

Abstract
A series of selectively fluorinated and other substituted UDP-d-galactose derivatives have been evaluated as substrates for Klebsiella pneumoniae UDP-d-galactopyranose mutase. This enzyme, which catalyses the interconversion of the pyranose and furanose forms of galactose as its UDP adduct, is a prospective drug target for a variety of microbial infections. We show that none of the 2″-, 3″- or 6″-hydroxyl groups of UDP-d-galactopyranose are essential for substrate binding and turnover. However, steric factors appear to play an important role in limiting the range of substitutions that can be accommodated at C-2″ and C-6″ of the sugar nucleotide substrate. Attempts to invert the C-2″ stereochemistry from equatorial to axial, changing d-galacto- to d-talo-configuration, in an attempt to exploit the higher percentage of furanose at equilibrium in the talo-series, met with no turnover of substrate.
Keywords: Fluorosugar nucleotides, UDP-d-galactopyranose mutase, Mechanism, Equilibrium
Introduction
The rise in the incidence of tuberculosis worldwide needs urgent attention. Resistance to current drugs is a major issue and very few new candidates are currently in clinical trials.1 Considerable effort is being made to identify new actives for development,2,3 but, given the issues of drug resistance, novel drug targets for the treatment of tuberculosis are also being sought.4 Given the precedent for effective drugs acting on mycobacterial cell wall biosynthesis,5,6 we were attracted by the notion of targeting cell wall biosynthetic enzymes that have not been the subject of drug development programmes to date. In particular, the central role played by d-galactofuranose (Galf) in cell wall integrity,5 and the absence of this 5-membered ring form of d-galactose in man, prompted us to consider d-galactofuranose biochemistry.
Originally confirmed in Escherichia coli,7 the key enzyme for the incorporation of galactofuranose into cell surface glycan is UDP-d-galactopyranose mutase, a novel flavoprotein that is capable of reversibly interconverting UDP-d-galactopyranose (UDP-d-Galp) and UDP-d-galactofuranose (UDP-d-Galf). The latter in turn serves as a donor substrate for a galactofuranosyltransferase-mediated incorporation of Galf into growing oligosaccharide chains.8 An orthologous UDP-d-galactopyranose mutase was subsequently identified in Klebsiella pneumoniae9 and in mycobacterial species.10 More recent studies have shown that the UDP-d-galactopyranose mutase-encoding glf gene also occurs in a wide variety of eukaryotic microbial and metazoal pathogens, including the human parasites Leishmania major and Trypanosoma cruzi, and the fungus Cryptococcus neoformans, among others.11 Hence, UDP-d-galactopyranose mutase may represent a potential drug target for a number of human microbial pathogens.
UDP-d-galactopyranose mutase has both a novel structure12 and a unique catalytic mechanism (Scheme 1).13 It has been proposed that the reduced form of flavin co-factor14 attacks the anomeric position of the UDP-d-galactose substrate displacing UDP, which is supported by positional isotope exchange experiments.15 The flavin is then able to drive cleavage of the O5-C1 bond resulting in opening of the sugar ring. Subsequent attack of O4 on C1 achieves formation of a furanose intermediate, which is intercepted by UDP with concomitant release of the flavin (Scheme 1). Other studies have shown that 1,4-anhydrogalactopyranose is not an intermediate16 in the mutase-catalysed reaction, but there is support for oxocarbenium ion character to the reaction mechanism.17 STD-NMR spectroscopy18,19 and site-directed mutagenesis studies20 have been employed in attempts to define the mode of substrate binding and to identify key substrate-binding residues. Numerous attempts have been made to develop inhibitors of UDP-d-galactopyranose mutase,21 with recent success leading not only to in vitro enzyme inhibition but also inhibition of the growth of Mycobacterium smegmatis (a non-pathogenic, fast-growing relation of M. tuberculosis that is often used for initial whole organism screening).22
Scheme 1.

Proposed role of flavin in the mechanism of action of UDP-d-galactopyranose mutase
Despite the wealth of information that is now available about UDP-d-galactopyranose mutase, we still lack definitive information about the molecular basis of substrate binding and specificity. A better understanding of the substrate tolerance of this enzyme could be exploited for the generation of potential inhibitors of therapeutic value. Building on our interest in the use of synthetic and unnatural sugar nucleotides to assess enzyme specificity,23 herein we report the generation of sugar nucleotides to probe substrate specificity of UDP-d-galactopyranose mutase. We set out to extend the observations of Barlow and Blanchard24 on the effect of turnover and equilibrium position of substituting fluorine for either the 2- or 3-OH groups on UDP-d-galactopyranose (similar studies in the furanose series have been reported by Zhang and Liu25). Specifically, we report investigations of 2- or 6-substitution of the galactopyranose unit of UDP-d-Galp on the kinetics of mutase action and the equilibrium position of the enzyme-catalysed reaction. These studies have relied on fluorosugar nucleotides, in particular, given that the C-F bond has interesting properties26,27 that have attracted considerable attention from the pharmaceutical sector.28
Results and discussion
Preparation of sugar nucleotides
The enzymatic synthesis of uridine 5′-diphospho-α-d-galactopyranose (UDP-Gal) using galactokinase (GalK; E.C. 2.7.1.6) and galactose-1-phosphate uridylyltransferase (GalPUT; E.C. 2.7.7.12) was first reported by Whitesides and coworkers.29 Subsequent use of these enzymes in recombinant form by Wang and coworkers enabled straightforward immobilisation of these catalysts.30 Adopting the Wang approach, we demonstrated the utility of GalK and GalPUT, acting together and GalPUT alone, for effecting transformation of a range of natural and unnatural reducing sugars and sugar-1-phosphates, respectively, to their UDP-sugar adducts.31 In particular, the catholic nature of GalPUT enables its use to prepare UDP-d-galactofuranose from synthetic α-d-galactofuranose-1-phosphate.32I In the current study, a range of sugar nucleotides (vide infra) were prepared using the GalK/GalPUT procedure, as described previously.31 In the case of UDP-6-deoxy-6-fluoro-d-galactose, both commercial 6-deoxy-6-fluoro-d-galactose and racemic 6-deoxy-6-fluoro-galactose from de novo synthesis34 were efficiently processed to give UDP-6-deoxy-6-fluoro-d-galactose.
Evaluation of sugar nucleotides as substrates for UDP-galactopyranose mutase
Fluorinated UDP-d-galactose derivatives
Using selectively 2″- and 3″-fluorinated substrates, the work of Barlow and Blanchard24 clearly shows that the UDP-d-galactopyranose mutase-catalysed reaction reaches an equilibrium position that reflects that of the corresponding reducing sugar. In addition, fluorine impacts on the rate of reaction in a manner that is dependent on its position with respect to the anomeric centre, as one would expect for a reaction mechanism that possesses carbenium ion character. We have extended this study to the corresponding 6″-fluorinated substrate (for consistency, we also report data produced in parallel for the 2″- and 3″-fluorinated substrates) (Table 1). Overall, these studies show that none of the 2″-, 3″- or 6″-hydroxyl groups of UDP-d-Galp are essential for substrate binding and turnover. As with the Barlow and Blanchard work, as one ‘walks’ the fluorine atom around the galactose ring the rate of mutase-catalysed reaction is affected: a drop of over 50,000 fold for the 2″-fluoro compound, nearly 400 fold for the 3″-fluoro compound and only a 10 fold drop for the 6″-fluoro compound. The equilibrium position reached for the 6″-fluoro compound reflects that of the corresponding reducing sugar, as previously reported for the 2″- and 3″-fluorinated substrates.24
Table 1.
Kinetic parameters for turnover of fluorinated galacto-nucleotides by Klebsiella pneumoniae UDP-d-galactopyranose mutase.a
| Compound* |
Km μM |
kcat min−1 |
Relative kcat(UDP-Galp)/kcat |
Mutase reaction: furanose observed % |
Reducing sugar: furanose observed % |
|---|---|---|---|---|---|
| UDP-d-Galactopyranose24,31 | 805 | 2120 | 1 | 8.1 | 7.724 |
| UDP-d-Galactofuranose31 | 16 | 2210 | 0.96 | 8.1 | 7.724 |
| UDP-2-Deoxy-2-fluoro-d-galactopyranose21a,24,31 | 203 | 0.04 | 53100 | 2.3 | 3.224 |
| UDP-3-deoxy-3-fluoro-d-galactopyranose24,31 | 280 | 5.5 | 386 | 1.9 | 2.324 |
| UDP-6-deoxy-6-fluoro-d-galactopyranose24,31 | 200 | 211 | 10.1 | 12.5 | 13.035 |
The error in the data is typically 5-10%, or less
For UDP-4-deoxy-4-fluoro-d-galactopyranose see ref 21a
2″-Modified UDP-d-galactose derivatives
Clearly 2″-fluorination of UDP-d-Galp has a profound impact on substrate turnover. We therefore looked further at 2″-substituted substrate analogues, focussing on the azide, amine and acetamide series (Table 2). The 2″-azido and 2″-acetamido compounds were not mutase substrates in our hands, whereas the 2″-amino compound proved to be a weak substrate (slightly increased Km; kcat reduced ~14 fold). It is conceivable that the lack of observed turnover for the azide and acetamide is due to a complete lack of furanose at equilibrium for these sugars. The older literature36 suggests that galactosamine and N-acetyl galactosamine exist exclusively in the pyranose form at equilibrium in aqueous solution (although this probably reflects the accuracy of available techniques at the time). However, this is not the case, with around 5% furanose being measurable in both cases by 600 MHz 1H NMR spectroscopy (Table 2). We therefore conclude that steric constraints around the 2″-position of the substrate impact on recognition.
Table 2.
Kinetic parameters for turnover of 2″-substituted d-galacto-nucleotides by Klebsiella pneumoniae UDP-d-galactopyranose mutase.a
| Compound |
Km μM |
kcat min−1 |
Relative kcat(UDP-Galp)/kcat |
Mutase reaction: furanose observed % |
Reducing sugar:36 furanose observed % |
|---|---|---|---|---|---|
| UDP-d-Galactopyranose24,31 | 805 | 2120 | 1 | 8.1 | 7.7 |
| UDP-2-Deoxy-2-fluoro-d-galactopyranose24,31 | 203 | 0.04 | 53100 | 2.3 | 3.2 |
| UDP-2-Azido-2-deoxy-d-galactopyranose31 | - | - | - | ND | 1.2* |
| UDP-2-Amino-2-deoxy-d-galactopyranose31 | 1140 | 83 | 13.8 | 4.8 | 5.0* |
| UDP-2-Acetamido-2-deoxy-d-galactopyranose31 | - | - | - | ND | 5.1* |
The error in the data is typically 5-10%, or less
ND - not detectable
This study
6″-Variants of UDP-d-galactose derivatives
6″-Modified substrates for the mutase also produced intriguing results. Replacement of -OH by -F is permissible (the 6″-OH group is not essential for substrate binding), but -OH to -H (giving d-fucose) apparently renders the molecule a non-substrate (Table 3).II In contrast, whilst removal of the C-6 hydroxymethyl group altogether (giving l-arabinose) increases Km ~4 fold, kcat is also increased slightly (Table 3).III These data suggest a need for hydrogen bonding at the 6″-position. Removal of the hydroxymethyl group altogether, giving l-arabinose, results in an active substrate. It is plausible that the loss of the bulky hydroxymethyl group could allow space for the inclusion of a water molecule, providing potential for hydrogen bonding. In contrast, steric restrictions would disallow hydrogen bonding to occur on going from galactose to the 6″-deoxy compound.
Table 3.
Kinetic parameters for turnover of 6″-variants of d-galacto-nucleotides by Klebsiella pneumoniae UDP-galactopyranose mutase.a
| Compound | Km μM | kcat min−1 | Relative kcat(UDP-Galp)/kcat |
Mutase reaction: furanose observed % |
Reducing sugar:36 furanose observed % |
|---|---|---|---|---|---|
| UDP-d-Galactopyranose24,31 | 805 | 2120 | 1 | 8.1 | 7.7 |
| UDP-6-Deoxy-6-fluoro-d-galactopyranose31 | 200 | 211 | 10.1 | 12.5 | 13.0 |
| UDP-6-Deoxy-d-galactopyranose (d-Fuc)31 | - | - | - | ND | 3.7* |
| UDP-l-Arabinopyranose31 | 3150 | 2530 | 0.84 | 6.4 | 4.5 |
The error in the data is typically 5-10%, or less
ND - not detectable
This study
From galactose to talose
UDP-d-galactopyranose mutase is an awkward enzyme to assay. In the forward direction, one only has an 8% conversion (to furanose) to play with. Despite the accessibility of UDP-d-Galf31 to assay the reverse reaction (to pyranose), the 1,2-cis arrangement of UDP and the 2-hydroxyl on the furanose ring renders UDP-d-Galf prone to degradation via 1,2-cyclic phosphate intermediates. Hence a more robust pyranose-based substrate that on exposure to UDP-d-galactopyranose mutase gives a substantial proportion of the UDP-d-Galf equivalent would be highly desirable. Given the need to leave the pyranose C-4 axial in order for the mutase to act, d-talose attracted our attention. d-Talose was of interest to us due to the fact that a substantial amount (29%) of the reducing sugar is in the furanose form at equilibrium (Table 4). We were therefore motivated to prepare and evaluate UDP-d-talopyranose. In addition, considering the potential electronic demands that might be made by placing the 2-hydroxyl group axial, we also considered the corresponding fluoro compound,26 UDP-2-deoxy-2-fluoro-d-talopyranose, where the corresponding reducing sugar shows 13% furanose at equilibrium39 (Table 4).
Table 4.
Reducing sugar anomeric composition at equilibrium in aqueous solution
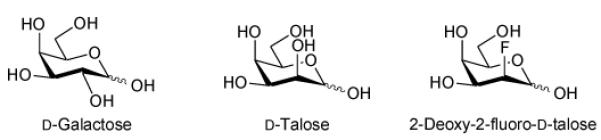
| Compound | α-pyranose | β-pyranose | α-furanose | β-furanose | Reducing sugar: furanose observed %36 |
Mutase reaction: furanose observed % |
|---|---|---|---|---|---|---|
| d-Galactose | 31.8 | 60.5 | 3.1 | 4.6 | 7.7 | 8.1 |
| d-Talose | 42.0 | 29.0 | 16.0 | 13.0 | 29.0 | 0* |
| 2-Deoxy-2-fluoro-d-talose | 50.0 | 37.0 | 8.0 | 5.0 | 13.039 | 0* |
this study
Chemoenzymatic synthesis of UDP-2-deoxy-2-fluoro-d-talose
The literature synthesis of 2-deoxy-2-[18F]fluoro-d-talose from 1,2-O-isopropylidene-α-d-galactofuranose40 employing nucleophilic fluorination (K18F) proceeded in 7 steps and 24% overall yield.39 We were motivated to develop a shorter synthesis of 2-deoxy-2-fluoro-d-talose (4) that avoided using a galactofuranose system since, beyond the parent compound which is accessible from commercial galactono-1,5-lactone,31 such derivatives can be complicated to prepare. In our hands, attempts to introduce the fluorine by an SN2 reaction using TBAF or DAST on an α-d-galactopyranose substrate were unsuccessful, with either no reaction occurring, or the formation of elimination products. We previously experienced similar issues when attempting to synthesise 3-deoxy-3-thio-galactopyranose derivatives.41 This prompted us to explore a strategy involving electrophilic fluorination of glycals using Selectfluor™, which has been extensively studied.42,43 Treatment of protected d-galactal with Selectfluor™ gives solely the d-galacto-configured product42 whilst d-glucal gives a mixture of the d-manno- and d-gluco-isomers.44,45 Accordingly, our approach involved the preparation of a 2-deoxy-2-fluoro-d-mannose derivative from appropriately protected d-glucal and subsequent C-4 inversion to give the desired d-talo-configured compound. The previously reported 1-O-acetyl-3,6-di-O-benzyl-2-deoxy-2-fluoro-α,β-d-mannopyranose (1)46, which was prepared from 3,6-di-O-benzyl-d-glucal, was converted to the C-4 triflate ester followed by inversion of stereochemistry using potassium nitrite in DMF47 to give fluorotalose derivative (2) (Scheme 1). Of the reaction conditions investigated, the optimum temperature proved to be 50 °C where complete conversion was achieved after 4 days: at room temperature no reaction occurred, while at 80 °C substrate decomposition was observed. In the 1H NMR spectrum of the product, coupling between both H-3 and H-4 (J3,4 3.0 Hz) and between H-4 and H-5 (J4,5 <1 Hz) were small, as expected for the C-4 axial compound (2). The benzyl protecting groups were removed by catalytic hydrogenation with palladium on carbon. Attempted sodium hydroxide-mediated de-O-acetylation of (3) resulted in decomposition, presumably due to elimination of HF. However, de-O-acetylation was successfully achieved under acidic conditions using Dowex 50WX8 (H) resin in water at 80 °C. The 1H NMR spectrum of the product was consistent with that reported previously for 2-deoxy-2-fluoro-d-talose (4).39 Thus, our approach yielded 2-deoxy-2-fluoro-d-talose (4) in 4 steps and in 27% overall yield from (1).
Both 2-deoxy-2-fluoro-d-talose (4) and commercially available d-talose were tested in parallel as substrates for the GalK/GalPUT system.31 As can be seen in Table 5, in the conversion to the corresponding UDP adduct 2-deoxy-2-fluoro-d-talose was a good substrate for the GalK/GalPUT system – comparable to the natural substrate galactose and much better than talose. This might be attributable to the presence of a bulky C-2 axial substituent (hydroxyl) in talose, compared with the slightly smaller C-2 axial substituent (i.e. fluorine) in 2-deoxy-2-fluoro-talose, although replacement of O by F is a relatively conservative change in terms of both electronegativity and size.26
Table 5.
One-pot enzymatic synthesis catalysed by GalK and GalPUT.31
| Substrate | Product | Yield (%)a |
|---|---|---|
| d-Galactose | UDP-d-Galp | 78 |
| d-Talose | UDP-d-Talp | 15 |
| 2-Deoxy-2-fluoro-d-talose | UDP-2F-d-Talp | 71 |
After 24 h incubation
Unfortunately, despite repeated attempts, we were unable to demonstrate by HPLC the turnover of either UDP-d-talose or UDP-2-deoxy-2-fluoro-d-talose by UDP-d-galactopyranose mutase. At this stage it is unclear whether this is due to steric constraints associated with recognition of the 2″- position of the sugar nucleotide (vide supra) or if the C-OH/C-F dipole impacts on attack on the substrate by the reduced flavin.
Conclusions
With the aid of a range of sugar nucleotide analogues we have demonstrated that none of the 2″-, 3″- or 6″-hydroxyl groups of UDP-d-Galp are essential for substrate binding and turnover by UDP-d-galactopyranose mutase. However, there are clear constraints regarding permissible substitutions at C-6″ of UDP-Galp. At C-2″, whether in the equatorial d-galacto- or axial d-talo-configuration, again steric constraints come into play. As with correlating UDP-d-galactopyranose mutase and co-factor structure with mechanism of action, the precise detail of how and why substrate substitution impacts on binding and turnover remains to be resolved. Intriguingly, the recently reported plant UDP-l-arabinopyranose mutase does not require flavin for activity.48 Further work is clearly required in order to understand these enigmatic ring contraction processes.
Experimental
Unless stated all chemical and biochemical reagents were supplied by Sigma Chemicals, Poole, Dorset, UK. Thin-layer chromatography (TLC) was performed on aluminium-backed, pre-coated silica gel plates (Silica Gel 60 F254, Merck). Detection was typically effected under ultraviolet (UV) light, where applicable, followed by treatment with H2SO4 in EtOH (5% v/v) and charring at ~180 °C. Column chromatography was performed on silica gel (40-70 μm, BDH-Merck). Optical rotations were measured at 18 °C using a Perkin-Elmer 141 polarimeter. 1H and 13C NMR spectra were recorded on a Bruker Avance spectrometer at 600 MHz or on a Varian Unity Plus spectrometer at 400 and 100.6 MHz, respectively. Chemical shifts are expressed as parts per million (ppm, δ) and are relative to the solvent as an internal reference [CDCl3: δ 7.27 (s) for 1H; δ 77.0 (t) for 13C; D2O: δ 4.67 (s) for 1H]. Resonance assignments were made with the aid of COSY and HSQC experiments when necessary. 31P NMR spectra were recorded on a Bruker Avance DPX-300 spectrometer at 121.5 MHz using 85% H3PO4 (δ 0.00) as an external reference. 19F NMR spectra were recorded on a JEOL Lambda spectrometer at 376 MHz using CFCl3 (δ 0.00) as an external reference. Low-resolution (LR) electrospray-ionisation (ESI) mass spectra (MS) were obtained on a Waters ZQ4000 mass spectrometer. High-resolution (HR) electrospray-ionisation mass spectra were obtained on a Finnigan MAT 900 XLT mass spectrometer. Anion-exchange HPLC was performed on a BioCAD™ SPRINT™ system (PerSeptive Biosystems). Anhydrous CH2Cl2, THF and pyridine were distilled under N2 according to Armarego and Chai.49
Production of sugar nucleotides
Over-expression and purification of GalK and GalPUT were performed essentially as described previously.30,31 Purified GalK and GalPUT were dialysed against HEPES buffer pH 8.0 prior to use. The 2-, 3- and 6-mono-fluorinated variants of UDP-d-galactopyranose, UDP-d-fucopyranose and UDP-l-arabinopyranose were prepared as described previously.31
Synthesis of 2-deoxy-2-fluoro-d-talose
1-O-Acetyl-3,6-di-O-benzyl-2-deoxy-2-fluoro-α,β-d-mannopyranose (1)46
Compound (1) was prepared, on a gram scale, according to the literature46 as predominantly the α-anomer: 1H NMR (CDCl3): α-anomer; δ 7.25-7.39 (m, 10H, 2 × OCH2Ph), 6.24 (dd, J1,2 2.0 , J1,F 6.4 Hz, H-1), 4.78 (d, 1H, JAB 11.6 Hz, OCH2Ph), 4.69 (d, 1H, JAB 11.6 Hz, OCH2Ph), 4.66 (ddd, 1H, J1,2 2.0, J2,3 2.4, J2,F 48.8 Hz, H-2), 4.62 (d, 1H, JAB 12.0 Hz, OCH2Ph), 4.55 (d, 1H, JAB 12.0 Hz, OCH2Ph), 4.08 (dd, 1H, J3,4 = J4,5 = 9.6 Hz, H-4), 3.80-3.86 (m, 1H, H-5), 3.64-3.79 (m, 3H, H-3, H-6a, H-6b), 2.71 (br s, 1H, 4-OH), 2.08 (s, 3H, OAc); 13C NMR (CDCl3): α-anomer; δ 168.4 (OC(O)Me), 137.7, 137.4 (2 × Cipso), 128.5, 128.3, 127.9, 127.7 (2 × OCH2Ph), 90.9 (d, J1,F 31.1 Hz, C-1), 84.9 (d, J2,F 179.8 Hz, C-2), 77.0 (d, J3,F 16.7 Hz, C-3), 73.6 (OCH2Ph), 73.4 (C-5), 72.0 (OCH2Ph), 69.6 (C-6), 67.2 (C-4), 20.8 (OC(O)Me); HRESIMS found m/z 422.1977 [M + NH4]+. Calcd for C22H29FNO6 422.1973.
1-O-Acetyl-3,6-di-O-benzyl-2-deoxy-2-fluoro-α,β-d-talopyranose (2)
To a stirred solution of a 10:1 α/β mixture of compound (1) (140 mg, 0.35 mmol) and pyridine (0.5 mL) in CH2Cl2 (5 mL) under N2 at −78 °C, Tf2O (70 μL, 0.42 mmol) was added dropwise. The reaction was allowed to warm to 0 °C and stirred at this temperature for 1 h. The reaction was diluted with CH2Cl2 (20 mL), washed with cold satd NaHCO3 (25 mL), cold 0.5 M HCl (25 mL), cold water (25 mL), cold satd NaCl (25 mL), dried (MgSO4), filtered and concentrated. To the residue in DMF (4 mL) was added KNO2 (317 mg, 3.7 mmol) and the reaction was stirred at 50 °C under N2 and monitored by TLC analysis (EtOAc/hexane 1:2). After 4 d, the reaction mixture was filtered and the solid was washed with CH2Cl2. The combined filtrate and washings were concentrated under reduced pressure. Purification of the crude product by flash chromatography (EtOAc/hexane 1:2) gave predominantly the α-anomer of (2) as a colourless gum (61 mg, 44%): Rf = 0.13 (EtOAc/hexane 1:2); 1H NMR (CDCl3): α-anomer; δ 7.30-7.38 (m, 10H, 2 × OCH2Ph), 6.32 (dd, 1H, J1,2 2.0, J1,F 8.0 Hz, H-1), 4.76 (d, 1H, JAB 11.6 Hz, OCH2Ph), 4.67 (d, 1H, JAB 11.6 Hz, OCH2Ph), 4.62 (ddd, 1H, J1,2 2.0, J2,3 3.0, J2,F 48.4 Hz, H-2), 4.59 (d, 1H, JAB 12.0 Hz, OCH2Ph), 4.55 (d, 1H, JAB 12.0 Hz, OCH2Ph), 4.10-4.16 (m, 1H, H-4), 3.98 (ddd, 1H, J4,5 <1, J5,6a 6.4, J5,6b 5.2 Hz, H-5), 3.83 (dd, 1H, J5,6a, J6a,6b 10.0 Hz, H-6a), 3.71 (dd, 1H, J6a,6b, J5,6b 5.2 Hz, H-6b), 3.63 (ddd, 1H, J2,3 = J3,4 = 3.0, J3,F 29.6 Hz, H-3), 2.48 (br s, 1H, 4-OH), 2.06 (s, 3H, OAc); 13C NMR (CDCl3): α-anomer; δ 168.3 (OC(O)Me), 137.8, 136.9 (2 × Cipso), 128.6, 128.3, 128.2, 127.9, 127.8 (2 × OCH2Ph), 91.2 (d, J1,F 32.6 Hz, C-1), 86.2 (d, J2,F 179.8 Hz, C-2), 73.7 (OCH2Ph), 72.4 (C-5), 71.4 (d, J3,F 15.1 Hz, C-3), 70.1 (OCH2Ph), 68.7 (C-6), 66.0 (C-4), 20.8 (OC(O)Me); HRESIMS found m/z 422.1977 [M + NH4]+. Calcd for C22H29FNO6 422.1973.
1-O-Acetyl-2-deoxy-2-fluoro-α,β-d-talopyranose (3)
To a solution of (2) (50 mg, 0.12 mmol) in MeOH (5 mL) was added AcOH (1 drop). The solution was flushed with Ar, and then Pd on C (10% w/w, 5 mg) was added. The reaction was flushed with H2 and stirred at room temperature and atmospheric pressure overnight. The reaction was filtered and the filtrate was concentrated under reduced pressure. Purification of the crude product by flash chromatography (EtOAc → EtOAc/MeOH 5:1) gave a 10:1 α/β mixture of (3) as a colourless amorphous mass (23 mg, 82%): Rf = 0.10 (EtOAc); 1H NMR (CD3OD): α-anomer; δ 6.13 (dd, 1H, J1,2 2.0, J1,F 9.2 Hz, H-1), 4.44 (ddd, 1H, J1,2 2.0, J2,3 <2, J2,F 48.4 Hz, H-2), 3.76-3.83 (m, 2H, H-3, H-4), 3.61-3.74 (m, 1H, H-3), 3.65 (dd, 1H, J5,6a 4.8, J6a,6b 11.6 Hz, H-6a), 3.61 (dd, 1H, J6a,6b, J,5,6b 5.2 Hz, H-6b), 1.99 (s, 3H, OAc); 13C NMR (CD3OD): α-anomer; δ 170.3 (OC(O)Me), 92.7 (d, J1,F 33.4 Hz, C-1), 89.2 (d, J2,F 178.3 Hz, C-2), 75.3, 69.2 (C-4, C-5), 67.0 (d, J3,F 16.7 Hz, C-3), 62.6 (C-6), 20.7 (OC(O)Me); LRESIMS 247 ([M + Na]+, 16%), 205 (100%). HRESIMS found m/z 242.1033 [M + NH4]+. Calcd for C8H17FNO6 242.1034.
2-Deoxy-2-fluoro-α,β-d-talose (4)39
Dowex 50WX8 (H) ion-exchange resin (25 mg) was added to the acetate compound (3) (10 mg, 0.04 mmol) in water (5 mL). The reaction was stirred overnight at 80 °C after which time TLC analysis (CH2Cl2/MeOH 8:1) showed complete consumption of the starting material. The reaction was filtered, the resin washed with water and the combined filtrate and washings were concentrated under reduced pressure to give 2-deoxy-2-fluoro-d-talose (4) as a colourless amorphous mass (6.0 mg, 74%): Rf = 0.16 (CH2Cl2/MeOH 8:1); LRESIMS m/z 205 ([M + Na]+, 100%). HRESIMS found m/z 200.0927 [M + NH4]+. Calcd for C6H15FNO5 200.0929. 1H NMR (D2O): δ 5.50 (dd, J1,2 3.0, J1,F 13 Hz, H-1β furanose), 5.45 (d, J1,2 9.0 Hz, H-1α), 4.88 (d, J1,F 21.0 Hz, H-1β pyranose), 4.75 (d, J2,F 52.0 Hz, H-2β pyranose), 4.63 (d, J2,F 48.0 Hz, H-2α pyranose). The 1H NMR spectroscopic data for compound (4) are consistent with those reported previously.39
General protocol for the synthesis of sugar nucleotides using GalK/GalPUT
A typical procedure for the enzymatic synthesis of sugar nucleotides from the corresponding reducing sugars ran as follows: Uridine 5′-triphosphate (UTP, 11mg, 20 μmol), adenosine 5′-triphosphate (ATP, 0.03 mg, 0.05 μmol), phospho(enol)pyruvic acid (PEP, 4 mg, 20 μmol), uridine 5′-diphosphoglucose (0.03 mg, 0.05 μmol) and monosaccharide (3 mg, 16 μmol) were dissolved in buffer (300 μL, sodium phosphate pH 8.0, 5 mM KCl, 10 mM MgCl2). The pH of the reaction mixture was re-adjusted to 8.0, as required, and GalU (2 U), pyruvate kinase (20 U, 20 μL), inorganic pyrophosphatase (2 U, 20 μL), GalK (2.4 mg, approx. 100 U) and GalPUT (2.4 mg, approx. 100 U) were added, giving a total reaction volume of 600 μL. The reaction was flushed with N2 and incubated at 30 °C and monitored by HPLC analysis.
HPLC analysis was carried out using a PorosHQ50 anion exchange column (3.9 mL, Applied Biosystems) at a flow rate of 12 mL min−1 with detection at 265 nm. Aliquots of reaction mixture (10–50 μL) were applied to the column, which had been equilibrated with water (2 column volumes). Elution with water (2 column volumes) was followed sequentially by a linear gradient of NH4HCO3 (5 mM→54 mM, over 20 column volumes), a linear gradient of NH4HCO3 (54 mM→189 mM, over 2 column volumes), washing with NH4HCO3 (1 M, 3 column volumes), followed by water (2 column volumes). Under these conditions retention times for sugar nucleotides were typically 3-4 min.
After 24 h incubation, addition of MeOH (400 μL) to the reaction mixture was employed to precipitate protein, which was remove by centrifugation. The supernatant was filtered through a 0.45 μm syringe filter and the filtrate was applied to a Poros HQ20 anion exchange column (2.1 mL, Applied Biosystems) which had been equilibrated with water (2 column volumes). The column was eluted, at 30 mL min–1 with detection at 265 nm, with water (3 column volumes) followed by a linear gradient of NH4HCO3 (30 mM→79 mM over 6 column volumes), and then washed with NH4HCO3 (1 M, 3 column volumes), followed by water (2 column volumes). Appropriate fractions were combined and freeze-dried to give the desired sugar nucleotides as their triammonium salts.
Uridine 5′-diphospho-2-deoxy-2-fluoro-α-d-talopyranose
The title compound was prepared using the standard procedure detailed above. 1H NMR (D2O): δ 7.64 (d, 1H, J5,6 8.0 Hz, H-6), 5.80 (d, 1H, J1′,2′ 3.6 Hz, H-1′), 5.66 (m, 1H, H-5), 5.57 (br dd, 1H, J1″,P = J1″,F = 7.8 Hz, H-1″), 4.48-4.62 (m, 1H, H-2″), 4.12-4.16 (m, 2H, H-2′, H-3′), 3.92-4.08 (m, 4H, H-4′, H-5′a, H-5′b, H-5″), 3.82 (ddd, 1H, J2″,3″ = J3″,4″ = 3.0, J3″,F 34.0 Hz, H-3″), 3.72-3.75 (m, 1H, H-4″), 3.61 (dd, 1H, J5″,6″a 7.6, J6″a,6″b 11.6 Hz, H-6″a), 3.54 (dd, 1H, J5″, 6″b 4.8 Hz, J6″a,6″b , H-6″b); 19F NMR (D2O): δ –202.32 (ddd, J 49.3, 33.8, 8.0 Hz, 2-F); 31P NMR (D2O): δ –7.69 (d, 1P, J 20.2 Hz, Pβ), −10.17 (d, 1P, J 20.2 Hz, Pα); LRESIMS 567 ([M - H]−, 21%), 61 (100%). HRESIMS found m/z 567.0437 [M - H]−. Calcd for C15H22FN2O16P2 567.0434.
Uridine 5′-diphospho-α-d-talopyranose50
The known title compound was prepared using the standard procedure detailed above. 1H NMR (D2O): δ 7.77 (d, 1H, J6,5 8.0 Hz, H-6), 5.85 (d, 1H, J1′,2′ 4.0 Hz, H-1′), 5.80 (d, 1H, J5,6 8.0 Hz, H-5), 5.47 (dd, 1H, J1″,2″ 1.6, J1″,P 8.0 Hz, H-1″), 4.19-4.22 (m, 2H, H-2′, H-3′), 4.19-4.22 (m, 2H, H-2′, H-3′), 4.11-4.14 (m, 1H, H-4′), 4.02-4.09 (m, 2H, H-5′a, H-5′b), 3.96-4.00 (m, 1H, H-5″), 3.78-3.85 (m, 3H, H-2″, H-3″, H-4″), 3.68 (dd, 1H, J5″,6″a, 7.2, J6″a,6″b 12.0 Hz, H-6″a), 3.61 (dd, 1H, J5″ ,6″b, J6″a,6″b 4.8 Hz, H-6″b); 31P NMR (D2O): δ −7.84 (d, 1P, J 20.4 Hz, Pβ), −10.13 (d, 1P, J 20.4 Hz, Pα); LRESIMS 565 ([M - H]−, 1%), 75 (100%). HRESIMS found m/z 565.0473 [M - H]−. Calcd for C15H23N2O17P2 565.0478. The 1H NMR spectroscopic data for this compound are consistent with those reported previously.50
Production of UDP-galactopyranose mutase
Recombinant K. pneumoniae UDP galp mutase was purified from Escherichia coli as described previously.14
Determination of mutase reaction kinetic parameters
The kinetic parameters of those compounds that could serve as substrates were determined by preparing a series of samples containing the purified mutase and the specific compound being tested over a range of appropriate substrate concentrations in a total volume of 100 μL of mutase buffer (50 mM MOPS, 10 mM sodium dithionite, 2 mM MgCl2 adjusted to pH 7.4). Each sample was incubated at 37 °C with the amount of enzyme and the length of incubation used to control conversion to be within 20%. The reaction mixture was then quenched by placing the reaction mixture in boiling water for 30 seconds. A sample of the reaction mixture was then applied to a Phenomenex Luna 5μ C18 HPLC column (4.6 × 250 mm), eluting in an isocratic manner at 1 mL min−1 with 6 column volumes of potassium phosphate buffer (50 mM potassium phosphate and 2.5 mM TBAMS adjusted to pH 6.9)51 with detection at 265 nm. Kinetic parameters were deduced by fitting data to the Michaelis-Menten equation using GraFit version 5 (Erithacus Software Ltd).
Determination of mutase reaction equilibria
The equilibrium constant, Keq, was calculated from the ratio of product to substrate at equilibrium. The ratios were determined by integration of the corresponding pyranose and furanose peaks from HPLC chromatograms. Specifically, a 100 μL incubation mixture containing an appropriate amount of the mutase enzyme and 50 μM of substrate in mutase buffer was allowed to reach equilibrium, as determined by the constant product/substrate ratio judged by HPLC.
Scheme 2.

Reagents: (a) Tf2O, pyr, CH2Cl2; (b) KNO2, DMF, 50 °C, 4 days, [44% over 2 steps]; (c) Pd-C, H2, MeOH [82%]; (d) Dowex 50WX8 (H+), H2O, 80 °C, 16 h [74%]; (e) GalK/GalPUT, UTP (see Table 5).
Acknowledgements
We thank Dr Monica Tello for assistance with protein expression, Dr Martin Rejzek for advice and assistance with HPLC analyses and Professor Andrea Vasella for helpful suggestions. This work was supported by the UK Engineering and Physical Sciences Research Council (Grant ref: GR/S82053/02), the Biotechnology and Biological Sciences Research Council (core support grant to the John Innes Centre), and through a Wellcome Trust International Research Collaboration grant.
Footnotes
Using this procedure hundreds of mgs of UDP-Galf can be prepared (T. L. Lowary, personal communication), a key step forward for the characterisation of mycobacterial galactofuranosyltransferase activities.33
Again, in contrast to our observations (Table 3), the recent literature claims that “L-fucose mainly exists as the α- and β-pyran forms in solution with trace amounts of the furan forms.”37
The reverse biotransformation of UDP-Araf to UDP-Arap by UDP-galactopyranose mutase has been reported by Zhang and Liu.38
References
- 1.Duncan K, Barry CE. Curr. Opin. Microbiol. 2004;7:460–465. doi: 10.1016/j.mib.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Ballell L, Field RA, Duncan K, Young RJ. Antimicrob. Agents Chemother. 2005;49:2153–2163. doi: 10.1128/AAC.49.6.2153-2163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janin YL. Bioorg. Med. Chem. Lett. 2007;15:2479–2513. doi: 10.1016/j.bmc.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 4.Duncan K. Curr. Pharm. Design. 2004;10:3185–3194. doi: 10.2174/1381612043383223. [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee D. Curr. Opin. Chem. Biol. 1997;1:579–588. doi: 10.1016/s1367-5931(97)80055-5. [DOI] [PubMed] [Google Scholar]
- 6.Goude R, Parish T. Future Microbiol. 2008;3:299–313. doi: 10.2217/17460913.3.3.299. [DOI] [PubMed] [Google Scholar]
- 7.Nassau PM, Martin SL, Brown RE, Weston A, Monsey D, McNeil MR, Duncan K. J. Bacteriol. 1996;178:1047–1052. doi: 10.1128/jb.178.4.1047-1052.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wing C, Errey JC, Mukhopadhyay B, Blanchard JS, Field RA. Org. Biomol. Chem. 2006;4:3945–3950. doi: 10.1039/b609455d. [DOI] [PubMed] [Google Scholar]
- 9.Koplin R, Brisson J-R, Whitfield C. J. Biol. Chem. 1997;272:4121–4128. doi: 10.1074/jbc.272.7.4121. [DOI] [PubMed] [Google Scholar]
- 10.Weston A, Stern RJ, Lee RE, Nassau PM, Monsey D, Martin SL, Scherman MS, Besra GS, Duncan K, McNeil MR. Tubercule and Lung Disease. 1998;78:123–131. doi: 10.1016/s0962-8479(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 11.Beverley SM, Owens KL, Showalter M, Griffith CL, Doering TL, Jones VC, McNeil MR. Eukaryotic Cell. 2005;4:1147–1154. doi: 10.1128/EC.4.6.1147-1154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders DAR, Staines AG, McMahon SA, McNeil MR, Whitfield C, Naismith JH. Nature Struc. Biol. 2001;8:858–863. doi: 10.1038/nsb1001-858. [DOI] [PubMed] [Google Scholar]
- 13.Soltero-Higgin M, Carlsson EE, Gruer TD, Kiessling LL. Nature Struc. Mol. Biol. 2004;11:539–543. doi: 10.1038/nsmb772. [DOI] [PubMed] [Google Scholar]
- 14.Fullerton SWB, Daff S, Sanders DAR, Ingledew WJ, Whitfield C, Chapman SK, Naismith JH. Biochemistry. 2003;42:2104–2109. doi: 10.1021/bi027077f. [DOI] [PubMed] [Google Scholar]
- 15.Barlow JN, Girvin ME, Blanchard JS. J. Am. Chem. Soc. 1999;121:6968–6969. [Google Scholar]
- 16.Caravano A, Sinay P, Vincent SP. Bioorg. Med. Chem. Lett. 2006;16:1123–1125. doi: 10.1016/j.bmcl.2005.11.106. [DOI] [PubMed] [Google Scholar]
- 17.Itoh K, Huang Z, Liu H-W. Org. Lett. 2007;9:879–882. doi: 10.1021/ol0631408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan Y, Wen X, Sanders DAR, Pinto BM. Biochemistry. 2005;44:14080–14089. doi: 10.1021/bi0513406. [DOI] [PubMed] [Google Scholar]
- 19.Yuan Y, Bleile DW, Wen X, Sanders DAR, Itoh K, Liu H-W, Pinto BM. J. Am. Chem. Soc. 2007;130:3157–3168. doi: 10.1021/ja7104152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chad JM, Sarathy KP, Gruber TD, Addala E, Kiessling LL, Sanders DAR. Biochemistry. 2007;46:6723–6732. doi: 10.1021/bi7002795. [DOI] [PubMed] [Google Scholar]
- 21 (a).Burton A, Wyatt P, Boons G-J. J. Chem. Soc., Perkin Trans. 1997;1:2375–2382. [Google Scholar]; (b) Lee RE, Smith MD, Pickering L, Fleet GWJ. Tetrahedron Lett. 1999;40:8689–8692. [Google Scholar]; (c) Scherman MS, Winans KA, Stern RJ, Jones V, Bertozzi CR, McNeil MR. Antimicrob. Agents Chemother. 2003;47:378–382. doi: 10.1128/AAC.47.1.378-382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Soltero-Higgin M, Carlson EE, Philips JH, Kiessling LL. J. Am. Chem. Soc. 2004;126:10532–10533. doi: 10.1021/ja048017v. [DOI] [PubMed] [Google Scholar]; (e) Veerapen N, Yuan Y, Sanders DAR, Pinto BM. Carbohydr. Res. 2004;339:2205–2217. doi: 10.1016/j.carres.2004.07.012. [DOI] [PubMed] [Google Scholar]; (f) Ghavami, Chen JJW, Pinto BM. Carbohydr. Res. 2004;339:401–407. doi: 10.1016/j.carres.2003.09.036. [DOI] [PubMed] [Google Scholar]; (g) Sadeghi-Khomami A, Blake AJ, Wilson C, Thomas NR. Org. Lett. 2005;7:4891–4894. doi: 10.1021/ol0517877. [DOI] [PubMed] [Google Scholar]; (h) Carlson EE, May JF, Kiessling LL. Chem. Biol. 2006;13:825–837. doi: 10.1016/j.chembiol.2006.06.007. [DOI] [PubMed] [Google Scholar]; (i) Carvano A, Dohi H, Sinay P, Vincent SP. Chem. Eur. J. 2006;12:3114–3123. doi: 10.1002/chem.200500991. [DOI] [PubMed] [Google Scholar]; (j) Desvergnes S, Desvergnes V, Martin OR, Itoh K, Liu H-W, Py S. Bioorg. Med. Chem.Lett. 2007;15:6443–6449. doi: 10.1016/j.bmc.2007.06.059. [DOI] [PubMed] [Google Scholar]; (k) Pan WD, Ansiaux C, Vincent SP. Tetrahedron Lett. 2007;48:4353–4356. [Google Scholar]; (l) Smellie IA, Bhakta S, Sim E, Fairbanks AJ. Org. Biomol. Chem. 2007;5:2257–2266. doi: 10.1039/b704788f. [DOI] [PubMed] [Google Scholar]; (m) Liautard V, Desvergnes V, Itoh K, Liu H-W, Martin OR. J. Org. Chem. 2008;73:3103–3115. doi: 10.1021/jo8001134. [DOI] [PubMed] [Google Scholar]
- 22.Dykhuizen EC, May JF, Tongpenyai A, Kiessling LL. J. Am. Chem. Soc. 2008;130:6706–6707. doi: 10.1021/ja8018687. [DOI] [PubMed] [Google Scholar]
- 23.For recent work see: Glycosyltransferases - Yang M, Proctor MR, Bolam DN, Errey JC, Field RA, Gilbert HJ, Davis BG. J. Am. Chem. Soc. 2005;127:9336–9337. doi: 10.1021/ja051482n. Sismey-Ragatz AE, Green DE, Rejzek M, Field RA, DeAngelis PL. J. Biol. Chem. 2007;282:28321–28327. doi: 10.1074/jbc.M701599200. Sugar nucleotide modifying enzymes - Tello M, Rejzek M, Wilkinson B, Lawson DM, Field RA. ChemBioChem. 2008;9:1295–1302. doi: 10.1002/cbic.200800021. Rejzek M, Kannathasan V. Sri, Wing C, Preston A, Westman EL, Lam JS, Naismith JH, Maskell DJ, Field RA. Org. Biomol. Chem. 2009 doi: 10.1039/b819607a. DOI: 10.1039/ B819607A.
- 24.Barlow JN, Blanchard JS. Carbohydr. Res. 2000;328:473–480. doi: 10.1016/s0008-6215(00)00135-x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang QB, Liu H-W. J. Am. Chem. Soc. 2001;123:6756–6766. doi: 10.1021/ja010473l. [DOI] [PubMed] [Google Scholar]
- 26.O’Hagan D. Chem. Soc. Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 27.Howard JAK, Hoy VJ, O’Hagan D, Smith GT. Tetrahedron. 1996;52:12613–12622. [Google Scholar]
- 28.Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 29.Heidlas JE, Lees WJ, Whitesides GM. J. Org. Chem. 1992;57:152–157. [Google Scholar]
- 30.Chen X, Fang JW, Zhang JB, Liu ZY, Shao J, Kowal P, Andreana P, Wang PG. J. Am. Chem. Soc. 2001;123:2081–2082. doi: 10.1021/ja005738v. [DOI] [PubMed] [Google Scholar]
- 31.Errey JC, Mukhopadhyay B, Kartha KPR, Field RA. Chem. Commun. 2004:2706–2707. doi: 10.1039/b410184g. [DOI] [PubMed] [Google Scholar]
- 32.De Lederkremer RM, Nahmad VB, Varela O. J. Org. Chem. 1994;59:690–692. [Google Scholar]
- 33.Rose NL, Completo GC, Lin SJ, McNeil M, Palcic MM, Lowary TL. J. Am. Chem. Soc. 2006;128:6721–6729. doi: 10.1021/ja058254d. [DOI] [PubMed] [Google Scholar]
- 34.Caravano A, Field RA, Percy JM, Rinaudo G, Roig R, Singh K. Org. Biomol. Chem. 2009 doi: 10.1039/b815342f. DOI: 10.1039/B815342F. [DOI] [PubMed] [Google Scholar]
- 35.Abraham RJ, Chambers EJ, Thomas WA. Magn. Reson. Chem. 1994;32:248–254. [Google Scholar]
- 36.Angyal SJ. Adv. Carbohydr. Chem. Biochem. 1984;42:15–68. [Google Scholar]; Angyal SJ. Adv. Carbohydr. Chem. Biochem. 1991;49:19–35. [Google Scholar]
- 37.Ryu K-S, Kim C, Park C, Choi B-S. J. Am. Chem. Soc. 2004;126:9180–9181. doi: 10.1021/ja047911j. [DOI] [PubMed] [Google Scholar]
- 38.Zhang QB, Liu H-W. Bioorg. Med. Chem. Lett. 2001;11:145–149. doi: 10.1016/s0960-894x(00)00616-8. [DOI] [PubMed] [Google Scholar]
- 39.Haradahira T, Kato A, Maeda M, Torii Y, Ichiya Y-I, Masuda K. Appl. Radiat. Isot. 1992;43:627–632. doi: 10.1016/0883-2889(92)90031-9. [DOI] [PubMed] [Google Scholar]
- 40.Morgenlie S. Acta Chem. Scand. 1973;27:3609–3610. [Google Scholar]
- 41.Turnbull WB, Field RA. J. Chem. Soc., Perkin Trans. 2000;1:1859–1866. [Google Scholar]
- 42.Dax K, Albert M, Ortner J, Paul BJ. Carbohydr. Res. 2000;327:47–86. doi: 10.1016/s0008-6215(00)00022-7. [DOI] [PubMed] [Google Scholar]
- 43.Nyffeler PT, Durón SG, Burkart MD, Vincent SP, Wong C-H. Angew. Chem. Int. Ed. 2005;44:192–212. doi: 10.1002/anie.200400648. [DOI] [PubMed] [Google Scholar]
- 44.Burkart MD, Zhang Z, Hung S-C, Wong C-H. J. Am. Chem. Soc. 1997;119:11743–11746. [Google Scholar]
- 45.Ortner J, Albert M, Weber H, Dax K. J. Carbohydr. Chem. 1999;18:297–316. [Google Scholar]
- 46.Vocadlo DJ, Withers SG. Carbohydr. Res. 2005;340:379–388. doi: 10.1016/j.carres.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 47.Albert R, Dax K, Link RW, Stutz AE. Carbohydr. Res. 1983;118:C5–C6. [Google Scholar]
- 48.Konishi T, Takeda T, Miyazaki Y, Ohnishi-Kameyama M, Hayashi T, O’Neill MA, Ishii T. Glycobiology. 2007;17:345–354. doi: 10.1093/glycob/cwl081. [DOI] [PubMed] [Google Scholar]
- 49.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 5th ed Butterworth-Heinemann; Sydney: 2003. [Google Scholar]
- 50.Shibaev VN, Eliseeva GI, Kraevskaya MA, Kochetkov NK. Bioorg. Khim. 1981;7:376–380. [Google Scholar]
- 51.Combes D, Meynial I, Paquet V. Analytical Chem. 1995;67:1627–1631. [Google Scholar]