

Abstract
Rather than being a constitutive enzyme as was first suggested, endothelial nitric oxide synthase (eNOS) is dynamically regulated at the transcriptional, posttranscriptional, and posttranslational levels. This review will focus on how changes in eNOS function are conferred by various posttranslational modifications. The latest knowledge regarding eNOS targeting to the plasma membrane will be discussed as the role of protein phosphorylation as a modulator of catalytic activity. Furthermore, new data are presented that provide novel insights into how disruption of the eNOS dimer prevents eNOS uncoupling and the production of superoxide under conditions of elevated oxidative stress and identifies a novel regulatory region we have termed the ‘flexible arm’.
Introduction
Nitric oxide (NO) is synthesized by a family of enzymes called NO synthases (NOS). There are three mammalian NOS isoforms: neuronal (nNOS), endothelial (eNOS), and inducible (iNOS). They share 50–60% homology at the amino acid level and have an N-terminal oxygenase domain with heme-, L-arginine-, tetrahydrobiopterin (BH4)-binding domains, a central calmodulin (CaM)-binding region, and a C-terminal reductase domain with NADPH, FAD, and FMN binding sites (Stuehr 1997). Under physiological conditions, the dominant NOS isoform in the vasculature is eNOS, which rather than being a constitutive enzyme as was first suggested, is dynamically regulated at the transcriptional, posttranscriptional, and posttranslational levels. This review will focus on the posttranslational modifications that regulate eNOS activity and will conclude with the identification of a new structural component of eNOS that regulates enzyme function under conditions of oxidative stress.
Membrane targeting of eNOS
In contrast to the other NOS isoforms, eNOS contains a myristoyl group that is covalently attached to the glycine residue found at the second amino acid in the N-terminal region of the protein (Sessa et al. 1993). The co-translational addition of this myristoyl group enables eNOS to localize to cellular membranes and also serves as a mechanism to ensure that eNOS is in close proximity with other factors that regulate its function. Mutational studies have shown that the myristoyl group is an absolute requirement for membrane localization and maximum eNOS activity (Sakoda et al. 1995). In cells, the loss of N-myristoylation confers a cytosolic location and reduced eNOS activity, but in isolated activity assays with maximal cofactors, eNOS activity is equal to the wild-type enzyme (Sessa et al. 1993). It is thought that myristoylation targets eNOS to the Golgi complex (Sessa et al. 1995), and although the myristoyl deficient eNOS can metabolize L-arginine and generate NO, it exhibits reduced coupling to external stimuli such as calcium ionophores (Sessa et al. 1995). eNOS is also subject to posttranslational palmitoylation on two cysteine residues (Cys-15 and Cys-26) within the oxygenase domain of the enzyme. This modification is reversible and requires the prior N-myristoylation and membrane anchoring of eNOS. It is believed that palmitoylation stabilizes the association of eNOS with the membrane and may also be required for correct localization of the enzyme within specialized lipid domains (Liu et al. 1995). The role of palmitoylation in eNOS activation is controversial. Reports have indicated that agonist-induced activation of eNOS is associated with depalmitoylation and translocation from membranes (Yeh et al. 1999). However, this study is contradicted by a similar report (Liu et al. 1995). Mutation of the palmitoylation sites has been shown to decrease agonist-stimulated eNOS activity, but to a lesser extent than for the myristoyl-deficient mutant (Shaul et al. 1996). The palmitoyl-deficient mutant also has an altered subcellular distribution in that it is virtually absent from the plasma membrane (Robinson & Michel 1995, Garcia-Cardena et al. 1996b). As with myristoyl-deficient eNOS, it should be noted that the palmitoyl-deficient enzyme is not catalytically inferior to the wild-type enzyme as the purified enzymes are kinetically identical (Liu et al. 1996). One particular site in the plasma membrane that is of great importance for eNOS function is the caveolae. Caveolae are specialized invaginations of the plasma membrane that are present in most cell types but are particularly enriched within the membranes of endothelial cells (EC), adipocytes, fibroblasts, and smooth muscle cells. It is thought that caveolae are crucial sites for the origination and integration of many signal transduction pathways (Anderson 1993). Studies indicate that, in the plasma membrane, eNOS is located in caveolae (Shaul et al. 1996) and that this location is important for eNOS activity. The localization of eNOS within the caveolae renders the enzyme inactive due to the interaction of eNOS with caveolin-1 ( Ju et al. 1997). This interaction requires that eNOS be both myristoylated and palmitoylated, at least in vivo (Sessa et al. 1993). The mechanism by which caveolin-1 reduces eNOS activity is by preventing CaM binding when calcium levels are low ( Ju et al. 1997, Michel et al. 1997) and caveolin-1 over-expression reduces basal eNOS activity. The interaction sequence for caveolin-1 in human eNOS has been located to amino acids 350–358, and when this region is mutated, eNOS activity is no longer affected by the level of caveolin-1 expression (Garcia-Cardena et al. 1997). However, it should be noted that there is also evidence for additional caveolin-1 binding sites, particularly interactions within the electron transport domain of eNOS, but the exact residues involved are yet to be described (Ghosh et al. 1998). Elevation of intracellular calcium levels or exposure of EC to shear stress causes caveolin-1 to become displaced by the active calcium/CaM complex. This event may also coincide with depalmitoylation (Robinson & Michel 1995) and translocation of eNOS to the cytosol (Michel et al. 1993). However, as mentioned previously, the depalmitoylation/translocation hypothesis has not been confirmed by all groups (Liu et al. 1995), and although the translocation of eNOS from membrane to cytosol has been demonstrated in a number of studies, its biological significance is not yet clear. It is also worth noting that eNOS has also been found within the Golgi complex both in vitro and in vivo (Sessa et al. 1995, Andries et al. 1998). Artificially restricting eNOS expression to the Golgi reveals that eNOS expression on the cytoplasmic leaflet of the cis Golgi membrane results in an enzyme that is active but exhibits blunted capacity to generate NO versus the plasma membrane pool of eNOS (Fulton et al. 2004). Interestingly, targeting eNOS to regions of trans Golgi resulted in dramatically less activity suggesting that distinct regions of the Golgi are supportive or inhibitory to eNOS activity (Jagnandan et al. 2005). While the exact function of the Golgi eNOS is unclear at the present time, acute modulation of cholesterol levels dramatically impacts the activity of eNOS at the plasma membrane but does not alter the output of the Golgi eNOS (Zhang et al. 2006). It may also be that the Golgi is the location for re-palmitoylation after agonist-induced activation and depalmitoylation when the enzyme is recycled to the plasma membrane via the Golgi. Indeed, the discovery of at least five enzymes including DHHC-21 that are localized to the Golgi and promote eNOS palmitoylation provide strong evidence in support of this hypothesis (Fernandez-Hernando et al. 2006). Interestingly, recent data have shown that, at least under conditions of oxidative stress, eNOS trafficking to the plasma membrane is positively regulated by caveolin-1 (Tian et al. 2010). This enrichment of eNOS in caveolae is called the compartmentation effect (Garcia-Cardena et al. 1996b, Shaul et al. 1996) while the inhibitory effect of caveolin-1 is known as the clamp effect (Garcia-Cardena et al. 1996a, Michel et al. 1997, Feron et al. 1998). Thus, changes in the expression of caveolin-1 can alter the balance of eNOS regulation and consequently alter NO generation. It has also been demonstrated that eNOS can interact with the 90 kDa heat-shock protein 90 (Hsp90). Hsp90 is a molecular chaperone that can modulate protein folding and activity. Hsp90 appears to increase eNOS activity by facilitating the CaM-induced displacement of caveolin-1 from eNOS (Gratton et al. 2000), which can be inhibited with the Hsp90 inhibitor, geldanamycin (Garcia-Cardena et al. 1998).
Phosphorylation of eNOS
eNOS can be regulated by multiple phosphorylation sites at tyrosine (Y), serine (S), and threonine (T) residues. The Y sites so far identified localize to Y81 and Y567, the S sites localize to S114, S615, S633, and S1177, and the T site localizes to T495 (using the human sequence nomenclature). The role of each phosphorylation site in regulating eNOS activity will be discussed below.
Tyrosine phosphorylation and eNOS
Both eNOS and nNOS differ from iNOS in their primary dependence on the presence of the calcium/CaM. Thus, eNOS activity can be acutely regulated by increases in the concentration of intracellular calcium that occurs secondary to agonists such as bradykinin (Gosink & Forsberg 1993), estradiol (Goetz et al. 1999), and vascular endothelial growth factor (VEGF) (Papapetropoulos et al. 1997). Fluid shear stress (FSS) can also activate eNOS in a different manner. The initial response of eNOS to FSS is calcium-dependent (Fleming & Busse 1995, Corson et al. 1996), but the long-term response appears to be less dependent on changes in intracellular calcium levels. This long-term activation is reduced by tyrosine kinase inhibitors (Ayajiki et al. 1996, Corson et al. 1996) and recent data have shown that the phosphorylation of tyrosine (Y) residues regulate the ability of eNOS to produce NO (Fulton et al. 2005, 2008). The respective residues have been identified as Y81 (Fulton et al. 2005) and Y657 (Fisslthaler et al. 2008). Interestingly these residues exert opposing effects on eNOS activity. Y81 is phosphorylated by pp60src and activates the enzyme (Fulton et al. 2005, 2008) while Y567 is phosphorylated by proline-rich tyrosine kinase 2 (PYK2) and appears to attenuate eNOS activity (Fisslthaler et al. 2008). Both pp60src and PYK2 are activated by shear, so it is not readily apparent why it is necessary to have opposing phosphorylation events that activate and inhibit eNOS activity. One possibility is that PYK2 activation may limit the generation of peroxynitrite from eNOS in response to sustained increases in flow (Fisslthaler et al. 2008). It has been noted that tyrosine phosphorylation appears to be most prominent in primary EC and may be lost when cells are cultured (Garcia-Cardena et al. 1996a, Fleming et al. 1998). This may account for the studies that have found no eNOS tyrosine phosphorylation (Corson et al. 1996, Venema et al. 1996, 1997, Dimmeler et al. 1999).
Serine and threonine phosphorylation and eNOS
Considerably more is known about the serine phosphorylation of eNOS. eNOS becomes rapidly serine phosphorylated when EC are exposed to FSS leading to phosphorylation of S1177 (human enzyme) that is mediated by AKT1 (Fulton et al. 1999, Gallis et al. 1999) and other kinases such as protein kinase A (PKA) and AMPK (Chen et al. 1999, Michell et al. 2001). Increases in intracellular calcium levels activate CaM that in turn activates CaM kinase II, which also phosphorylates S1177 (Fleming et al. 2001). AKT1 signaling is thus important for both agonist and FSS activation of eNOS (Dimmeler et al. 1999). Although the AKT1-mediated activation of eNOS by FSS has been reported to be independent of changes in intracellular calcium (Dimmeler et al. 1999), it is more likely that the phosphorylation of S1177 renders the enzyme more responsive to much lower levels of calcium (McCabe et al. 2000). FSS has also been shown to stimulate the phosphorylation of eNOS at S633 within an internal autoinhibitory (AI) loop (see section The AL loop of eNOS) and S1177 by a PKA-dependent mechanism (Boo et al. 2003). Mimicking this effect through increased expression of the active catalytic subunit of PKA also leads to the dephosphorylation of T495 (Boo et al. 2003). Using eNOS point mutants where S633 was mutated to either A (to prevent phosphorylation) or D (to mimic the charge of the phosphate group), it was then shown that NO production in S633D-expressing HEK-293 or bovine aortic EC was higher than that obtained from either wild-type eNOS or the S633A eNOS mutant (Boo et al. 2003). Importantly, S633D expressing cells were also able to generate NO in the presence of the calcium chelator, BAPTA-AM (Boo et al. 2003). In vitro analysis of mutants of eNOS containing phosphomimics (aspartic acid mutations) at S615 or S1177 has also demonstrated that phosphorylation of these sites significantly lowers the EC50 for both CaM binding and enzyme activation from the wild-type values of 180±2 and 397±23 nM to 109±2 and 258±11 nM respectively (Tran et al. 2008) for S615 and to 109±2 and 258±11 nM respectively for S1177 (Tran et al. 2009). Although introducing aspartic acid at both S617 and S1177 did not enhance the Vmax of eNOS, the double phosphorylation mutant protein does exhibit a further reduction in the EC50 for both CaM binding and enzyme activation to 77±2 and 130±5 nM. Similarly, in cultured human aortic EC, the introduction of an alanine at aa615 (S615A, phospho-null) into the phosphomimic S1177D mutant eNOS significantly attenuates NO synthesis at resting calcium states (Ritchie et al. 2010). Conversely, mimicking the phosphorylation of S615 by introducing an aspartic acid (S615D) enhances NO generation (Ritchie et al. 2010). These data suggest that there is cooperation between AKT1-mediated phosphorylation of S615 and S1177 to enhance NO generation from eNOS at resting calcium concentrations.
Other factors such as bradykinin also stimulate NO synthesis by increasing the phosphorylation of S1177. However, many of these agents also induce phosphorylation of eNOS at S615 and S633 while stimulating the dephosphorylation of eNOS at T495 (Harris et al. 2001). eNOS can be phosphorylated at T495 by PKC and AMPK and this is thought to be associated with eNOS inhibition (Chen et al. 1999, Michell et al. 2001). In fact, the paradigm in the literature is that there is a yin–yang relationship between T495 and S1177 with phosphorylation of one being associated with dephosphorylation of the other. However, this is not always the case as a recent study indicated that increases in S1177 could maintain NO signaling despite enhanced T495 phosphorylation (Li et al. 2007, Hsu et al. 2010). eNOS can also be phosphorylated at S114 and this is thought to produce a decrease in enzyme activity (Li et al. 2007). More recently, the MAPK Erk1/2 was shown to be the kinase responsible for S114 phosphorylation and this alters the ability of eNOS to bind the prolyl isomerase Pin1, which negatively impacts eNOS activity (Ruan et al. 2011). The regulation of eNOS phosphorylation also involves the activity of the protein phosphatase 2A (PP2A; Greif et al. 2002, Church & Fulton 2006), PP1 (Schmitt et al. 2009), and calcineurin (PP2B; Kou et al. 2002), but the contribution of individual phosphatases to the site-specific dephosphorylation of eNOS remains largely unexplored.
The AI loop of eNOS
Within the three NOS isoforms, there are substantial differences in the rate of electron flow between reductase and oxygenase domains with eNOS having the lowest rate. Studies have shown that this is due to sequences located within the reductase domain since chimeras in which the eNOS oxygenase domain was fused to the iNOS or nNOS reductase domain exhibited higher electron flow than wild-type eNOS while the opposite was true with chimeras in which the iNOS or nNOS oxygenase domains were fused to the eNOS reductase domain (Nishida & Ortiz de Montellano 1999). By aligning the primary sequences of the NOS reductase domain, a 43–45 amino acid insert (at residue 595 of eNOS and 835 of nNOS) was identified that was absent in iNOS (Nishida & Ortiz de Montellano 1999). This region was termed the AI element (Nishida & Ortiz de Montellano 1999) as its deletion from either nNOS or eNOS leads to an increase in CaM-dependent NO synthesis in both enzymes such that activity approached that found in iNOS (Nishida & Ortiz de Montellano 1999, Nishida & de Montellano 2001), although the Ca2+ independence found in iNOS was not observed. Synthetic peptides based on the 45 amino acid insert of eNOS 601SSPRPEQHKSYKIRFNSVSCSDPLVSSWRRKRK633 were found to prevent the binding of CaM and the activation of both eNOS and nNOS (Salerno et al. 1997). Other shorter peptides also exhibited an inhibitory effect leading the authors to conclude that the inhibitory peptide requires the RRKRK motif (Salerno et al. 1997). They also hypothesized that there was another region in the peptide that may interact with EERKSYKVRF and EQHKSYKIRF sequences that are present as insertions within the reductase domains of both eNOS and nNOS (Salerno et al. 1997). An additional AI region (aa1165–1178) was found in the reductase of eNOS but absent in iNOS. Disruption of this region alone slightly improves eNOS calcium sensitivity, but deletion of both the 45aa and the 14aa domains results in an eNOS enzyme that is constitutively active and profoundly calcium insensitive (Chen & Wu 2003, Church & Fulton 2006). It is also noteworthy that the major eNOS phosphorylation sites (S615, S633, and S1177) lie within these two domains. Indeed the generation of an eNOS truncation mutant resulting from the deletion of the last 27 amino acids of eNOS (1177–1205; the C-terminal 27 amino acids) produced an enzyme that had an EC50 for calcium that was five times less than the wild-type eNOS and a significant increase in catalytic activity (Lane & Gross 2002), which is very similar to the enzyme kinetics of the S1177D mutant eNOS which mimics the phosphorylation of S1177 (McCabe et al. 2000). Furthermore, it has been postulated that the AI element interacts with the FMN-binding domain to reduce electron flow (Lane & Gross 2002). However, as a full-length eNOS (or nNOS) protein has not as yet been crystallized, this possibility remains unproven. In addition, whether there are other ways to regulate the release of the AI element remains unknown.
BH4 and eNOS
BH4 is a cofactor essential for the catalytic activity of all three NOS isoforms (Kwon et al. 1989, Tayeh & Marletta 1989, Mayer & Hemmens 1997, Gross et al. 2000). Studies indicate that cellular BH4 levels have important consequences for the structure of NOS enzymes. BH4 binding causes NOS to shift its heme iron to a high-spin state, increases L-arginine binding, and, at least in some NOS isoforms, stabilizes the active dimeric form of the enzyme (Gross et al. 2000). Suboptimal concentrations of BH4 reduce the generation of NO and favor NOS ‘uncoupling’ leading to NOS-mediated reduction of oxygen and the formation of superoxide anions and hydrogen peroxide (H2O2) at the expense of NO. Enhanced BH4 degradation in arteries exposed to oxidative stress likely contribute to the pathogenesis of endothelial dysfunction in hypertension, hypercholesterolemia, diabetes, smoking, and ischemia–reperfusion (Milstien & Katusic 1999, Heitzer et al. 2000, Shinozaki et al. 2000, Tiefenbacher et al. 2000, Baker et al. 2001, Katusic 2001). Supporting this hypothesis, studies have shown that endothelial function can be normalized by BH4 supplementation in experimental animal models of insulin resistance and hypercholesterolemia (Laursen et al. 1997, Shinozaki et al. 1999, 2000, Tiefenbacher et al. 2000). BH4 is a potent reducing agent and exhibits antioxidant activity against both superoxide anions and hydroxyl radicals (Kojima et al. 1995). Oxidative stress can lead to excessive oxidation and depletion of BH4 and previous studies indicate that BH4 is a molecular target for oxidative stress and can cause ‘uncoupling’ of eNOS (Shinozaki et al. 2000, Tiefenbacher et al. 2000, Katusic 2001). Consistent with these findings, vitamin C has been shown to stimulate NOS activity via chemical stabilization of BH4 in cultured human umbilical vein EC (Huang et al. 2000, Heller et al. 2001). This effect of vitamin C appears to be independent of the chemical antagonism between vitamin C and superoxide anions (Baker et al. 2001).
The exact redox mechanism by which BH4 participates in the biosynthesis of NO is still not completely understood (Gross et al. 2000). However, accumulated evidence indicates that optimal concentrations of BH4 are fundamentally important for the normal function of eNOS in EC. Information gained from the work of Klatt et al. (1995) indicates that BH4 is intimately involved in maintaining nNOS dimers. In contrast, the work of Rodríguez-Crespo & Ortiz de Montellano (1996) suggests that BH4 may not be required for eNOS dimerization. However, the study of N-terminal deletions in bovine eNOS revealed that the deletion of 91 or 105 amino acids reduced the ability of the enzyme to dimerize (Rodríguez-Crespo et al. 1997). This suggests that some of the protein residues involved in dimer formation may be located in amino acids 52–105 of eNOS. The crystal structure for the heme domain of eNOS has been solved (Raman et al. 1998) and reveals that zinc ion is tetrahedrally coordinated by two pairs of symmetry-related cysteine residues (corresponding to C94 and C99 from each monomer) predicted to be located at the eNOS dimer interface (Fig. 1).
Figure 1.

Homology model mediated reconstruction of the flexible arm region of human eNOS. Cysteines 94 and 99 from adjacent subunits form a zinc-tetrathiolate cluster (ZnS4) that stabilizes the eNOS dimer. The ZnS4 cluster appears to maintain the N-terminal region in a rigid conformation. It is also apparent that the catalytic center of eNOS is separated from the ZnS4 cluster and no direct contacts are likely between them (A). Utilizing the Yasara modeling software, the ‘flexible arm’ region of human eNOS (corresponding to amino acids 105–125) was reconstructed. The ‘flexible arm’ connects the ZnS4 cluster with the rest of the molecule (A). Also shown are the relative locations of the active site cavity, the ZnS4 cluster, and the BH4 binding site to the ‘flexible arm’ region (A). The ‘flexible arm’ appears to be seated under the cavity in the dimeric conformation with an intact ZnS4 cluster. Molecular dynamic simulations predict that disruption of the ZnS4 cluster will result in closure of the entrance to the catalytic cavity (arrow) of eNOS (B). Zoomed region of the structures with molecular surface are shown representing human eNOS with an intact (left) and disrupted ZnS4 cluster (right) after molecular dynamic simulation (B). Flexible arm appeared in the substrate channel (blue) and blocked access to the heme after Zn removal.
Regulation of eNOS by dimerization
The active form of eNOS enzyme exists as two identical subunits that form a head to tail homodimer. However, the dimeric interface of the two eNOS monomers has not been entirely elucidated. It has been previously shown that cysteines 94 and 99 of eNOS form a zinc tetra-coordinated (ZnS4) cluster between each subunit (Fig. 1A). Zinc bound to the tetrathiolate cluster has also been shown to stabilize the dimer interface on the N-terminal region of eNOS (Raman et al. 1998, Hemmens et al. 2000). However, the molecular mechanisms by which the ZnS4 cluster regulates the catalytic activity of eNOS are poorly understood. Comparative analysis of bacterial NOS and cytochrome P450 structures, which are the closest homologues to NOS, reveal the ZnS4 cluster to be a unique redox-sensitive structural feature of eNOS (Fig. 1A). The bacterial NOS from Bacillus subtilis (bsNOS; Wang et al. 2007) also does not have a ZnS4 cluster (Fig. 2A). Interestingly, it has been recently shown that cytochrome P450 undergoes a structural rearrangement in the catalytic domain (Scott et al. 2003, Savino et al. 2009). Previous X-ray structures of P450 have been solved in a closed conformation, but recent studies have identified a structure with an open channel allowing substrate access (Scott et al. 2003, Savino et al. 2009; Fig. 2B). Interestingly, within this structure, located between amino acids 100–109 within Helix B′ (Honma et al. 2005), lies a flexible region of P450 (Fig. 2B). The N-terminal region of eNOS is also poorly described in the available crystal structures for eNOS, and more specifically, the N-terminal amino acids 105–125 is not present in the X-ray structure of human eNOS. This region, which we have termed the flexible arm, links the ZnS4 cluster with the rest of the protein structure. This flexible region appears to be conserved from mammalian P450s to NOS (Table 1) and it is possible that this sequence may be of importance in regulating the catalytic activity of eNOS.
Figure 2.

Flexible arm feature of eNOS compared to bacterial NOS (bsNOS) and cytochrome P450. Panel A demonstrates the structural similarity of bsNOS (magenta) and eNOS (yellow). The N-terminal end of bsNOS begins immediately after the flexible arm region (red) of eNOS. Thus, bsNOS is deficient in both the redox regulation. Panel B demonstrates the superposition of open (yellow) and closed (magenta) conformations of P450. This flexible region of P450 controls substrate access to the heme and thus serves the same function as the eNOS flexible arm. However, as the P450 lacks the ZnS4 cluster, it is not susceptible to redox regulation.
Table 1.
Alignment of conserved flexible region in endothelial NOS and mammalian P450s. Positively charged amino acids are indicated in bold
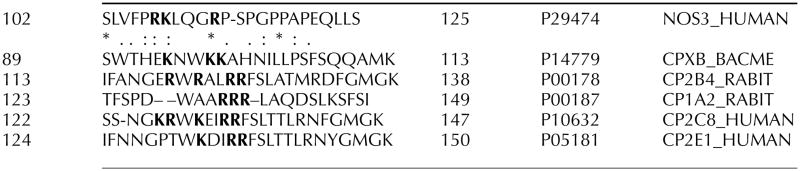
|
Regulation of NO production during conditions of oxidative stress is very important for cell survival. Previous studies have shown that increased oxidative stress is often associated with decreased NO levels (Wiseman et al. 2007, Yada et al. 2007, Gracia-Sancho et al. 2008). NO production, together with increased superoxide generation, leads to elevated peroxynitrite (ONOO−) formation (Beckman et al. 1990). ONOO− is a powerful oxidant and nitrative agent that has been shown to corrupt protein function, mediate cellular dysfunction, and induce apoptosis (Poderoso 2009). It has been shown that the ZnS4 cluster is highly sensitive to oxidants such as ONOO−, NO, and H2O2 (Zou et al. 2002, Ravi et al. 2004, Tummala et al. 2008), and the oxidation of the ZnS4 cluster results in monomerization of eNOS and inhibition of catalytic activity. Thus, the inhibition of NO synthesis through dimer dissociation could play a protective role within EC experiencing increased superoxide generation. Another consideration is that, even after dimer dissociation, the monomeric eNOS retains its heme and can continue to interact with reactive oxygen species (ROS) such as H2O2 (Porasuphatana et al. 2001, Woodward et al. 2009). This would result in the formation of reactive iron–oxygen compounds on the heme. Formation of highly oxidative heme states, in turn, could induce oxidation of surrounding proteins with further self-amplification of oxidative damage.
It is worthwhile noting that the effect of oxidants such as H2O2 on eNOS function is complex. It is now apparent that ROS such as the superoxide anion and H2O2 can act as second messenger molecules (Griendling et al. 2000) altering the function of specific proteins; although in most cases, the mechanisms by which they interact with their molecular targets is still unclear. With respect to eNOS, the overall published data indicate that H2O2 can both stimulate (Drummond et al. 2000) and inhibit (Wedgwood & Black 2005, Kumar et al. 2008) eNOS expression and stimulate (Thomas et al. 2001, Aschner et al. 2007, Tian et al. 2010) and inhibit (Miyamoto et al. 1996) eNOS activity. Of particular importance for eNOS is the kinase, pp60Src, that is the prototype non-receptor tyrosine kinase and is an important signaling molecule with many functions. pp60Src can phosphorylate phospholipase C (Marrero et al. 1995), complex with the epidermal growth factor receptor (Eguchi et al. 1998), the focal adhesion protein, paxillin (Ishida et al. 1999), the janus kinase-2 (Sayeski et al. 1999), and mediate the activation of MAP kinases (Abe et al. 1997). Furthermore, pp60Src is activated by H2O2 leading to an increase in phosphorylation at Y418 (the autophosphorylation site) and Y215 (the SH2-domain) that are inhibited by antioxidants (Abe et al. 1997, Griendling et al. 2000, Ushio-Fukai et al. 2001). The activation of pp60Src by H2O2 stimulates eNOS activity via PI3 kinase and AKT1 (Thomas et al. 2001). However, the effect of H2O2 on eNOS activity appears to be both concentration dependent and time dependent with lower levels stimulating activity (Aschner et al. 2007, Tian et al. 2010) and higher levels inhibiting the enzyme (Miyamoto et al. 1996). The effect of H2O2 on eNOS activity may also be developmentally regulated (Wedgwood & Black 2005). When taken together, previously published studies from a number of other groups suggest that there may also be differences in response to H2O2 depending on the vascular bed. Related to these responses are the endogenous cellular defense mechanisms including both small molecular weight antioxidants (vitamins C and E, reduced GSH, etc.) and antioxidant enzymes. These include CuZn- and Mn-SOD, glutathione peroxidase, and especially, catalase. Recent data has shown that these antioxidant defense mechanisms, especially catalase, can be compromised under conditions of endothelial dysfunction (Sharma et al. 2007). Thus, the effect of H2O2 on eNOS can be separated into either a physiologic response (if catalase levels are maintained) or a pathologic response (when catalase levels are compromised). Thus, under the physiologic conditions of acute increases in oxidative stress, eNOS will be activated through S1177 phosphorylation (Fig. 3, right), while under sustained oxidative stress, eNOS dimerization will be compromised and NO signaling will be attenuated (Fig. 3, left).
Figure 3.
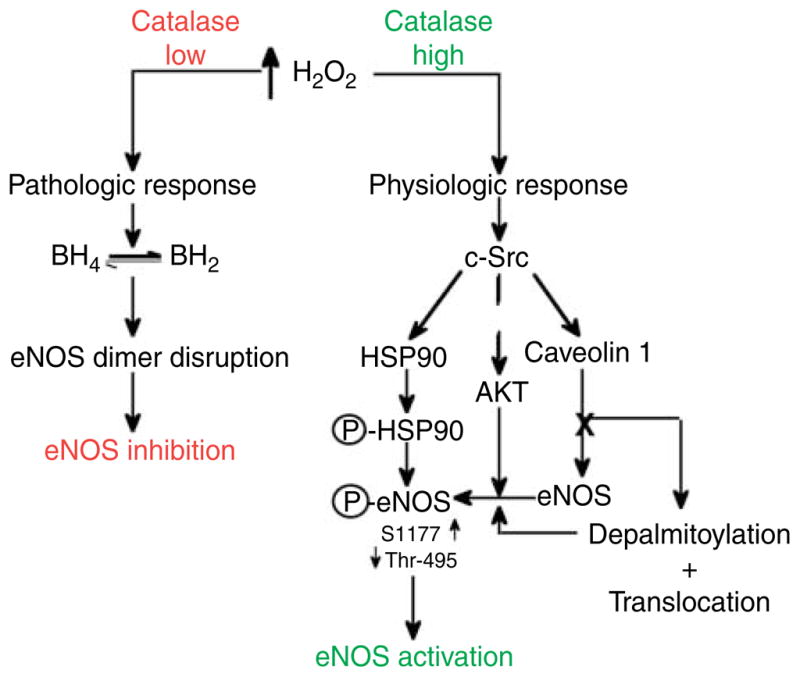
The effect on NO signaling in endothelial cells challenged by H2O2. The response of EC with regard to NO generation in response to oxidative stress can be divided into two pathways: a physiologic response (right) and a pathologic response (left). The physiologic/pathologic decision is then predicated on the level of H2O2 to which the cell is exposed, the duration of the stimulus, and the ability of the cell to metabolize it.
The flexible arm of eNOS: a new regulatory region
The molecular mechanism by which ZnS4 cluster oxidation results in eNOS inactivation and monomerization still remains under investigation (Chen et al. 2010). The ZnS4 cluster is formed by four covalent bonds at the dimeric interface of eNOS. The ZnS4 cluster is ~20 Å away from the catalytic center (Fig. 1A); this makes it unlikely that posttranslational modification of the cysteine residues in ZnS4 could directly affect eNOS activity. Despite the distance between the ZnS4 cluster and the eNOS catalytic center, enzymatic activity is inhibited by ZnS4 disruption suggesting that this region provides significant conformational changes within eNOS (Zou et al. 2002, Ravi et al. 2004, Tummala et al. 2008). Thus, other regions of the protein are implicated in regulating enzyme activity. The crystal structure of eNOS (PDB ID 3NOS; Fischmann et al. 1999) lacks the region located between amino acids 105–125 suggesting a general flexibility within this region. We have termed this sequence the ‘flexible arm’. Utilizing available structures of bovine eNOS (pdb ID – 1FOO), we reconstructed the 105–125 amino acid region of human eNOS (Fig. 1A). Then, to evaluate potential conformational changes in the flexible arm after ZnS4 disruption, we utilized a molecular dynamic (MD) approach. ZnS4 disruption was carried out by removing the Zn atom from the control structure. MD simulations were performed for both structures (with and without the Zn atom) using the same initial parameters. We identified structural changes in the flexible arm region when the normal and ZnS4-disrupted eNOS structures were super-imposed (Fig. 1B). The MD simulations predicted that disruption of the ZnS4 cluster results in the movement of the flexible arm (blue coil Fig. 1B) to block the access of substrate to the catalytic site (red arrow).
To test whether ZnS4 disruption affects access to the heme binding site, we purified three ZnS4 cluster mutants (C94A, C99A, and C94/99A) and identified a loss of a dimeric status in each of the mutant proteins as determined by analytic gel filtration (Fig. 4A). We then subjected the proteins to spectral analysis. We found that each protein contained a Soret band in the visible region characteristic of heme containing enzymes (Fig. 4B). The wild-type eNOS, in the presence of L-arginine and BH4 in the active site, has a Soret band maximum at 396 nm corresponding to high-spin iron (Berka et al. 2008). This absorption maximum is affected by a charge transfer from the sulfur of the cysteine at position 184 to the iron of the heme (see insert Fig. 4B). It has been previously described that NOS enzymes lacking L-arginine and BH4 exhibit a shift in the Soret band to an absorption maximum at 412 nm, indicative of a low-spin iron state (Berka et al. 2008). Interestingly, we observed the same Soret shift to 412 nm for all the ZnS4 mutants (Fig. 4B). This may be explained by hindrance of heme access for L-arginine and BH4 in ZnS4 cluster mutants. To test the accessibility of the heme in these mutants, we conducted cyanide and imidazole binding experiments. Cyanide is a very small molecule that can easily access the heme cavity, and by binding to the heme, it produces a strong absorption in the u.v.–VIS region of the enzyme’s spectrum (Yadav et al. 2003). We measured the iron–cyanide complex formation for the wild-type eNOS, as well as the C94A, C99A, and C94/99A eNOS mutants using increasing concentrations of cyanide. Our data indicate that cyanide binding to wild-type eNOS resulted in a binding-type equilibrium curve with saturation point around 10 mM KCN (Fig. 5A). Surprisingly, none of the ZnS4 cluster mutants exhibited detectable cyanide binding (Fig. 5B–D). In addition, we tested imidazole binding to the heme in ZnS4 mutants. Imidazole competes with L-arginine for binding to the heme. Our data indicate that although imidazole was able to dose dependently bind to the heme site in wild-type eNOS (Fig. 6A), it was unable to bind to any of the ZnS4 cluster mutants (Fig. 6B–D). It is possible that the disruption of the ZnS4 cluster alters either co-factor binding or the structure of the heme such that cyanide or imidazole cannot bind efficiently. To test this possibility, we utilized two other mutant proteins. The W445A eNOS mutant has an altered BH4 binding ability (Joshi & Bauer 2008) while the C184A mutant removes the cysteine thiolate that acts as the proximal heme ligand (Chen et al. 1994). Our data indicate that both C184A (Fig. 7A and C) and W445A (Fig. 7B and D) have efficient cyanide and imidazole binding. These data provide clear evidence that the heme is accessible to both cyanide and imidazole in these mutant proteins. Thus, alterations in either the heme environment or the alterations in BH4 binding are unlikely to explain the lack of cyanide or imidazole binding in the ZnS4 cluster mutant eNOS proteins. Although we did not investigate the access of other small molecules such as oxygen, NO, or H2O2, it is likely that the access to the heme would also be attenuated. This suggests that the disruption of ZnS4 cluster is more important in regulating accessibility to the heme versus altering BH4 binding affinity or an altered heme state.
Figure 4.

Human eNOS ZnS4 cluster mutants have reduced dimer levels. The dimer levels of human eNOS ZnS4 cluster mutants purified as described (Ravi et al. 2004) was assessed by analytical gel filtration (A), again as described (Ravi et al. 2004). Analysis indicates that the dimer (D) levels are decreased and monomer (M) levels are increased in ZnS4 mutants (A). Images are representative of n=3. The absorbance spectra of wild-type and ZnS4 mutants were also analyzed (B). The Soret absorption maximum in wild-type eNOS is found at 395 nm and corresponds to the heme bound to L-arginine and BH4 (B). A red shift in the Soret band maximum to 412 nm is observed for the ZnS4 mutants corresponding to L-arginine and BH4 dissociation (B). The data presented are the mean from three independent experiments. Shown in the insert is a scheme depicting the metal to ligand charge transfer (MLCT) and ‘Soret band’ interference (B).
Figure 5.

Human eNOS ZnS4 cluster mutants have reduced heme accessibility for cyanide. U.V.–visible spectra of the cyanide–iron complex were measured on a Shimadzu spectrophotometer in the region from 350 to 600 nm. Recombinant wild-type (A) and the ZnS4 cluster mutants of eNOS (B–D) were dissolved in PBS to final concentration 0·1 mg/ml. The initial spectrum was obtained before addition of KCN and then the spectrum was recorded after each addition of 1 μl KCN solution (125 mM stock). The addition of 1 μl KCN stock solution was repeated until the reaction reached saturation, as determined by no further changes in absorption at 412 nm. There is a dose-dependent increase in KCN binding to the wild-type protein (A) that does not occur in any of the ZnS4 cluster mutants (B–D). The data presented are the mean±S.D. from three independent experiments.
Figure 6.

Human eNOS ZnS4 cluster mutants have reduced heme accessibility for imidazole. U.V.–visible spectra of the imidazole–iron complex were measured on a Shimadzu spectrophotometer in the region from 350 to 600 nm. Recombinant wild-type (A) and the ZnS4 cluster mutants of eNOS (B–D) were dissolved in PBS to final concentration 0·1 mg/ml. The initial spectrum was obtained before addition of imidazole and then the spectrum was recorded after each addition of 1 μl imidazole solution (100 mM stock). The addition of 1 μl imidazole stock solution was repeated until the reaction reached saturation, as determined by no further changes in absorption at 430 nm. There is a dose-dependent increase in imidazole binding to the wild-type protein (A) that does not occur in any of the ZnS4 cluster mutants (B–D). The data presented are the mean±S.D. from three independent experiments.
Figure 7.

Endothelial NOS mutants with altered heme and BH4 binding do not exhibit altered heme accessibility. Recombinant mutants of eNOS with altered heme (C184A) and BH4 binding (W445A) were exposed to increasing concentrations of potassium cyanide (KCN, 0–12 mM) (A and B) or imidazole (C and D) as described in Fig. 6 and the changes in absorbance between 350 and 600 nm determined. There is a dose-dependent increase in both KCN and imidazole binding in the C184A- and W445A-eNOS mutant proteins. The data presented are the mean±S.D. from three independent experiments.
We next determined the effect of the ZnS4 cluster mutants on NO and superoxide generation compared to the wild-type protein. We found almost a total loss of NO production in C94A, C99A, and C94/99A mutants (Fig. 8A) and a significant reduction in superoxide production (Fig. 8B), suggesting that a functional ZnS4 cluster is required for both NO synthesis and eNOS uncoupling. These data are consistent with previous studies (Zou et al. 2002, Ravi et al. 2004). Further MD simulations predicted that the flexible arm movement is driven by formation of salt bridges between the flexible arm and the substrate channel wall. These simulations further predict that interactions between three positively charged amino acids situated in the flexible arm (Arg107, Lys108, and Arg112; Fig. 9) and negatively charged amino acids from the adjacent surface (Asp264, Glu272, Asp478, and Glu75) are the driving force in the closure of the substrate channel (Fig. 9). To test this, we generated a flexible arm mutant in which all the possible sites of salt bridges predicted from our MD simulation were mutated in combination with a disrupted ZnS4 mutation. Thus, to the C94A/C99A mutant, we added R107A, K108A, and R112A mutations. We then determined how the flexible arm mutations affected dimerization using analytical gel filtration. Our data indicate that this mutant exhibits partial eNOS dimer restoration (Fig. 10A), suggesting that interactions of the flexible arm with the wall of the substrate channel can overcome the need for the ZnS4 cluster in forming a dimer. Our data also indicate that the Soret band of the flexible arm mutant has a maximum at 395 nm, indicative of BH4 and L-arginine binding to the heme (Fig. 10B). In addition, the mutation of positive amino acids on the flexible arm prevents the substrate channel from being blocked as both cyanide binding and imidazole were able to dose dependently bind to the protein (Fig. 10C and D). Indeed the equilibrium curve obtained for KCN binding (Fig. 10C) was similar to that of the wild-type protein (Fig. 5A). Furthermore, although the C94A/C99A mutant lacks the ability to generate NO, mutation of the flexible arm restores NO production to levels found in the wild-type enzyme (Fig. 10E). However, superoxide generation by the flexible arm mutant was increased by twofold compared with the WT eNOS (Fig. 10F), indicating that a significant amount of eNOS uncoupling is occurring. This also resulted in the flexible arm mutant generating significant peroxynitrite (Fig. 10G) that resulted in enhanced self-nitration of eNOS (Fig. 10H). The increase in superoxide generation in the flexible arm mutant may be due either to increased BH4 dissociation from the heme pocket or perhaps an increase in oxygen intake due to the inability of the mutant proteins to close the substrate channel. However, the generation of NO and superoxide in close proximity results in the production of ONOO−, which results in increased self-nitration of the flexible arm mutant protein. Thus, our data suggest that the main role of the flexible arm is to regulate the heme access of molecular oxygen in order to prevent the enhanced superoxide generation and subsequent ONOO− formation that ZnS4 cluster disruption would otherwise produce. Thus, under L-arginine and BH4-saturated conditions, the flexible arm is stabilized in the open position allowing access to the heme. Decreases in L-arginine or BH4 levels are known to increase superoxide generation and contribute to oxidation of the ZnS4 cluster. This leads to the release of the flexible arm and closure of the heme cavity. This mechanism would limit superoxide generation by eNOS. Thus, the redox-sensitive nature of the ZnS4 cluster allows conformational changes within eNOS that preserves enzyme integrity under conditions of increased oxidative stress. As the oxidation of cysteines in the ZnS4 is reversible, the ZnS4 can be rapidly regenerated when the redox state of the cell returns to a reduced state. When the ZnS4 cluster reforms, the flexible arm region of eNOS is restored to its normal position below the substrate access cavity, allowing L-arginine and BH4 to bind heme and restoring the capacity to generate NO. This process allows eNOS to rapidly cycle between activated and inhibited conformations in response to the cellular redox environment and represents a new mechanism by which eNOS activity can be regulated.
Figure 8.

NO and superoxide production by wild-type and the ZnS4 cluster mutants of eNOS. Recombinant wild-type eNOS and the ZnS4 cluster mutants (C94A, C99A, and C9499A) were incubated with L-arginine and the appropriate co-factors for 30 min at 37 °C and NO (A) and superoxide levels (B) determined as described (Grobe et al. 2006). There is a significant reduction in both NO (A) and superoxide (B) generation in the ZnS4 cluster mutants. Data are presented as mean±S.E.M.; n=3. *P<0·05 versus wild-type eNOS. †P<0·05 versus C94A mutant.
Figure 9.
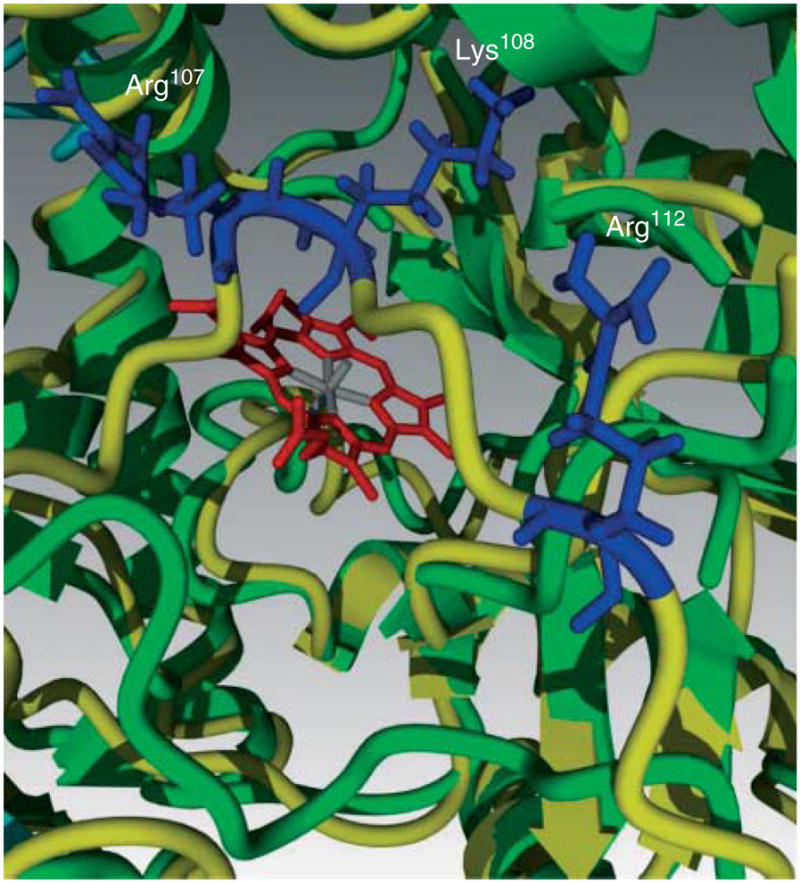
Molecular dynamic simulations identify the amino acids in the flexible arm of human eNOS responsible for closure of the heme cavity. Superposition of intact eNOS (green) and with disrupted ZnS4 (yellow) represents the flexible arm movement. Molecular dynamic simulations predict conformational changes in the flexible arm region that are driven by salt bridge formation between positively charged amino acids (blue) of the flexible arm (Arg107, Lys108, and Arg112) and negatively charged amino acids on the wall of the substrate channel.
Figure 10.

The human eNOS flexible arm mutant undergoes self-nitration. Purified wild-type recombinant eNOS was compared to the flexible arm mutant contained on a disrupted ZnS4 cluster background (C94A/C99A/R107A/K108A/R112A). The flexible arm mutant has dimer (D) and monomer (M) levels, as determined by analytic gel filtration, that are similar to those seen with wild-type eNOS (wt eNOS, A). The absorbance spectrum of the flexible arm mutant contains a Soret band at 395 nm that is comparable with wild-type eNOS (B). The flexible arm mutation also allows both cyanide (C) and imidazole (D) to access the heme. The flexible arm mutant produces NO at levels comparable to the wild-type protein (E). However, the flexible arm mutant significantly generates greater levels of both superoxide (F) and ONOO− (G) compared with the wild-type protein. Western blot analysis using an antibody specific for 3-NT residues indicates that the flexible arm mutant undergoes increased self-nitration compared with either wild-type eNOS or the ZnS4 cluster mutants (C94A, C99A, and C94A/99A, H). A representative image is shown. Data are presented as mean±S.E.M.; n=3. *P<0·05 versus wild-type eNOS.
Conclusions
The mechanisms by which eNOS is posttranslationally regulated have been under active investigation for nearly two decades. However, with our discovery of the flexible arm as a regulatory control element in eNOS, it is clear that new control mechanisms are still to be identified and even previously identified regulatory mechanisms are still far from being fully elucidated. Thus, there is still much to be learned regarding how eNOS is regulated under both physiologic and pathologic conditions.
Acknowledgments
Funding
This review was supported in part by grants HL60190 (to S M B), HL67841 (to S M B), HL72123 (to S M B), HL70061 (to S M B), HL084739 (to S M B), R21HD057406 (to S M B), HL085827 (to D F), and HL092446 (to D F) all from the National Institutes of Health, by a grant from the Foundation Leducq (to S M B), and a Seed Award from the Cardiovascular Discovery Institute of the Medical College of Georgia (to S K). R R and F V F were supported in part by NIH training Grant, 5T32HL06699.
Footnotes
This paper is one of three papers that form part of a Thematic review section on eNOS activation and NO function. The Guest Editor for this section was Ian Bird, University of Wisconsin-Madison, USA.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. Journal of Biological Chemistry. 1997;272:20389–20394. doi: 10.1074/jbc.272.33.20389. [DOI] [PubMed] [Google Scholar]
- Anderson R. Caveolae: where incoming and outgoing messengers meet. PNAS. 1993;90:10909–10913. doi: 10.1073/pnas.90.23.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries LJ, Brutsaert DL, Sys SU. Nonuniformity of endothelial constitutive nitric oxide synthase distribution in cardiac endothelium. Circulation Research. 1998;82:195–203. doi: 10.1161/01.RES.82.2.195. [DOI] [PubMed] [Google Scholar]
- Aschner JL, Foster SL, Kaplowitz M, Zhang Y, Zeng H, Fike CD. Heat shock protein 90 modulates endothelial nitric oxide synthase activity and vascular reactivity in the newborn piglet pulmonary circulation. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2007;292:L1515–L1525. doi: 10.1152/ajplung.00252.2006. [DOI] [PubMed] [Google Scholar]
- Ayajiki K, Kindermann M, Hecker M, Fleming I, Busse R. Intracellular pH and tyrosine phosphorylation but not calcium determine shear stress-induced nitric oxide production in native endothelial cells. Circulation Research. 1996;78:750–758. doi: 10.1161/01.RES.78.5.750. [DOI] [PubMed] [Google Scholar]
- Baker TA, Milstien S, Katusic ZS. Effect of vitamin C on the availability of tetrahydrobiopterin in human endothelial cells. Journal of Cardiovascular Pharmacology. 2001;37:333–338. doi: 10.1097/00005344-200103000-00012. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. PNAS. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berka V, Wang LH, Tsai AL. Oxygen-induced radical intermediates in the nNOS oxygenase domain regulated by L-arginine, tetrahydrobiopterin, and thiol. Biochemistry. 2008;47:405–420. doi: 10.1021/bi701677r. [DOI] [PubMed] [Google Scholar]
- Boo YC, Sorescu GP, Bauer PM, Fulton D, Kemp BE, Harrison DG, Sessa WC, Jo H. Endothelial NO synthase phosphorylated at SER635 produces NO without requiring intracellular calcium increase. Free Radical Biology and Medicine. 2003;35:729–741. doi: 10.1016/S0891-5849(03)00397-6. [DOI] [PubMed] [Google Scholar]
- Chen PF, Wu KK. Structural elements contribute to the calcium/calmodulin dependence on enzyme activation in human endothelial nitric-oxide synthase. Journal of Biological Chemistry. 2003;278:52392–52400. doi: 10.1074/jbc.M305469200. [DOI] [PubMed] [Google Scholar]
- Chen PF, Tsai AL, Wu KK. Cysteine 184 of endothelial nitric oxide synthase is involved in heme coordination and catalytic activity. Journal of Biological Chemistry. 1994;269:25062–25066. [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Letters. 1999;443:285–289. doi: 10.1016/S0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, Tsai AL, Zweier JL. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. 2010;49:3129–3137. doi: 10.1021/bi9016632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church JE, Fulton D. Differences in eNOS activity because of subcellular localization are dictated by phosphorylation state rather than the local calcium environment. Journal of Biological Chemistry. 2006;281:1477–1488. doi: 10.1074/jbc.M505968200. [DOI] [PubMed] [Google Scholar]
- Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circulation Research. 1996;79:984–991. doi: 10.1161/01.RES.79.5.984. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Drummond G, Cai H, Davis M, Ramasamy S, Harrison D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circulation Research. 2000;86:347–354. doi: 10.1161/01.RES.86.3.347. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Numaguchi K, Iwasaki H, Matsumoto T, Yamakawa T, Utsunomiya H, Motley ED, Kawakatsu H, Owada KM, Hirata Y, et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Journal of Biological Chemistry. 1998;273:8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- Fernandez-Hernando C, Fukata M, Bernatchez PN, Fukata Y, Lin MI, Bredt DS, Sessa WC. Identification of Golgi-localized acyl transferases that palmitoylate and regulate endothelial nitric oxide synthase. Journal of Cell Biology. 2006;174:369–377. doi: 10.1083/jcb.200601051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feron O, Saldana F, Michel JB, Michel T. The endothelial nitric-oxide synthase-caveolin regulatory cycle. Journal of Biological Chemistry. 1998;273:3125–3128. doi: 10.1074/jbc.273.6.3125. [DOI] [PubMed] [Google Scholar]
- Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, et al. Structural characterization of nitric oxide synthase isoforms reveals striking active-site conservation. Nature Structural Biology. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Loot AE, Mohamed A, Busse R, Fleming I. Inhibition of endothelial nitric oxide synthase activity by proline-rich tyrosine kinase 2 in response to fluid shear stress and insulin. Circulation Research. 2008;102:1520–1528. doi: 10.1161/CIRCRESAHA.108.172072. [DOI] [PubMed] [Google Scholar]
- Fleming I, Busse R. Control and consequences of endothelial nitric oxide formation. Advances in Pharmacology. 1995;34:187–206. doi: 10.1016/S1054-3589(08)61086-8. [DOI] [PubMed] [Google Scholar]
- Fleming I, Bauersachs J, Fisslthaler B, Busse R. Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circulation Research. 1998;82:686–695. doi: 10.1161/01.RES.82.6.686. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circulation Research. 2001;88:E68–E75. doi: 10.1161/hh1101.092677. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Babbitt R, Zoellner S, Fontana J, Acevedo L, McCabe TJ, Iwakiri Y, Sessa WC. Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt-versus calcium-dependent mechanisms for nitric oxide release. Journal of Biological Chemistry. 2004;279:30349–30357. doi: 10.1074/jbc. M402155200. [DOI] [PubMed] [Google Scholar]
- Fulton D, Church JE, Ruan L, Li C, Sood SG, Kemp BE, Jennings IG, Venema RC. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. Journal of Biological Chemistry. 2005;280:35943–35952. doi: 10.1074/jbc.M504606200. [DOI] [PubMed] [Google Scholar]
- Fulton D, Ruan L, Sood SG, Li C, Zhang Q, Venema RC. Agonist-stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of eNOS phosphorylation at Tyr83. Circulation Research. 2008;102:497–504. doi: 10.1161/CIRCRESAHA.107.162933. [DOI] [PubMed] [Google Scholar]
- Gallis B, Corthals GL, Goodlett DR, Ueba H, Kim F, Presnell SR, Figeys D, Harrison DG, Berk BC, Aebersold R, et al. Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. Journal of Biological Chemistry. 1999;274:30101–30108. doi: 10.1074/jbc.274.42. 30101. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Stern DF, Liu J, Sessa WC. Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. Journal of Biological Chemistry. 1996a;271:27237–27240. doi: 10.1074/jbc.271.44.27237. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: implications for nitric oxide signaling. PNAS. 1996b;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cardena G, Martasek P, Masters BS, Skidd PM, Couet J, Li S, Lisanti MP, Sessa WC. Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. Journal of Biological Chemistry. 1997;272:25437–25440. doi: 10.1074/jbc.272.41.25437. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Gachhui R, Crooks C, Wu C, Lisanti M, Stuehr D. Interactions between caveolin 1 and the reductase domain of endothelial nitric-oxide synthase. Consequences for catalysis. Journal of Biological Chemistry. 1998;273:22267–22271. doi: 10.1074/jbc.273.35.22267. [DOI] [PubMed] [Google Scholar]
- Goetz R, Thatte H, Prabhakar P, Cho M, Michel T, Golan D. Estradiol induces the calcium-dependent translocation of endothelial nitric oxide synthase. PNAS. 1999;96:2788–2793. doi: 10.1073/pnas.96.6.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosink E, Forsberg E. Effects of ATP and bradykinin on endothelial cell Ca2+ homeostasis and formation of cGMP and prostacyclin. American Journal of Physiology. 1993;265:C1620–C1629. doi: 10.1152/ajpcell.1993.265.6.C1620. [DOI] [PubMed] [Google Scholar]
- Gracia-Sancho J, Lavina B, Rodriguez-Vilarrupla A, Garcia-Caldero H, Fernandez M, Bosch J, Garcia-Pagan JC. Increased oxidative stress in cirrhotic rat livers: a potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology. 2008;47:1248–1256. doi: 10.1002/hep.22166. [DOI] [PubMed] [Google Scholar]
- Gratton JP, Fontana J, O’Connor DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. Journal of Biological Chemistry. 2000;275:22268–22272. doi: 10.1074/jbc. M001644200. [DOI] [PubMed] [Google Scholar]
- Greif DM, Kou R, Michel T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 2002;41:15845–15853. doi: 10.1021/bi026732g. [DOI] [PubMed] [Google Scholar]
- Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:2175–2183. doi: 10.1161/01.ATV.20.10.2175. [DOI] [PubMed] [Google Scholar]
- Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, Fineman JR, Black SM. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2006;290:L1069–L1077. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- Gross S, Jones C, Hattori Y, Raman C. Tetrahydrobiopterin: an essential cofactor of nitric oxide synthase with an elusive role. In: Jignarro L, editor. Nitric Oxide Biology and Pathobiology. San Diego, CA: Academic Press; 2000. pp. 167–187. [Google Scholar]
- Harris MB, Ju H, Venema VJ, Liang H, Zou R, Michell BJ, Chen ZP, Kemp BE, Venema RC. Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. Journal of Biological Chemistry. 2001;276:16587–16591. doi: 10.1074/jbc. M100229200. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Munzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circulation Research. 2000;86:E36–E41. doi: 10.1161/01.RES.86.2.e36. [DOI] [PubMed] [Google Scholar]
- Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. Journal of Biological Chemistry. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- Hemmens B, Goessler W, Schmidt K, Mayer B. Role of bound zinc in dimer stabilization but not enzyme activity of neuronal nitric-oxide synthase. Journal of Biological Chemistry. 2000;275:35786–35791. doi: 10.1074/jbc.M005976200. [DOI] [PubMed] [Google Scholar]
- Honma W, Li W, Liu H, Scott EE, Halpert JR. Functional role of residues in the helix B′ region of cytochrome P450 2B1. Archives of Biochemistry and Biophysics. 2005;435:157–165. doi: 10.1016/j.abb.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Hsu JH, Oishi P, Wiseman DA, Hou Y, Chikovani O, Datar S, Sajti E, Johengen MJ, Harmon C, Black SM, et al. Nitric oxide alterations following acute ductal constriction in the fetal lamb: a role for superoxide. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2010;298:L880–L887. doi: 10.1152/ajplung.00384.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A, Vita JA, Venema RC, Keaney JF., Jr Ascorbic acid enhances endothelial nitric-oxide synthase activity by increasing intracellular tetrahydrobiopterin. Journal of Biological Chemistry. 2000;275:17399–17406. doi: 10.1074/jbc.M002248200. [DOI] [PubMed] [Google Scholar]
- Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. Journal of Clinical Investigation. 1999;103:789–797. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagnandan D, Sessa WC, Fulton D. Intracellular location regulates calcium-calmodulin-dependent activation of organelle-restricted eNOS. American Journal of Physiology. Cell Physiology. 2005;289:C1024–C1033. doi: 10. 1152/ajpcell.00162.2005. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Bauer JA. Preliminary computational modeling of nitric oxide synthase 3 interactions with caveolin-1: influence of exon 7 Glu298Asp polymorphism. Acta Biochimica et Biophysica Sinica. 2008;40:47–54. doi: 10.1111/j.1745-7270.2008.00369.x. [DOI] [PubMed] [Google Scholar]
- Ju H, Zou R, Venema VJ, Venema RC. Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. Journal of Biological Chemistry. 1997;272:18522–18525. doi: 10.1074/jbc.272.30.18522. [DOI] [PubMed] [Google Scholar]
- Katusic ZS. Vascular endothelial dysfunction: does tetrahydrobiopterin play a role? American Journal of Physiology. Heart and Circulatory Physiology. 2001;281:H981–H986. doi: 10.1152/ajpheart.2001.281.3.H981. [DOI] [PubMed] [Google Scholar]
- Klatt P, Schmidt K, Lehner D, Glatter O, Bachinger HP, Mayer B. Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and L-arginine in the formation of an SDS-resistant dimer. EMBO Journal. 1995;14:3687–3695. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Ona S, Iizuka I, Arai T, Mori H, Kubota K. Antioxidative activity of 5, 6 ,7,8-tetrahydrobiopterin and its inhibitory effect on paraquat-induced cell toxicity in cultured rat hepatocytes. Free Radical Research. 1995;23:419–430. doi: 10.3109/10715769509065263. [DOI] [PubMed] [Google Scholar]
- Kou R, Greif D, Michel T. Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. Journal of Biological Chemistry. 2002;277:29669–29673. doi: 10.1074/jbc.M204519200. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sun X, Wedgwood S, Black SM. Hydrogen peroxide decreases endothelial nitric oxide synthase promoter activity through the inhibition of AP-1 activity. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2008;295:L370–L377. doi: 10.1152/ajplung.90205.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon NS, Nathan CF, Stuehr DJ. Reduced biopterin as a cofactor in the generation of nitrogen oxides by murine macrophages. Journal of Biological Chemistry. 1989;264:20496–20501. [PubMed] [Google Scholar]
- Lane P, Gross SS. Disabling a C-terminal autoinhibitory control element in endothelial nitric-oxide synthase by phosphorylation provides a molecular explanation for activation of vascular NO synthesis by diverse physiological stimuli. Journal of Biological Chemistry. 2002;277:19087–19094. doi: 10.1074/jbc.M200258200. [DOI] [PubMed] [Google Scholar]
- Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.CIR.95.3.588. [DOI] [PubMed] [Google Scholar]
- Li C, Ruan L, Sood SG, Papapetropoulos A, Fulton D, Venema RC. Role of eNOS phosphorylation at Ser-116 in regulation of eNOS activity in endothelial cells. Vascular Pharmacology. 2007;47:257–264. doi: 10.1016/j.vph.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Garcia-Cardena G, Sessa WC. Biosynthesis and palmitoylation of endothelial nitric oxide synthase: mutagenesis of palmitoylation sites, cysteines-15 and/or -26, argues against depalmitoylation-induced translocation of the enzyme. Biochemistry. 1995;34:12333–12340. doi: 10.1021/bi00038a029. [DOI] [PubMed] [Google Scholar]
- Liu J, Garcia-Cardena G, Sessa WC. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: implications for caveolae localization. Biochemistry. 1996;35:13277–13281. doi: 10.1021/bi961720e. [DOI] [PubMed] [Google Scholar]
- Marrero MB, Schieffer B, Paxton WG, Schieffer E, Bernstein KE. Electroporation of pp60c-src antibodies inhibits the angiotensin II activation of phospholipase C-gamma 1 in rat aortic smooth muscle cells. Journal of Biological Chemistry. 1995;270:15734–15738. doi: 10.1074/jbc.270.26.15734. [DOI] [PubMed] [Google Scholar]
- Mayer B, Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends in Biochemical Sciences. 1997;22:477–481. doi: 10.1016/S0968-0004(97)01147-X. (erratum appears in Trends Biochem Sci 1998 Feb;23(2):87) [DOI] [PubMed] [Google Scholar]
- McCabe TJ, Fulton D, Roman LJ, Sessa WC. Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. Journal of Biological Chemistry. 2000;275:6123–6128. doi: 10.1074/jbc.275.9.6123. [DOI] [PubMed] [Google Scholar]
- Michel T, Li G, Busconi L. Phosphorylation and subcellular translocation of endothelial nitric oxide synthase. PNAS. 1993;90:6252–6256. doi: 10.1073/pnas.90.13.6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. Journal of Biological Chemistry. 1997;272:15583–15586. doi: 10.1074/jbc.272.25. 15583. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. Journal of Biological Chemistry. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochemical and Biophysical Research Communications. 1999;263:681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Akaike T, Yoshida M, Goto S, Horie H, Maeda H. Potentiation of nitric oxide-mediated vasorelaxation by xanthine oxidase inhibitors. Proceedings of the Society for Experimental Biology and Medicine. 1996;211:366–373. doi: 10.3181/00379727-211-43982. [DOI] [PubMed] [Google Scholar]
- Nishida CR, de Montellano PR. Control of electron transfer in nitric-oxide synthases. Swapping of autoinhibitory elements among nitric-oxide synthase isoforms. Journal of Biological Chemistry. 2011;276:20116–20124. doi: 10.1074/jbc.M101548200. [DOI] [PubMed] [Google Scholar]
- Nishida CR, Ortiz de Montellano PR. Autoinhibition of endothelial nitric-oxide synthase. Identification of an electron transfer control element. Journal of Biological Chemistry. 1999;274:14692–14698. doi: 10.1074/jbc.274.21. 14692. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Garcia Cardena G, Madri J, Sessa W. Nitric oxide production contributes to the angiogenic proterties of vascular endothelial growth factor in human endothelial cells. Journal of Clinical Investigation. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poderoso JJ. The formation of peroxynitrite in the applied physiology of mitochondrial nitric oxide. Archives of Biochemistry and Biophysics. 2009;484:214–220. doi: 10.1016/j.abb.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Porasuphatana S, Tsai P, Pou S, Rosen GM. Involvement of the perferryl complex of nitric oxide synthase in the catalysis of secondary free radical formation. Biochimica et Biophysica Acta. 2001;1526:95–104. doi: 10.1016/s0304-4165(01)00114-3. [DOI] [PubMed] [Google Scholar]
- Raman CS, Li H, Martasek P, Kral V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/S0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. PNAS. 2004;101:2619–2624. doi: 10.1073/pnas.0300464101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie SA, Kohlhaas CF, Boyd AR, Yalla KC, Walsh K, Connell JM, Salt IP. Insulin-stimulated phosphorylation of endothelial nitric oxide synthase at serine-615 contributes to nitric oxide synthesis. Biochemical Journal. 2010;426:85–90. doi: 10.1042/BJ20091580. [DOI] [PubMed] [Google Scholar]
- Robinson LJ, Michel T. Mutagenesis of palmitoylation sites in endothelial nitric oxide synthase identifies a novel motif for dual acylation and subcellular targeting. PNAS. 1995;92:11776–11780. doi: 10.1073/pnas.92.25.11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Crespo I, Ortiz de Montellano PR. Human endothelial nitric oxide synthase: expression in Escherichia coli, coexpression with calmodulin, and characterization. Archives of Biochemistry and Biophysics. 1996;336:151–156. doi: 10.1006/abbi.1996.0543. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Crespo I, Moënne-Loccoz P, Loehr TM, Ortiz de Montellano PR. Endothelial nitric oxide synthase: modulations of the distal heme site produced by progressive N-terminal deletions. Biochemistry. 1997;36:8530–8538. doi: 10.1021/bi970192j. [DOI] [PubMed] [Google Scholar]
- Ruan L, Torres CM, Qian J, Chen F, Mintz JD, Stepp DW, Fulton D, Venema RC. Pin1 prolyl isomerase regulates endothelial nitric oxide synthase. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31:392–398. doi: 10.1161/ATVBAHA.110.213181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoda T, Hirata K, Kuroda R, Miki N, Suematsu M, Kawashima S, Yokoyama M. Myristoylation of endothelial cell nitric oxide synthase is important for extracellular release of nitric oxide. Molecular and Cellular Biochemistry. 1995;152:143–148. doi: 10.1007/BF01076076. [DOI] [PubMed] [Google Scholar]
- Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM, Martasek P, Roman LJ, Masters BS, Jones CL, et al. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. Journal of Biological Chemistry. 1997;272:29769–29777. doi: 10.1074/jbc.272.47. 29769. [DOI] [PubMed] [Google Scholar]
- Savino C, Montemiglio LC, Sciara G, Miele AE, Kendrew SG, Jemth P, Gianni S, Vallone B. Investigating the structural plasticity of a cytochrome P450: three-dimensional structures of P450 EryK and binding to its physiological substrate. Journal of Biological Chemistry. 2009;284:29170–29179. doi: 10.1074/jbc.M109.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayeski PP, Ali MS, Hawks K, Frank SJ, Bernstein KE. The angiotensin II-dependent association of Jak2 and c-Src requires the N-terminus of Jak2 and the SH2 domain of c-Src. Circulation Research. 1999;84:1332–1338. doi: 10.1161/01.RES.84.11.1332. [DOI] [PubMed] [Google Scholar]
- Schmitt CA, Heiss EH, Aristei Y, Severin T, Dirsch VM. Norfuraneol dephosphorylates eNOS at threonine 495 and enhances eNOS activity in human endothelial cells. Cardiovascular Research. 2009;81:750–757. doi: 10.1093/cvr/cvn326. [DOI] [PubMed] [Google Scholar]
- Scott EE, He YA, Wester MR, White MA, Chin CC, Halpert JR, Johnson EF, Stout CD. An open conformation of mammalian cytochrome P450 2B4 at 1.6-A resolution. PNAS. 2003;100:13196–13201. doi: 10.1073/pnas. 2133986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa WC, Barber CM, Lynch KR. Mutation of N-myristoylation site converts endothelial cell nitric oxide synthase from a membrane to a cytosolic protein. Circulation Research. 1993;72:921–924. doi: 10.1161/01.RES.72.4.921. [DOI] [PubMed] [Google Scholar]
- Sessa WC, Garcia-Cardena G, Liu J, Keh A, Pollock JS, Bradley J, Thiru S, Braverman IM, Desai KM. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. Journal of Biological Chemistry. 1995;270:17641–17644. doi: 10.1074/jbc.270.30. 17641. [DOI] [PubMed] [Google Scholar]
- Sharma S, Grobe AC, Wiseman DA, Kumar S, Englaish M, Najwer I, Benavidez E, Oishi P, Azakie A, Fineman JR, et al. Lung antioxidant enzymes are regulated by development and increased pulmonary blood flow. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2007;293:L960–L971. doi: 10.1152/ajplung.00449.2006. [DOI] [PubMed] [Google Scholar]
- Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG, Michel T. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. Journal of Biological Chemistry. 1996;271:6518–6522. doi: 10.1074/jbc.271.11.6518. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Kashiwagi A, Nishio Y, Okamura T, Yoshida Y, Masada M, Toda N, Kikkawa R. Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/ imbalance in insulin-resistant rat aorta. Diabetes. 1999;48:2437–2445. doi: 10.2337/diabetes.48.12.2437. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, Masada M, Toda N, Kikkawa R, Kashiwagi A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circulation Research. 2000;87:566–573. doi: 10.1161/01.RES.87.7.566. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ. Structure–function aspects in the nitric oxide synthases. Annual Review of Pharmacology and Toxicology. 1997;37:339–359. doi: 10.1146/annurev.pharmtox.37.1.339. [DOI] [PubMed] [Google Scholar]
- Tayeh MA, Marletta MA. Macrophage oxidation of L-arginine to nitric oxide, nitrite, and nitrate. Tetrahydrobiopterin is required as a cofactor. Journal of Biological Chemistry. 1989;264:19654–19658. [PubMed] [Google Scholar]
- Thomas S, Chen K, Keaney J., Jr Hydrogen peroxide activates endothelial nitric oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. Journal of Biological Chemistry. 2001;277:6017–6024. doi: 10.1074/jbc.M109107200. [DOI] [PubMed] [Google Scholar]
- Tian J, Hou Y, Lu Q, Wiseman DA, Vasconcelos Fonsesca F, Elms S, Fulton DJ, Black SM. A novel role for caveolin-1 in regulating endothelial nitric oxide synthase activation in response to H2O2 and shear stress. Free Radical Biology & Medicine. 2010;49:159–170. doi: 10.1016/j.freeradbiomed.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiefenbacher CP, Bleeke T, Vahl C, Amann K, Vogt A, Kubler W. Endothelial dysfunction of coronary resistance arteries is improved by tetrahydrobiopterin in atherosclerosis. Circulation. 2000;102:2172–2179. doi: 10.1161/01.CIR.102.18.2172. [DOI] [PubMed] [Google Scholar]
- Tran QK, Leonard J, Black DJ, Persechini A. Phosphorylation within an autoinhibitory domain in endothelial nitric oxide synthase reduces the Ca(2+) concentrations required for calmodulin to bind and activate the enzyme. Biochemistry. 2008;47:7557–7566. doi: 10.1021/bi8003186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran QK, Leonard J, Black DJ, Nadeau OW, Boulatnikov IG, Persechini A. Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. Journal of Biological Chemistry. 2009;284:11892–11899. doi: 10.1074/jbc.M806205200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala M, Ryzhov V, Ravi K, Black SM. Identification of the cysteine nitrosylation sites in human endothelial nitric oxide synthase. DNA and Cell Biology. 2008;27:25–33. doi: 10.1089/dna.2007.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21:489–495. doi: 10. 1161/01.ATV.21.4.489. [DOI] [PubMed] [Google Scholar]
- Venema VJ, Marrero MB, Venema RC. Bradykinin-stimulated protein tyrosine phosphorylation promotes endothelial nitric oxide synthase translocation to the cytoskeleton. Biochemical and Biophysical Research Communications. 1996;226:703–710. doi: 10.1006/bbrc.1996.1417. [DOI] [PubMed] [Google Scholar]
- Venema VJ, Zou R, Ju H, Marrero MB, Venema RC. Caveolin-1 detergent solubility and association with endothelial nitric oxide synthase is modulated by tyrosine phosphorylation. Biochemical and Biophysical Research Communications. 1997;236:155–161. doi: 10.1006/bbrc.1997.6921. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Lawson RJ, Buddha MR, Wei CC, Crane BR, Munro AW, Stuehr DJ. Bacterial flavodoxins support nitric oxide production by Bacillus subtilis nitric-oxide synthase. Journal of Biological Chemistry. 2007;282:2196–2202. doi: 10.1074/jbc.M608206200. [DOI] [PubMed] [Google Scholar]
- Wedgwood S, Black SM. Endothelin-1 decreases endothelial NOS expression and activity through ETA receptor-mediated generation of hydrogen peroxide. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2005;288:L480–L487. doi: 10.1152/ajplung.00283.2004. [DOI] [PubMed] [Google Scholar]
- Wiseman DA, Wells SM, Hubbard M, Welker JE, Black SM. Alterations in zinc homeostasis underlie endothelial cell death induced by oxidative stress from acute exposure to hydrogen peroxide. American Journal of Physiology. Lung Cellular and Molecular Physiology. 2007;292:L165–L177. doi: 10.1152/ajplung.00459.2005. [DOI] [PubMed] [Google Scholar]
- Woodward JJ, Chang MM, Martin NI, Marletta MA. The second step of the nitric oxide synthase reaction: evidence for ferric-peroxo as the active oxidant. Journal of the American Chemical Society. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- Yada T, Kaji S, Akasaka T, Mochizuki S, Ogasawara Y, Tanemoto K, Yoshida K, Kajiya F. Changes of asymmetric dimethylarginine, nitric oxide, tetrahydrobiopterin, and oxidative stress in patients with acute myocardial infarction by medical treatments. Clinical Hemorheology and Microcirculation. 2007;37:269–276. [PubMed] [Google Scholar]
- Yadav J, Sagami I, Shimizu T. Cyanide binding study of neuronal nitric oxide synthase: effects of inhibitors and mutations at the substrate binding site. Journal of Inorganic Biochemistry. 2003;95:25–30. doi: 10.1016/S0162-0134(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Yeh DC, Duncan JA, Yamashita S, Michel T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. Journal of Biological Chemistry. 1999;274:33148–33154. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Church JE, Jagnandan D, Catravas JD, Sessa WC, Fulton D. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:1015–1021. doi: 10.1161/01.ATV.0000216044. 49494.c4. [DOI] [PubMed] [Google Scholar]
- Zou MH, Shi C, Cohen RA. Oxidation of the zinc–thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. Journal of Clinical Investigation. 2002;109:817–826. doi: 10.1172/JCI200214442. [DOI] [PMC free article] [PubMed] [Google Scholar]