

Abstract
The present investigation reports the design and evaluation of six-hour extended release film-coated matrix tablets of cephalexin using different grades of hydrophilic polymer hydroxypropylmethylcellulose (HPMC) employing direct compression method. The preformulation studies performed included the physical compatibility studies, Differential Scanning Calorimetry analysis, drug characterization using Fourier Transform Infra Red spectroscopic analysis and particle size analysis using sieve method. The tablets were evaluated for weight variation, hardness, thickness and friability. Results of the studies indicate that the polymers used have significant release-retarding effect on the formulation. The dissolution profile comparison of the prepared batches P1 to P8 and market preparation (Sporidex AF 375) was done by using Food and Drug Administration-recommended similarity factor (f2) determination. The formulation P8 (10% HPMC K4M, 15% HPMC 15cps) with a similarity factor (f2) of 77.75 was selected as the optimized formulae for scale-up batches. The dissolution data of the best formulation P8 was fitted into zero order, first order, Higuchi and Korsemeyer-Peppas models to identify the pharmacokinetics and mechanism of drug release. The results of the accelerated stability study of best formulation P8 for three months revealed that storage conditions were not found to have made any significant changes in final formulation F3. The release of cephalexin was prolonged for 6 h by using polymer combinations of HPMC and a twice daily matrix tablet was formulated.
Keywords: Cephalexin, differential scanning calorimetry, Fourier transform infra red spectroscopic, hydroxy propyl methyl cellulose, matrix tablets, release kinetics, similarity factor (f2)
INTRODUCTION
Oral delivery of drugs is by far the most preferable route of drug delivery due to the ease of administration, patient compliance and flexibility in formulation. Extended-release oral drug formulations have been used since the 1960s to enhance performance and increase patient compliance.[1] By incorporating the dose for 24 h into one tablet from which the drug is slowly released, peaks of high plasma concentration and troughs of low plasma concentration can be prevented.[2] This helps to avoid the side-effects associated with high concentrations and the lack of activity associated with low concentrations giving better overall therapy. In biopharmaceutics, scientists generally are faced with an engineering problem; to develop drug delivery systems that hit a desired target. The target in pharmacokinetics is generally a plasma/blood drug concentration that lies between the minimum effect concentration (MEC) and minimum toxic concentration (MTC). Cephalexin is a semisynthetic antibiotic derived from cephalosporin C and is almost completely absorbed from the gastrointestinal tract with a bioavailability of 95%. Cephalexin has a half life of around 1.1 h.[3–5] To maintain the therapeutic range, the drug should be administered three to four times a day, which leads to saw tooth kinetics resulting in ineffective therapy.[6] Addressing this problem, we attempted to formulate extended-release tablets of cephalexin, which can provide a constant effective drug level for six hours, based on calculations considering pharmacokinetic parameters.
MATERIALS AND METHODS
Materials
The chemicals used in the experiment are: Cephalexin monohydrate (Aurobindo Pharma Ltd., India), microcrystalline cellulose PH102 (Weiming Industries, China), lactose anhydrous (DMV- Fonterra Excipients, Germany), HPMC 15cps, HPMC K4M, HPMC K15M, HPMC K100M (Feicheng Ruitai Fine chemicals, China), colloidal silicon dioxide (Degussa India Pvt. Ltd., India), magnesium stearate (Ferro Corporation, USA), Instacoat universal (Ideal Curves Pvt. Ltd., India), Sporidex AF 375 (Ranbaxy laboratories Pvt. Ltd., India). All the other reagents used were of analytical grade.
Methods
Preformulation studies
Preformulation investigations are done to characterize properties of raw materials including their physico-chemical, biopharmaceutical, and mechanical properties, as well as compatibility.
Physical drug excipient compatibility studies
The physical compatibility studies were coupled with the stability studies at higher temperature and humidity conditions. The drug excipient compatibility study protocol included the preparation of homogenous physical mixture in 1:1 ratio of drug and all possible excipients to be used in the formulation. The physical mixtures were sealed into 15-ml USP Type III flint glass vials and stored in a stability chamber (Servewell instruments Pvt. Ltd., Bengaluru, India) for 30 days at 40°C temperature and 75% relative humidity conditions. The initial state of the mixtures was noted and further evaluation for the possible occurrence of any interactions like physical or chemical changes, was performed after the 15th and 30th day.[7,8]
Differential scanning calorimetry
The DSC thermograms of pure drug, polymer and formulation were generated and investigated for presence of additional peaks or absence of peaks indicating possible polymer interactions or phase transformations. The thermal peaks give the melting points of the samples which can be used as a test for purity analysis and also for sample characterization by comparing with the standard melting points reported for corresponding samples.[7–9]
Drug characterization using FTIR spectroscopy
The authenticity of the drug cephalexin monohydrate was confirmed by comparing the absorption maxima with that of cephalexin monohydrate reference standard. The KBr pellet method was used to generate the IR spectrum (Shimadzu Corporation, Japan) of the raw material and the reference standard and spectrum was compared for identification and purity analysis of the sample. The overlaid spectrum confirms the authenticity of the raw material and is provided in Figure 1.[7,9]
Figure 1.
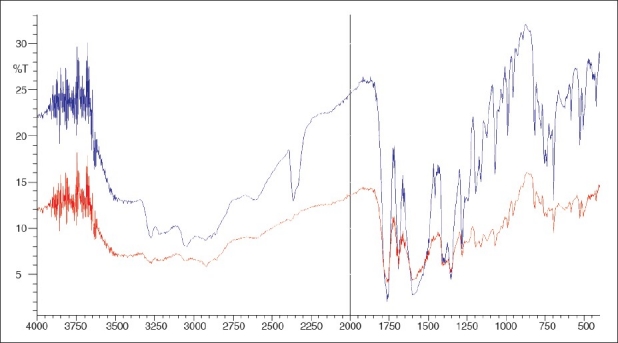
FTIR spectrum combined of cephalexin sample and cephalexin RS. The red spectrum and blue spectrum are of cephalexin RS and cephalexin sample respectively
Particle size analysis of drug
Active pharmaceutical ingredient (API) [cephalexin monohydrate] was analyzed for particle size distribution by means of sieving method using mechanical sieve shaker (Verder RETSCH Trading co. Ltd, China). A series of standard sieves namely 30#, 40#, 60#, 80# and 100# were stacked one above the other so that sieves with larger pore size (less sieve number) occupied top position followed by sieves of decreasing pore size (larger sieve number) towards the bottom. Weighed quantity of API was placed in sieve no. 40. Sieve shaker was set for 5 min at amplitude of 60. Remove the set up from the sieve shaker after 5 min and weigh the API retained in each mesh individually. The percentage retained in each sieve was calculated with the following formula.[9]
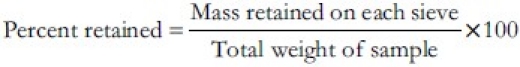
Determination of dosage for cephalexin extended-release tablets
Determination of the total dose required for a sustained release dosage form, requires the addition of the amount of drug needed to achieve the desired blood level quickly (the immediately available portion) to the sustaining portion.
DT = DI* + DM
Where,
DT, DI* and DM are total dose of drug required, corrected initial dose and maintenance dose/sustained dose respectively.
The sustaining portion is determined by multiplying the zero-order rate constant for sustained drug delivery, KR0, by the desired sustaining time, h. The desired zero order release rate is determined by the equation below;
KR0 = KE * Vd * DTC
Where,
KR0, KE, Vd and DTC are zero-order rate constant, first order elimination rate constant, volume of distribution and desired therapeutic concentration respectively. KE can be determined by the equation;
KE = 0.693/ t½
The maintenance dose is calculated using the equation below
DM = KR0 * T
Where ‘T’ is desired duration of extended release. Amount of drug released from maintenance dose during release of initial dose till peak plasma concentration (cmax) is calculated using the equation given below;
W = KR0 * Tmax
Where,
W and Tmax are amount of drug released from maintenance dose during release of initial dose till peak plasma concentration Cmax and time to reach Cmax respectively.
The corrected initial dose is calculated using the equation given below;[10]
DI* = DI – W
Where DI is the initial dose.
Determination of amount of cephalexin to be used in a tablet
Cephalexin is available as a hydrous molecule cephalexin monohydrate. Labeled claims are to be expressed in terms of the equivalent amount of anhydrous cephalexin present in the dosage form. The quality control department analyses the raw material and determines the assay value and percentage of water present in it. These variables can be used to determine the conversion factor, a term which decides the actual amount of cephalexin monohydrate to be used in order to meet the labeled claim specification of cephalexin present on anhydrous basis. The total amount of cephalexin to be used in the formulation can be calculated using the following formula:
Required dose = Labeled Claim × Conversion factror

Formulation of cephalexin matrix tablets using direct compression method
Manufacture of matrix tablets by direct compression method involves mainly three steps.
Sifting: Accurately weighed quantity of drug and excipients were passed through sieve no. 20 and 40 respectively.
Blending: Drug and excipients (excluding lubricant) were added in geometric proportions and mixed thoroughly for 15 min.
Compression: The blend was mixed slightly with lubricant and finally compressed in 15 * 8.5 mm punches.
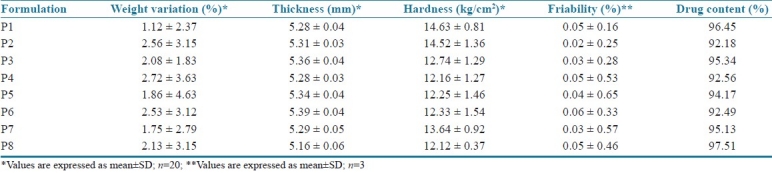
Total of eight batches (P1 to P8) were prepared and the formulae used are represented in Table 1. The pre-compression parameters of cephalexin granules like angle of repose, bulk density, tapped density, Carr's index and Hausner's ratio are summarized in Table 2. The prepared cephalexin matrix tablets were evaluated for thickness, hardness, friability, uniformity of weight and drug content. The thicknesses of tablets were measured by vernier caliper (Aerospace, China). Hardness of tablets was tested using a validated modified dial type hardness tester (Shivani Scientific Industries, India). Friability of tablets was determined by using Roche Friabilator (Electrolabs, India). The drug content of each batch was determined as per USP assay of cephalexin using HPLC (SCL-10AVP, Shimadzu Corporation, Japan).[10,11] The tablet evaluation results are summarized in Table 3.
Table 1.
Composition of batches P1 to P8
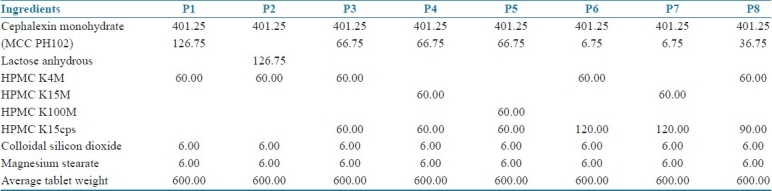
Table 2.
Precompression parameters of batches P1 to P8
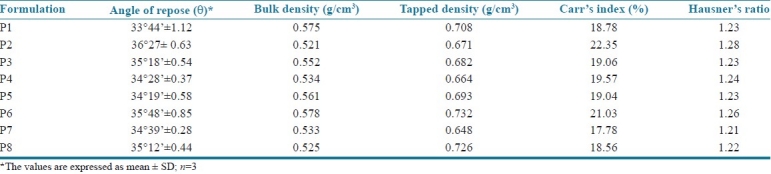
Table 3.
Post-compression parameters of batches P1 to P8
In vitro drug release study
Preparation of buffers and reagents
Preparation of 0.01N HCl: 8.5 ml of concentrated hydrochloric acid was accurately measured and diluted to 10 liter with demineralized water to obtain 0.01N HCl.[9]
Preparation of 0.2M mono basic potassium phosphate: About 27.22 g of mono basic potassium phosphate (KH2PO4) was dissolved and diluted to 1000 ml with demineralized water.[9]
Preparation of 0.2M sodium hydroxide solution: About 8.0 g of sodium hydroxide (NaOH) was dissolved and diluted to 1000 ml with demineralized water.[9]
Preparation of phosphate buffer (pH=6.8): 250 ml of mono basic potassium phosphate (KH2PO4) was placed in a 1000 ml volumetric flask, 112 ml of 0.2M NaOH was added and volume was made up to 1000 ml with demineralized water and pH adjusted to 6.8 using dilute NaOH solution.[9]
Procedure of dissolution study: The in vitro dissolution studies of cephalexin extended-release tablets were performed using USP Type I dissolution apparatus (basket type) (Electrolabs, India). The dissolution medium consisted of 900 ml of 0.01N HCl (1st h) and phosphate buffer (pH=6.8) during the rest of the study period maintained at 37±0.2°C. The speed of the paddle was set at 100 rpm. Aliquot of samples (5 ml) were withdrawn at specific time intervals and the same amount of buffer was replaced into the dissolution bowl to maintain the sink conditions.[9]
Preparation of cephalexin standard stock solution (200 μg/ml): A standard stock solution of cephalexin monohydrate was prepared by dissolving accurately weighed 20 mg of cephalexin reference standard in corresponding medium (0.01N HCl and phosphate buffer) and made up to volume in a 100 ml standard flask. Ten ml of the above solution was again diluted to 100 ml in a standard flask to get a solution of concentration 20 μg/ml which is used as the cephalexin working standard.[9]
Estimation of drug release using single external standard method: Five ml of sample withdrawn was diluted with corresponding medium and made up to volume in a 100 ml standard flask. The above solution was sonicated for 15 min and the absorbance was measured at 262 nm. The amount of cephalexin release was determined in a UV-1650 spectrophotometer (Shimadzu corp., Japan) at the wavelength of maximum absorbance 262 nm using the formula below:

The in vitro drug release specification was 20-40% at 1st h, 40-60% at 2nd h, 60-80% at 4th h and > 80% at 6th h determined from theoretical drug release and dissolution pattern of the marketed product. The in vitro drug release data is summarized in Table 4 and graphically represented in Figure 2.
Table 4.
Cumulative percentage drug release of P1 to P8
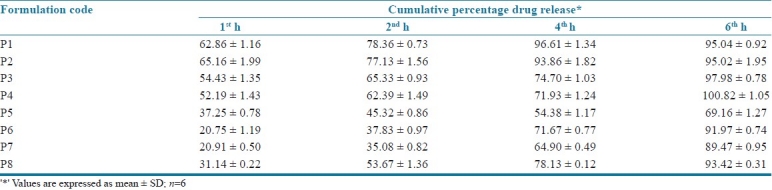
Figure 2.
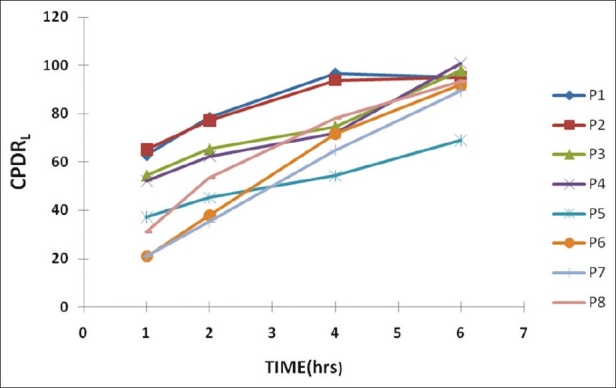
Cumulative percentage drug release of P1 to P8 (graphical representation)
Estimation of drug content using high-performance liquid chromatography
The assay of cephalexin by high-performance liquid chromatography (HPLC) was performed as per USP procedure. The procedure involves preparation of mobile phase, internal standard solution, standard and sample.
Preparation of mobile phase
1015 ml of a suitable mixture of water:acetonitrile:methanol: triethylamine (850:100:50:15) was prepared. One gram sodium 1-pentanesulfonate was dissolved in this mixture; adjusted with phosphoric acid to a pH of 3.0 ± 0.1 and degassed.[12]
Preparation of standard
Dissolved an accurately weighed quantity 20 mg of USP cephalexin reference standard with water in a 100 ml standard flask. The solution was sonicated and filtered to obtain the standard solution.
Assay preparation and procedure
Twenty tablets were weighed and finely powdered. An accurately weighed quantity of powder equivalent to 20 mg of cephalexin was transferred to a 100 ml of volumetric flask; water was added to the volume and mixed. The solution was sonicated and filtered to obtain a clear solution. Equal volumes (20 μl) of standard (five times) and assay samples (two times) were injected separately into the chromatographs and corresponding chromatograms were recorded and the responses for major peaks were noted. The program gives the mean standard and sample peak areas, which when substituted in the formula below gives the %w/w of cephalexin present in the sample assayed.[12] The drug content values are indicated in Table 3:

Comparison of dissolution profiles
The similarity in the drug release pattern of the marketed product and the formulation developed was determined by calculating the similarity factor (f2). The two products are said to be similar if the value of f2 lies between 50 and 100. The similarity factor and a similarity testing have been recommended for dissolution profile comparison in the Food and Drug Administration (FDA)'s Guidance for Industry. f2 is given by the formula given below:
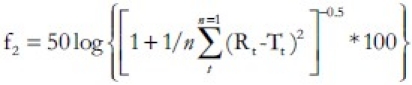
Where,
Rt = percentage of reference product dissolved at a specified time
Tt = percentage of test product dissolved at a specified time.
n = no: of sampling points.
The similarity factors of batches P1 to P8 are shown in Table 5. The formulation P8 was found to have a similarity factor of 77.75 and was chosen to be the scale up batch formulae. The drug release pattern of optimized product P8 and the theoretical release pattern were compared to determine the similarity represented by Figure 3. The graph shows good correlation between the dissolution profiles of marketed product, F3 and the theoretical release pattern.[13]
Table 5.
Similarity factor of formulation P1 to P8

Figure 3.
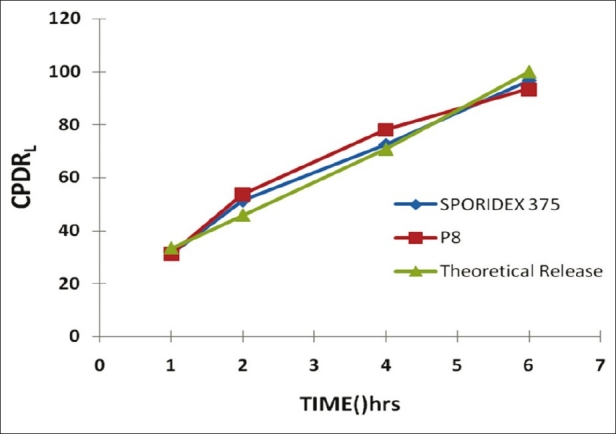
Comparative dissolution profiles of marketed product (Sporidex 375), P8 and theoretical release profile.
Kinetic modeling of in vitro drug release
To study the release kinetics, the data obtained from in vitro drug release studies of optimized formulation P8 was plotted in various kinetic models:
Zero order rate kinetics: Cumulative percentage of drug released vs. time
First order rate kinetics: Log cumulative percentage of drug remaining vs. time
Higuchi model : Cumulative percentage of drug released vs. square root of time
Korsmeyer Peppas model: Log cumulative percentage of drug released vs. log time
The plots were drawn using Microsoft Excel 2007 and the regression equations were obtained for each plot. The linearity of the plots was obtained from the value of regression coefficient (R). The model with the highest linearity (R value approaches unity) was chosen as the best-fit kinetic model. The Korsmeyer Peppas model is used to study the mechanism of drug and the slope of the plots gives the diffusional exponent ‘n’. When n<0.5, the drug diffuses through the polymeric matrix by a Fickian (Case I) diffusion mechanism. For 0.5<n<1, an anomalous (non-Fickian) mechanism occurs; n=1 indicates a zero-order (Case II) and n>1 indicates non-Fickian super Case II release mechanism.[13,14]
Film coating of tablets
Coating specifications
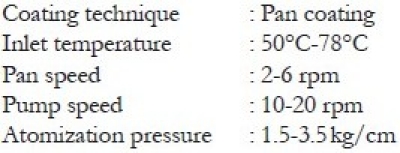
Preparation of coating solution
The compressed tablets were film-coated with Instacoat universal by using conventional coating pan (Jyothi Industries, Bengaluru). The amount of Instacoat required is about 3% of the weight of the tablet bulk to be coated and this amount forms 5% of the total reconstituted coating solution. The solvent system for reconstitution consists of IPA:MC in the ratio 35:65. The coating solution was prepared by dispersing specified quantity of Instacoat in isopropyl alcohol and stirred for 5 min. Then accurate quantity of methylene chloride was added and stirring was continued until a clear dispersion was formed. The targeted weight gain of the tablets after coating was fixed to be 2.5%.[10]
Fourier transform infra red study
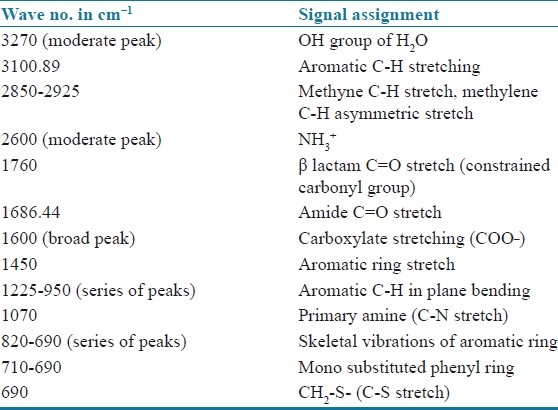
FTIR studies were used to study the interaction between the drug and excipients. The scanning was done from 4000 cm-1 to 400 cm-1. The spectral data of cephalexin standard and formulation P8 was interpreted in detail and the overlaid spectrum [Figure 4] shows similar peaks and the signal assignment is provided in Table 6 to give substantial evidence in support of chemical and physical compatibility between the drug and excipients used in the formulation.[9]
Figure 4.
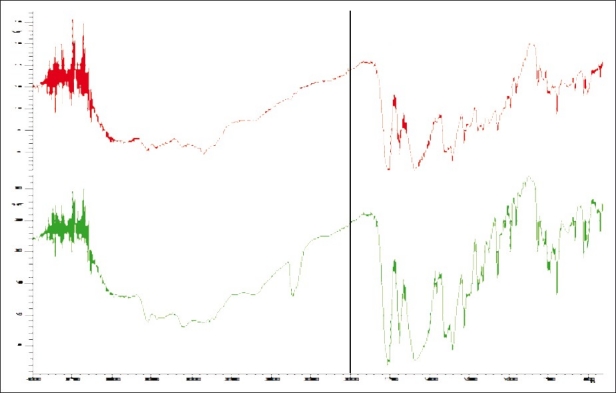
Combined Spectrum of cephalexin RS and P8. The red spectrum and green spectrum are of cephalexin RS and cephalexin ER 375 (P8) respectively
Table 6.
Spectral interpretation of cephalexin RS and P8
Accelerated stability studies
Short-term accelerated stability studies for a period of three months according to International Conference on Harmonization guidelines were performed on the optimized tablet formulations of blister-packed cephalexin extended-release tablets. The tablets were subjected to stability studies at 40°C/75%RH in a stability chamber for a period of three months. Initial evaluation of the tablets was done and at the end of first, second and third month the tablets were again analyzed for physical appearance, water content and in vitro drug release profile.[15]
RESULTS AND DISCUSSION
Physical drug excipient compatibility studies
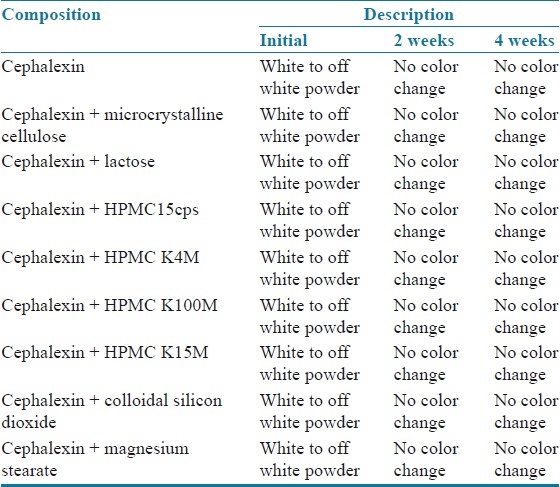
From the drug excipients’ compatibility study report shown in Table 7, it was observed that there were no incompatibilities or interaction between drug and excipients. Based on the physical compatibility result, the excipients were chosen for the formulation development.
Table 7.
Drug excipient compatibility report
Differential scanning calorimetry
The drug shows an exothermic peak at 190.72°C [Figure 5] and polymer HPMC 15cps shows a broad endothermic transition peak at 81.33°C [Figure 6]. The DSC thermogram of formulation P8 [Figure 7] shows an exothermic peak at 188.95°C (drug) and endothermic peak at 92.19°C (polymer) thus retaining the peak of drug and the polymer. The DSC thermograms of pure drug, polymer and formulation (P8) reveal that polymer interactions or phase transformations have not occurred and the drug and excipients are chemically compatible with each other.
Figure 5.
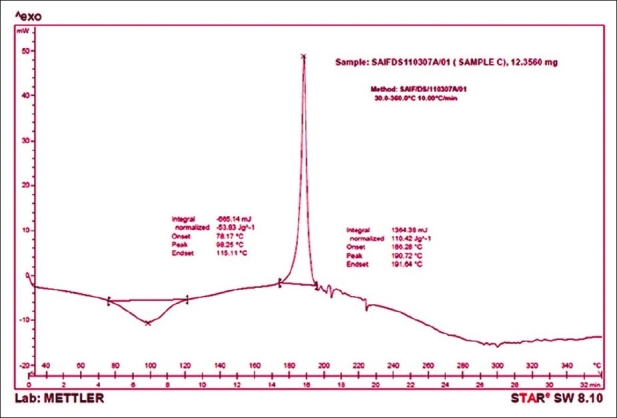
DSC thermogram of cephalexin
Figure 6.
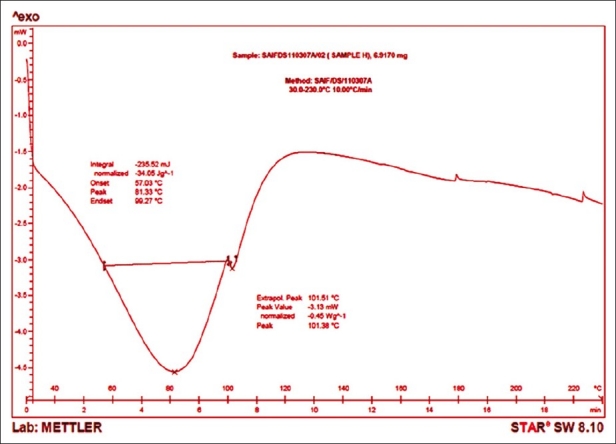
DSC thermogram of HPMC 15cps
Figure 7.
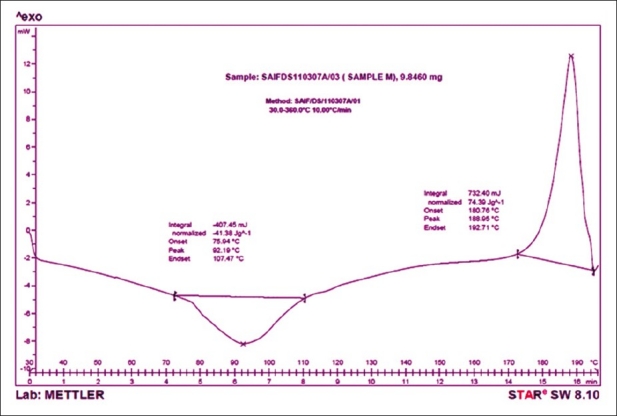
DSC thermogram of P8
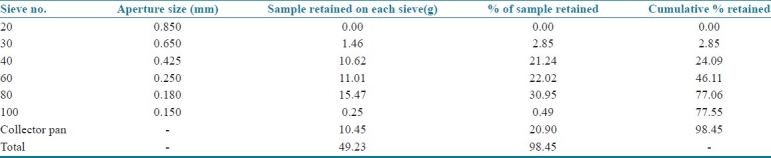
Particle size analysis of drug
Initial weight of powder: 50 g
Final weight of powder: 49.23 g
From the sieve analysis report shown in Table 8, it was concluded that 78% of the drug particles had a size less than 150 μm and as no powder was retained on sieve no. 20, it was found suitable for sifting of drug during the formulation development.
Table 8.
Particle size distribution of cephalexin
Determination of dosage for cephalexin extended-release tablets
The plasma half life (t1/2) of cephalexin was reported to be around 1 h and the volume of distribution (Vd) was almost 15 liters. For 125 mg dose the desired therapeutic concentration obtained was 4.5 mg/l. The drug release was sustained for duration of 6 h. Thus the desired zero order release rate is;
KR0 = 0.693 * 15 * 4.5
= 46.7 mg/h
The maintenance dose was calculated and is;
DM = 46.7* 6
= 280.7 mg
The Tmax of cephalexin was reported to be 1 h. Amount of drug released from maintenance dose during release of initial dose till peak plasma concentration (cmax) was calculated to be:
W = 46.7 * 1
= 46.7 mg
The corrected initial dose was calculated to be:
DI* = 125 – 46.7
= 78.3 mg
So the total dose becomes;
DT = 78.3 + 280.7
= 359 mg
The corrected dose of cephalexin taken was 375 mg for the preparation of matrix tablets.
Determination of amount of cephalexin to be used in a tablet
The quantity of cephalexin monohydrate to be used in the formulation of a tablet containing 375 mg of cephalexin anhydrous was calculated from the assay value and the water content determined by the Karl-Fischer method.
Assay value = 100.36%
%Water content = 6.55%
Correction factor=1.07; the required amount of cephalexin monohydrate was calculated using Equation 8 and was found to be 401.25 mg.
Kinetic modeling of in vitro drug release
The kinetic study reveals that the cephalexin extended-release tablets follow mixed order kinetics as the regression coefficients approach unity for first and zero orders. The n values from the Korsemeyer Peppas model show that the drug release pattern follows mainly the Fickian diffusion mechanism as the n value is below 0.5. The Higuchi plots also show good linearity indicating that the drug release is proportional to the square root of time and the drug release is at a slower rate as the distance of diffusion increases. The results are summarized in Table 9.
Table 9.
Kinetic study report of P8

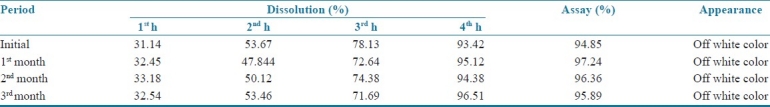
Accelerated stability studies report
The accelerated stability studies for three months as per ICH guidelines reveal that the formulation (P8) has not undergone any physical or chemical degradation during the period. There are no significant differences in the in vitro drug release and the drug content of the optimized formulation [Table 10].
Table 10.
Accelerated stability studies report of cephalexin ER 375 (P8)
CONCLUSION
The present research work was successful in improving the efficacy of cephalexin oral therapy as the drug release was extended for six hours thus reducing dosing frequency thereby improving patient compliance. The saw tooth kinetics of conventional therapy was completely avoided by the development of a new formulation. The study also revealed the applicability of different grades of hydroxypropylmethylcellulose as rate-controlling polymers in matrix tablets. The mixed polymer combination of grades HPMC K4M and HPMC 15cps was found to sustain the release of cephalexin for six hours and also met the release specifications. The higher viscosity grades of HPMC were not found suitable for small duration of sustained drug release t. The in vitro drug release profile of the newly developed tablets also shows good similarity with that of the innovator's product and ideal controlled release pattern. There were no formulation problems associated with the optimized batch (P8) of cephalexin matrix tablets. The tablet also passed the short-term accelerated stability studies indicating the physical and chemical stability of the product.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Pogula M, Nazeer S. Extended release formulation. Int J Pharm Tech. 2010;2:625–84. [Google Scholar]
- 2.Fyhr P, Downie K. Extended release drug delivery technology. Innov Pharm Tech [INTERNET] 2003. [Last cited on 2010 May 5]. pp. 80–6. Available from: http://www.iptonline.com .
- 3.Sweetman SC. 36th ed. Vol. 1. London, Chigago: Pharmaceutical Press; c2009. Martindale: The Complete Drug Reference; pp. 218–9. [Google Scholar]
- 4.San Fransisco, New York: Healthline Networks Inc; 1999. [Last cited on 2011 Jan 14]. Healthline: Connect to better health. Available from: http://www.healthline.com/ahfscontent/cephalexin . [Google Scholar]
- 5.California: RxList Inc; 1995. [Last cited on 2011 Jan 14]. RxList: The Internet Drug Index. Available from: http://www.rxlist.com/keflex-drug.htm . [Google Scholar]
- 6.Shin SC, Cho SJ. Kinetics of cephalexin release from Eudragit-Hydroxypropylcellulose membranes. Drug Dev Ind Pharm. 1996;22:299–305. [Google Scholar]
- 7.Qui Y, Chen Y, Zhang GG. Burlington, USA: Elsevier Inc; c2009. Developing solid oral dosage forms: Pharmaceutical theory and practice; pp. 125–45. [Google Scholar]
- 8.Moreton RC. Excipient interactions. In: Katdare A, Chaubal MV, editors. Excipient development for pharmaceutical, biotechnology, and drug delivery systems. New York: Informa Healthcare USA, Inc; c2006. pp. 101–4. [Google Scholar]
- 9.Niazi SK. New York: Informa Healthcare USA, Inc; c2007. Handbook of Preformulation; p. 56. (73, 251). [Google Scholar]
- 10.Chang RK, Robinson JR. Sustained drug release from tablets and particles through coating Sustained release dosage form. In: Lieberman HA, Lachman L, Schwartcz JB, editors. Theory and practice of industrial pharmacy. 2nd ed. Vol. 3. New York: Marcel Dekker, Inc; c1990. p. 200. (203-5, 235-8). [Google Scholar]
- 11.Shangraw RS. Compresssed tablets by direct compression. In: Lieberman HA, Lachman L, Schwartcz JB, editors. Theory and practice of industrial pharmacy. 2nd ed. Vol. 1. New York: Marcel Dekker, Inc; c1989. pp. 198–203. [Google Scholar]
- 12.Rockville, MD. USA: United States Pharmacopoeial Convention Inc; 2007. USP 30 NF 25 2007; p. 1689. [Google Scholar]
- 13.Costa P, Lobo JM. Modeling and comparison of dissolution profiles. Eur J Pharm. 2001;13:123–33. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- 14.Grassi M, Grassi M. Mathematical modelling and controlled drug delivery: Matrix systems. Curr Drug Deliv. 2005;2:97–116. doi: 10.2174/1567201052772906. [DOI] [PubMed] [Google Scholar]
- 15.Yoshioka S, Stella VJ. New York: Kluwer Academic Publishers; c2002. Stability of Drugs and Dosage Forms; pp. 205–13. [Google Scholar]