P, Pletneva EV, Gray HB, Winkler JR, Wolynes PG. Electrostatic effects on funneled landscapes and structural diversity in denatured protein ensembles. Proc Natl Acad Sci USA. 2009;106:1796–1801. doi: 10.1073/pnas.0813120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho SS, Weinkam P, Wolynes PG. Origins of barriers and barrierless folding in BBL. Proc Natl Acad Sci USA. 2008;105:118–123. doi: 10.1073/pnas.0709376104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang WY, Gruebele M. Folding at the speed limit. Nature. 2003;423:193–197. doi: 10.1038/nature01609. [DOI] [PubMed] [Google Scholar]
- 35.Loladze VV, Makhatadze GI. Removal of surface charge-charge interactions from ubiquitin leaves the protein folded and very stable. Protein Sci. 2002;11:174–177. doi: 10.1110/ps.29902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung MS, Garcia AE, Onuchic JN. Protein folding mediated by solvation: Water expulsion and formation of the hydrophobic core occur after the structural collapse. Proc Natl Acad Sci USA. 2002;99:685–690. doi: 10.1073/pnas.022387699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Escamilla AM, et al. Solvation in protein folding analysis: Combination of theoretical and experimental approaches. Proc Natl Acad Sci USA. 2004;101:2834–2839. doi: 10.1073/pnas.0304180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viguera AR, Vega C, Serrano L. Unspecific hydrophobic stabilization of folding transition states. Proc Natl Acad Sci USA. 2002;99:5349–5354. doi: 10.1073/pnas.072387799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berezovsky IN, Chen WW, Choi PJ, Shakhnovich EI. Entropic stabilization of proteins and its proteomic consequences. PLoS Comp Biol. 2005;1:e47. doi: 10.1371/journal.pcbi.0010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaenicke R, Bohm G. The stability of proteins in extreme environments. Curr Opin Struct Biol. 1998;8:738–748. doi: 10.1016/s0959-440x(98)80094-8. [DOI] [PubMed] [Google Scholar]
- 41.Sanchez-Ruiz JM, Makhatadze GI. To charge or not to charge? Trends Biotechnol. 2001;19:132–135. doi: 10.1016/s0167-7799(00)01548-1. [DOI] [PubMed] [Google Scholar]
- 42.Wang W, Hecht MH. Rationally designed mutations convert de novo amyloid-like fibrils into monomeric beta-sheet proteins. Proc Natl Acad Sci USA. 2002;99:2760–2765. doi: 10.1073/pnas.052706199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandelin E, Nordlund A, Andersen PM, Marklund SS, Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J Biol Chem. 2007;282:21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
- 44.Oliveberg M, Fersht AR. New approach to the study of transient protein conformations: The formation of a semiburied salt link in the folding pathway of barnase. Biochemistry. 1996;35:6795–6805. doi: 10.1021/bi9529317. [DOI] [PubMed] [Google Scholar]
- 45.Waldburger CD, Schildbach JF, Sauer RT. Are buried salt bridges important for protein stability and conformational specificity? Nat Struct Biol. 1995;2:122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- 46.Baud F, Karlin S. Measures of residue density in protein structures. Proc Natl Acad Sci USA. 1999;96:12494–12499. doi: 10.1073/pnas.96.22.12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones S, Thornton JM. Principles of protein-protein interactions. Proc Natl Acad Sci USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 49.Schreiber G, Buckle AM, Fersht AR. Stability and function: Two constraints in the evolution of barstar and other proteins. Structure. 1994;2:945–951. doi: 10.1016/s0969-2126(94)00096-4. [DOI] [PubMed] [Google Scholar]
- 50.Schreiber G, Fersht AR. Rapid, electrostatically assisted association of proteins. Nat Struct Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 51.Eriksson SK, Kutzer M, Procek J, Grobner G, Harryson P. Tunable membrane binding of the intrinsically disordered dehydrin lti30, a cold-induced plant stress protein. Plant Cell. 2011;23:2391–2404. doi: 10.1105/tpc.111.085183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linse S, et al. The role of protein surface charges in ion binding. Nature. 1988;335:651–652. doi: 10.1038/335651a0. [DOI] [PubMed] [Google Scholar]
- 53.Leinartaite L, Saraboji K, Nordlund A, Logan DT, Oliveberg M. Folding catalysis by transient coordination of Zn2+ to the Cu ligands of the ALS-associated enzyme Cu/Zn superoxide dismutase 1. J Am Chem Soc. 2010;132:13495–13504. doi: 10.1021/ja1057136. [DOI] [PubMed] [Google Scholar]
- 54.Parge HE, Getzoff ED, Scandella CS, Hallewell RA, Tainer JA. Crystallographic characterization of recombinant human CuZn superoxide dismutase. J Biol Chem. 1986;261:16215–16218. [PubMed] [Google Scholar]
- 55.Danielsson J, Kurnik M, Lang L, Oliveberg M. Cutting off functional loops from homodimeric enzyme superoxide dismutase 1 (SOD1) leaves monomeric beta-barrels. J Biol Chem. 2011;286:33070–33083. doi: 10.1074/jbc.M111.251223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fersht AR. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: WH Freeman and Co; 1999. [Google Scholar]
- 57.Tanford C. Protein denaturation. Adv Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.