

Abstract
Cathepsin L mutants with the ability to condense silica from solution have been generated and a 1.5 Å crystal structure of one of these chimeras allows us to rationalize the catalytic mechanism of silicic acid condensation.
Silica materials are of interest in biotechnology and drug delivery1 but controllable silicate formation is very difficult. Yet a variety of sponge species make highly ordered specific glass structures called spicules2-5. Harnessing the processes that underlie this biosynthesis has considerable application. The enzyme silicatein α forms part of the organic filament found in spicules which in situ condenses silicate (Figure 1a), although the exact form of the natural substrate is not known5. Both wild type and recombinant silicatein α have been shown to catalyse the condensation of siloxanes such as tetraethoxysilane4, 6 in solution and at surfaces7. Silicatein α has also been shown to have the ability to cause the deposition of other compounds at surfaces, including titanium phosphate, titanium oxide, zirconium oxide and l-lactide8, 9.
Fig 1.
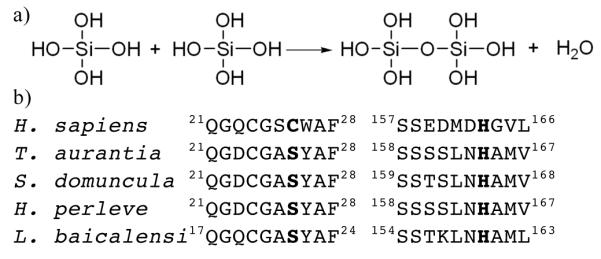
(a) The chemical reaction catalyzed by the organic filament of the sponge spicule. (b) Conservation of residues flanking catalytic serine and histidine in silicatein-α from various sponge species. Shown is the amino acid sequence around the catalytic serine/cysteine and histidine in human cathepsin L (UniProtKB/TrEMBL entry P07711) and silicatein α from: Tethya aurantia (O76238); Suberites domuncula (Q2MEV3); Hymeniacidon perleve (Q2HYF6); Lubomirskia baicalensi (Q2PC18).
Above 100 ppm, condensation of silicic acid occurs spontaneously, presumably as the concentration of nucleophilic ionised molecules is high enough. There are two possible mechanisms for enzyme catalysis i) stabilise at the active site one molecule of deprotonated silicic acid (the nucleophile) which will then react with another molecule of silicic acid or ii) stabilise a protonated silicic acid (the electrophile) which will then react with another molecule of silicic acid. Neither the wild type nor recombinant silicatein α are amenable to biophysical study due to low levels of protein expression and inclusion body formation when recombinantly expressed in E. coli 4, 7, 8.
Sequence comparison identifies a significant degree of similarity between silicatein α and the human cysteine protease cathepsin L4 with an overall 52 % identity and 65 % similarity. The most notable differences between the two sequences are the presence of a loop of 4 amino acids in cathepsin L10 that is absent in silicatein α, a large number of hydroxyl containing residues (serines, threonines and tyrosines) in silicatein α that are not present in cathepsin L and the substitution of the catalytic cysteine (C25) in cathepsin L11 for serine (S25) in silicatein α. This serine residue has been shown by mutagenesis to be essential for the function of silicatein α12. The other residues in the catalytic triad of cathepsin L (H163 and N187) are similarly conserved in silicatein α. A mechanism has been proposed for polymerisation of Si(OEt)4 in which a covalent protein silicate intermediate is formed, this active electrophile is then decomposed by water yielding a more active nucleophile12. Whether this mechanism operates in vivo is unresolved. Interestingly, there is also a change in the flanking residues either side of the catalytic serine and histidine and this difference is conserved across several different silicateins, (Figure 1b).
We have made a series of cathepsin L mutants that increasingly match the sequence features that are unique to silicatein α (Table 1). Mutants were made using a combination of the Quikchange™ mutagenesis technique and conventional PCR. The mutant proteins were then recombinantly expressed as the pro-protein and purified from Pichia pastoris as described previously13. Mature cathepsin-L/silicatein α chimeras were then obtained via published procedures14.
| Cathepsin L Constructa |
Mutations | Match to silicatein α |
|---|---|---|
| C2S | C25S | catalytic serine |
| AS2 | S24A W26Y | Residues flanking catalytic serine |
| AS2H | M161L, D162N, G164A, V165M |
Residues flanking catalytic histidine |
| LOOP |
173ESTESDNN180 to ISNNQ |
To replicate loop |
| 2SER | E159S, D160S | match serines |
| 4SER | E153S, P154S | match serines |
the mutations are additive from top to bottom, 5 thus 4SER has all the preceding mutations.
The various mutant proteins were assayed for silica condensation activity (detected by precipitation of silicate) using water glass as a substrate, (Figure 1a). Mutation of the catalytic cysteine to serine (the C25S mutant) did not confer significant levels of condensing activity. The C25S mutant would have been expected to be sufficient from the predicted mechanism of silicatein α 6. In order to obtain significant condensing activity, it was found to be necessary to mutate the residues either side of the catalytic S25 residue (Table 1, Figure 2) before any significant activity was observed. Additional mutations to cathepsin L to further increase its resemblance to silicatein α did not significantly increase the ability to condense silica; in fact many of these additional mutations tended to decrease condensation activity. It is likely therefore that these residues play other functions in the sponge spicule such as in the association of silicatein α with silicatein β and galectin15, 16, or in the templating of silica into specific morphologies, rather than underlie catalysis per se. While the various active mutant proteins were found to readily precipitate silica from the natural sodium silicate substrate we could not detect any precipitate from tetraethoxysilane substrate, which can be utilised by silicatein-α4, 6.
Figure 2.
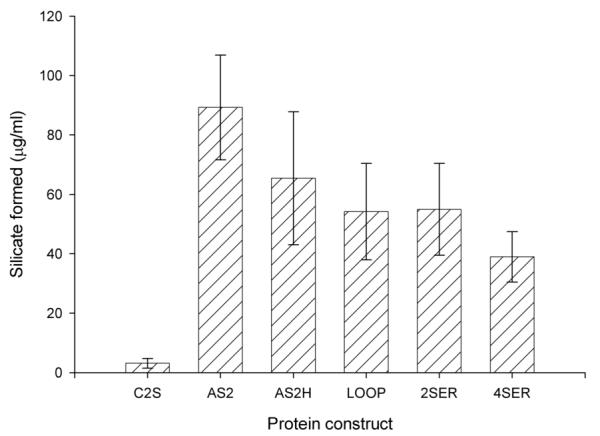
Silica condensing activity of mutant proteins. To 0.1 mg / mL of protein solution in 0.1 M sodium phosphate buffer pH 7 was added 4.5 mM sodium silicate. Samples were incubated at room temperature for 24 hours precipitated silica was collected by centrifugation and then assayed using the Merck spectroquant silicon assay kit as described17. Shown is the average value of 5 separate incubations with the error as shown.
In an effort to understand the catalytic basis of silica condensation we have solved the X-ray crystal structure of the 4SER mutant (Figure 3a). Crystals of the 4SER mutant were obtained by hanging drop vapour diffusions. The structure was determined to 1.5 Å using cathepsin L (PDB 1MHW) as a model for molecular replacement19, 20. Unsurprisingly the 4SER chimera is identical to cathepsin L with a RMSD of only 0.5 Å between the two structures. Full experimental details are given in supporting material.
Figure 3.

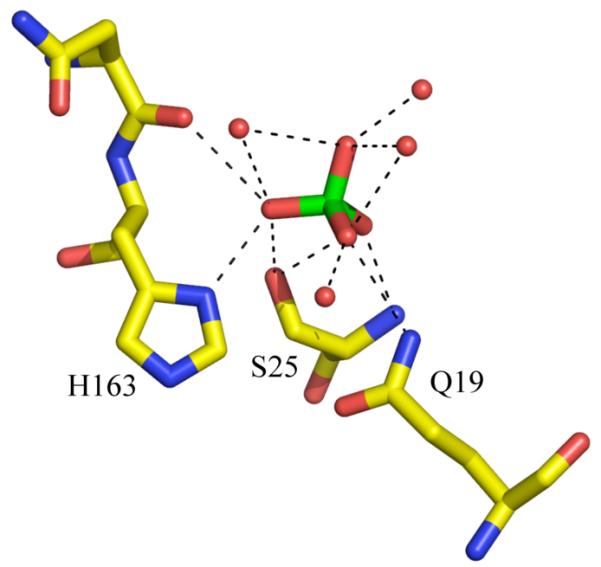
(a) The structure 4SER chimera shown in cartoon. The sulphate at the active site is shown as space filling spheres. The two “catalytic” residues H163 and S25 are shown in sticks. Carbon atoms are coloured yellow, oxygen red, sulphur green and nitrogen blue. (b) Close up view active site of the 4SER chimera showing the close contacts (< 3.5 Å) as dashed lines. The sulphate molecule is shown in sticks, the colour scheme remains as part (a). The extensive interactions mediated by water would allow extensive proton shuttling. Figures produced with PYMOL18.
Figure 3b shows the active site architecture of the 4SER mutant with that of the C25S pro-cathepsin L crystal structure21 (PDB 1CJL). The C25S mutant of cathepsin is itself inactive as a protease indicating the nucleophilicity of the OG atom of S25 is low. In 4SER the distance between the OG atom on S25 and ND1 on H163 has increased to 3.6 Å (beyond hydrogen bonding distance), further decreasing the nucleophilicity of S25. Mutation of the flanking residues perturbs a cluster of residues that sit behind S25 (Figure 2). The mutations remove a hydrogen bond (S24A) and decrease van der Waal interaction (W26Y). The mutations are both to smaller residues than the native enzyme. The net result would be to allow the S25 to move away from the H163 and to enlarge the volume at the active site.
An understanding of the mechanism of silica condensation by the chimera is aided by the presence a sulphate ion at the active site. (A second sulphate is at a crystal contact 17 Å distant from S25). The crystal structure reveals it would be difficult to fit the bulky Si(OEt)4 at the active site and without fairly significant conformational adjustment. This would seem to explain the lack of activity of the chimera against Si(OEt)4 and would suggest that silicatein α must have a larger active site than 4SER which would allow it to accommodate the bulky Si(OEt)4. We believe the tetrahedral sulphate ion at the active site is a good mimic for the tetrahedral silicic acid. The sulphate is hydrogen bonded to S25. This is consistent with S25 making a nucleophilic attack on Si(OH)4 to generate a covalent enzyme intermediate. However, such a high energy intermediate would seem chemically unlikely and prone to immediate hydrolysis back to Si(OH)4.
The sulphate is hydrogen bonded to H163, the residue that activates the protein nucleophile in proteases. We favour a mechanism in which H163 binds and stabilises the deprotonated form of Si(OH)4 at the active site. The extensive network of water molecules and hydrogen bonds (Figure 3b) would permit significant proton shuttling, such that the negative charge may residue on different oxygen. This deprotonated enzyme bound species is a sufficient nucleophile that it will attack Si(OH)4 solution species initiating polymerisation (Figure 4). Loss of a Si(OH)4 hydroxyl group carried out by protonation and elimination of water could also promote the reaction but we have no evidence for an acid. It is also unlikely that at pH 7 Si(OH)4 will be a sufficient base to first deprotonate H163 (whose pKa is likely to be lower than 7 due to the presence of D187) and then for the hydroxyl group of the second Si(OH)4 to perform a nucleophilic attack.
Figure 4.
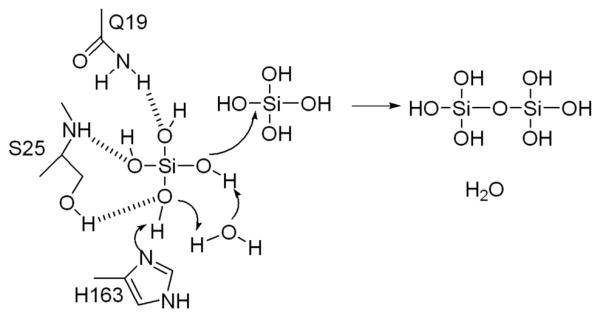
The chemical mechanism proposed to operate for the polymerisation of silicic acid. We propose H163 deprotonates Si(OH)4, the extensive water network could permit proton shuttling. We have illustrated one possibility.
In our proposed mechanism, the role of C25S and flanking mutations are simply to create a sufficiently sized pocket that will allow recognition of the tetrahedral Si(OH)4 molecule in such a way that H163 can deprotonate it. The deprotonated Si(OH)4 protein complex can be thought of as template for condensation reaction. In this proposal there is no need to involve a high energy covalent intermediate. The presence of a specific Si(OH)4 transporter in sponge22 suggests the true substrate in vivo is indeed silicic acid not high energy silicon alkoxides. This being so, the simple acid base activation mechanism we propose seems a good model for the biological process.
Supplementary Material
Electronic Supplementary Information (ESI) available: Molecular biology, protein purification and crystallographic details
Acknowledgement
This work was supported by the Leverhulme Trust, grant F/00 273/H. The Scottish Structural Proteomics Facility is funded by the Scottish Funding Council and Biotechnology and Biological Sciences Research Council.
Footnotes
The final crystal structure and experimental have been deposited with the Protein Data Bank, entry 2VHS.
Notes and references
- 1.Giri S, Trewyn BG, Lin VS. Nanomed. 2007;2:99–111. doi: 10.2217/17435889.2.1.99. [DOI] [PubMed] [Google Scholar]
- 2.Muller WE, Belikov SI, Tremel W, Perry CC, Gieskes WW, Boreiko A, Schroder HC. Micron. 2006;37:107–120. doi: 10.1016/j.micron.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Schroder HC, Boreiko O, Krasko A, Reiber A, Schwertner H, Muller WE. J Biomed Mater Res B Appl Biomater. 2005;75:387–392. doi: 10.1002/jbm.b.30322. [DOI] [PubMed] [Google Scholar]
- 4.Shimizu K, Cha J, Stucky GD, Morse DE. Proc Natl Acad Sci U S A. 1998;95:6234–6238. doi: 10.1073/pnas.95.11.6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver JC, Morse DE. Microsc Res Tech. 2003;62:356–367. doi: 10.1002/jemt.10401. [DOI] [PubMed] [Google Scholar]
- 6.Cha JN, Shimizu K, Zhou Y, Christiansen SC, Chmelka BF, Stucky GD, Morse DE. Proc Natl Acad Sci U S A. 1999;96:361–365. doi: 10.1073/pnas.96.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahir MN, Theato P, Muller WE, Schroder HC, Janshoff A, Zhang J, Huth J, Tremel W. Chem Commun (Camb) 2004:2848–2849. doi: 10.1039/b410283e. [DOI] [PubMed] [Google Scholar]
- 8.Curnow P, Kisailus D, Morse DE. Angew Chem Int Ed Engl. 2006;45:613–616. doi: 10.1002/anie.200502738. [DOI] [PubMed] [Google Scholar]
- 9.Tahir MN, Theato P, Muller WE, Schroder HC, Borejko A, Faiss S, Janshoff A, Huth J, Tremel W. Chem Commun (Camb) 2005:5533–5535. doi: 10.1039/b510113a. [DOI] [PubMed] [Google Scholar]
- 10.Smith SM, Gottesman MM. J Biol Chem. 1989;264:20487–20495. [PubMed] [Google Scholar]
- 11.Mason RW, Green GD, Barrett AJ. Biochem J. 1985;226:233–241. doi: 10.1042/bj2260233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, Shimizu K, Cha JN, Stucky GD, Morse DE. Angewandte Chemie-International Edition. 1999;38:780–782. doi: 10.1002/(SICI)1521-3773(19990315)38:6<779::AID-ANIE779>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 13.Fairhead M, van der Walle CF. Prot. Pept. Lett. 2008 doi: 10.2174/092986608783330468. In Press. [DOI] [PubMed] [Google Scholar]
- 14.Menard R, Carmona E, Takebe S, Dufour E, Plouffe C, Mason P, Mort JS. J Biol Chem. 1998;273:4478–4484. doi: 10.1074/jbc.273.8.4478. [DOI] [PubMed] [Google Scholar]
- 15.Curnow P, Bessette PH, Kisailus D, Murr MM, Daugherty PS, Morse DE. J Am Chem Soc. 2005;127:15749–15755. doi: 10.1021/ja054307f. [DOI] [PubMed] [Google Scholar]
- 16.Schroder HC, Boreiko A, Korzhev M, Tahir MN, Tremel W, Eckert C, Ushijima H, Muller IM, Muller WE. J Biol Chem. 2006;281:12001–12009. doi: 10.1074/jbc.M512677200. [DOI] [PubMed] [Google Scholar]
- 17.Muller WE, Krasko A, Le Pennec G, Schroder HC. Microsc Res Tech. 2003;62:368–377. doi: 10.1002/jemt.10402. [DOI] [PubMed] [Google Scholar]
- 18.DeLano WL. DeLano Scientific. San Carlos; CA, USA: 2002. [Google Scholar]
- 19.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Acta Cryst. D. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 20.Storoni LC, McCoy AJ, Read RJ. Acta Cryst. D. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 21.Coulombe R, Grochulski P, Sivaraman J, Menard R, Mort JS, Cygler M. Embo J. 1996;15:5492–5503. [PMC free article] [PubMed] [Google Scholar]
- 22.Schroder HC, Perovic-Ottstadt S, Rothenberger M, Wiens M, Schwertner H, Batel R, Korzhev M, Muller IM, Muller WEG. Biochem. J. 2004;381:665–673. doi: 10.1042/BJ20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.