

Abstract
Objective:
Schisandrin B (Sch B) is the most abundant, active dibenzocyclooctadiene derivative isolated from the fruit of Schisandra chinensis (Turcz) Baillon (Schisandraceae). (–)Sch B was found to be the most potent stereoisomer of Sch B in producing cytoprotective action in H9c2 cardiomyocytes. The elucidation of biochemical mechanism underlying the cytoprotection of (–)Sch B has attracted much interest in the area of preventive medicine. Here, we examined whether the (–)Sch B-induced enhancement of glutathione antioxidant and heat shock responses and the associated cytoprotection against hypoxia/reoxygenation-induced apoptosis are mediated by reactive oxygen species (ROS) arising from cytochrome P-450 (CYP)-catalyzed metabolism of (–)Sch B in H9c2 cardiomyocytes.
Materials and Methods:
The effects of CYP inhibitor (1-aminobenzotriazole, ABT) and antioxidant (dimethylthiouracil, DMTU) on (–)Sch B-induced ROS production and associated increases in cellular-reduced glutathione (GSH) level as well as heat shock protein (Hsp) 25/70 production were investigated in H9c2 cardiomyocytes. The (–)Sch B-induced ROS generation was monitored with or without ABT/DMTU for 6 h in situ, while (–)Sch B-induced cellular GSH level and Hsp 25/70 production, as well as cytoprotection were measured at 16 h post-(–)Sch B exposure.
Results:
The results indicated that (–)Sch B caused a dose-dependent increase in ROS production in H9c2 cardiomyocytes, which was completely suppressed by pre- and co-treatment with ABT or DTMU. The incubation with (–)Sch B for 6 h caused dose-dependent increases in cellular GSH level and Hsp 25/70 production, as well as protection against hypoxia/reoxygenation-induced apoptosis at 16-h post-drug exposure in H9c2 cardiomyocytes. All these cellular responses were abrogated by treatment with ABT or DMTU.
Conclusion:
The results suggest that ROS arising from the CYP-catalyzed metabolism of (–)Sch B elicit glutathione antioxidant and heat shock responses, thereby protecting against oxidant-induced apoptosis in H9c2 cardiomyocytes.
KEY WORDS: Cytochrome P-450, glutathione, heat shock protein, reactive oxygen species, Schisandrin B
Introduction
Schisandrin B (Sch B) is the most abundant, active dibenzocyclooctadiene derivative isolated from the fruit of Schisandra chinensis (Turcz) Baillon (Schisandraceae), a traditional Chinese herb commonly used as astringent and clinically used for the treatment of viral and chemical hepatitis.[1] Early studies in our laboratory have demonstrated the ability of Sch B to protect against myocardial ischemia/reperfusion (I/R) injury.[2,3] The cardioprotection afforded by Sch B pretreatment was associated with the enhancement in tissue glutathione antioxidant status, particularly in the mitochondrion.[2–4] Recent studies also showed that heat shock protein (Hsp)25 and Hsp70 expression contributes at least partly to the cardioprotection afforded by Sch B pretreatment against I/R injury.[5] In regard to the action on mitochondria, Sch B treatment was found to decrease the sensitivity of Ca2+-induced mitochondrial permeability transition and mitochondrial membrane potential in hypoxia/reoxygenation-challenged H9c2 cardiomyocytes.[6] However, the biochemical mechanism(s) underlying the Sch B-induced glutathione antioxidant and/or heat shock responses in cardiomyocytes remains to be established. In this connection, it has been suggested that reactive oxygen species (ROS) generated from cytochrome P-450 (CYP)-catalyzed reaction with Sch B may trigger the glutathione antioxidant responses in rat hearts.[7]
The Sch B preparation used in our previous animal and cell studies was found to (±)γ-schisandrin [(±)g-Sch] and (–)schisandrin B [(–)Sch B] in a ratio of 2:8 (w/w).[7,8] (–)Sch B [Figure 1] was found to be more potent than (±)γ-Sch in eliciting the glutathione antioxidant response and protecting against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes.[6] To define the role of CYP-mediated ROS generation in the cytoprotective action of (–)Sch B, we here examined the effects of 1-aminobenzotriazole (ABT, a broad spectrum inhibitor of CYP) and antioxidant (dimethylthiouracil, DMTU) on (–)Sch B-induced ROS production and the subsequent glutathione antioxidant and heat shock responses in H9c2 cardiomyocytes. The effects of ABT and DMTU pretreatment on (–)Sch B-induced protection against hypoxia/reoxygenation-induced apoptosis were also investigated.
Figure 1.
Chemical structure of (–)schisandrin B
Materials and Methods
Cell Culture Materials and Chemicals
H9c2 cell line was purchased from ATTC (Rockville, MD). Cell culture medium and fetal bovine serum (FBS) were obtained from GIBCO BRL Life Technologies (Grand Island, NY). Reduced glutathione (GSH), oxidized glutathione (GSSG), glutathione reductase (GR), ABT and DMTU were purchased from Sigma (St. Louis, MO). 2’,7’-dichlorofluorescein diacetate (DCFDA) was purchased from Fluka (Buchs, Switzerland). (–)Sch B was prepared as previously described.[6] All other chemicals were of analytical grade.
Cell Culture
H9c2 cells, cultured as monolayers in Dulbecco's modified Eagle's medium (DMEM) (GIBCO BRL), is a permanent cell line derived from cardiac myoblast,[8] which was supplemented with 10% (v/v) FBS. The medium contained glucose (4.5 g/L) and glutamine (4.5 mM), supplemented with NaHCO3 (17 mM), penicillin (100 IU/mL), and streptomycin (100 μg/mL). The cells were grown under an atmosphere of 5% (v/v) CO2 in air at 37°C. The medium was replaced by fresh medium every 2 or 3 days. A stock of cells was grown in a 75 cm2 culture flask and split before confluence at a subcultivation ratio of 1:10. Cells used for experiments were seeded at a density of 3.75 × 104 cells/well in a 12-well culture plate, and cells in each well were allowed to grow to achieve 60-80% confluence within 24 h prior to drug treatment.
Drug Treatment
To investigate the effects of CYP inhibitor/antioxidants on (–)Sch B-induced ROS production, cells were pre-incubated with ABT (10 mM final concentration, for 2 h) or DMTU (20 mM, 1 h)) prior to the addition of (–)Sch B at 7.5 or 15 μM. Vehicle [phosphate-buffered saline (PBS)] was added for the respective control. Then cells were coincubated with ABT/DMTU and (–)Sch B, and the ROS generation was monitored in situ for 6 h at 37°C.
To investigate the effects of (–)Sch B on cellular GSH and Hsp 25/70 levels, cells were incubated with (–)Sch B at 7.5 or 15 μM for 6 h. Then the drug-containing medium was removed, and the cells were cultured in fresh medium (without drug) for 16 h. Preliminary time-course studies indicated that 6 h of drug exposure followed by 16 h of drug-free culture caused maximum increases in cellular GSH and Hsp 25/70 levels in H9c2 cells. Exposure of cells to (–)Sch B at concentrations higher than 15 μM produced cytotoxic effect.
Measurement of ROS Generation
Cells (1×105) were seeded on a 96-well black multi-titer plate with clear bottom, and they were allowed to grow overnight. The cells were washed twice with PBS and then incubated with 0.1% BSA in PBS containing 5 μM (final concentration) DCFDA (dissolved in 0.2% (v/v) ethanol) for 30 min at 37°C. The cells were washed twice with PBS again and then incubated with fresh medium containing 500 μM NADPH (final concentration), 7.5 or 15 μM (–)Sch B, with or without ABT or DMTU pre- and co-treatment, as described above. DMTU was used for scavenging ROS generated in the cells. The fluorescence intensity of each well was measured at an excitation wavelength of 485 nm and an emission wavelength of 530 nm by Victor V2 Multi-label Counter (Perkin Elmer, Turku, Finland) every minute for 6 h at 37°C. The fluorescence intensity of drug-treated cells was normalized with reference to the respective time-matched vehicle (i.e., DMSO) control. The extent of drug-induced ROS production was estimated by computing the area under the curve (AUC) plotting normalized fluorescence intensity against time (min), and expressed in arbitrary unit.
Measurement of Cellular GSH Level
For the measurement of cellular GSH level, cells were seeded at a density of 3.75 × 104 cells/well in a 12-well culture plate, and they were allowed to grow overnight. After the drug treatment, cells were centrifuged at ×540g for 15 min at 4°C. The cells were washed once with PBS. An aliquot of 100 μL 3% (v/v) 5-sulfosalicyclic acid (SSA) were added to cells. After shaking for 10 min at 4°C, the mixtures were then centrifuged at ×540g for 15 min at 4°C. The supernatant was used for GSH measurement. GSH levels were determined enzymatically using 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB) and GR, in a protocol modified from Griffith.[9] To measure GSSG levels, aliquots (50 μL) of SSA supernatants were mixed with 5 μL of 20% (w/v) 2-vinylpyridine and 5 μL 60% (v/v) triethanolamine in microcentrifuge tubes. Each tube was allowed to stand at room temperature for 1 h. A reaction mixture containing 0.63 mM DTNB and 0.053 mM NADPH in phosphate buffer (0.1 M, with 5 mM Na2EDTA; pH 7.5) was pre-incubated at 30°C for 5 min. An aliquot (30 μL) of SSA sample supernatant (total glutathione) or an aliquot (30 mL) of GSSG sample was added to a well of a 96-well microtiter plate, and 180 μL of prewarmed reaction mixture, containing 0.525 U/mL GR, was next added. Absorbance changes at 412 nm were monitored spectrophotometrically for 5 min. The concentrations of total glutathione and GSSG were estimated from calibration curves using GSH and GSSG [dissolved in 3% (w/v) SSA) as standards, and expressed as nmol/mg protein. GSH levels were estimated by subtracting twice the amount of GSSG from that of total glutathione.
Measurement of Cellular Hsp 25 and Hsp 70 Levels
Cells (2.5 × 105 cells/well) were seeded on 6-well plate, and they were allowed to grow overnight. After drug treatment, cells were washed once with PBS. The cells were then incubated with 300 μL of SDS-lysis buffer (20 mM Tris-HCl, 2 mM EDTA, 3 mM EGTA, 1% Triton X-100, 10% glycerol, 2 mM dithiothreitol, 5 μg/mL leupeptin, 5 μg/mL aproptinin, 5 μg/mL pepstain, 1 μM phenylmethylsulphonyl fluoride, pH 7.5) for 20 min at 4°C on a shaker. The cell lysates were further processed by passing through a 26G ½ syringe for 10 times, and the protein concentrations in cell lysates were determined by using the DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Hsp 25 and Hsp 70 levels were estimated by Western blot analysis using specific antibodies (anti-Hsp 25, catalog # SPA-801; anti-Hsp 70, catalog # SPA-812) from StressGen (Vancouver, BC, Canada) following SDS-PAGE analysis of the cell lysates, using a separating gel of 10% (w/v) acrylamide.[10] Hsp 25/27 and Hsp 70 (human recombinant proteins from StressGen) and actin (prepared from bovine muscle, Sigma) were used as markers. The immuno-stained protein bands were revealed by an enhanced chemiluminescence reaction (Amersham ECL+). All immuno-blots were analyzed by densitometry, and the amounts (arbitrary units) of Hsp were normalized with reference to actin levels (arbitrary units) in the sample.
In Vitro Hypoxia/Reoxygenation Challenge
Cells used for this experiment were seeded at 2.5 × 105 cells on a 40 mm2 culture plate, and they were allowed to grow overnight. After drug treatment, the cells were washed twice with Krebs-Ringer Bicarbonate buffer (KRB, 115 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24 mM NaHCO3, 10 mM HEPES, pH 7.4). Aliquots (2.5 mL) of KRB supplemented with 0.01% bovine serum albumin were added to the cells immediately prior to the hypoxia. A Billups-Rothenberg modular incubator chamber (Billups-Rothenberg, Inc., Del Mar, CA) was used to produce an in vitro hypoxia/reoxygenation challenge. In essence, cells were placed in the sealed chamber, and the chamber was flushed with nitrogen for 15 min at flow rate of 20 mL/min. After closing all sealable connectors, the chamber was transferred to an incubator and the cells in the chamber were subjected to a 2-h period of hypoxia at 37°C. Reoxygenation was initiated by opening the chamber and then replacing the KRB with fresh DMEM medium. The cells were then cultured in the incubator under an atmosphere of 5% CO2 in air at 37°C for 16 h. Preliminary time course studies showed that a 2-h period of hypoxia followed by 16 h of reoxygenation was optimal for achieving a 1-2-fold increase in apoptotic cell death in H9c2 cells.
Apoptosis Assay
Apoptotic cell death was measured by Annexin V-FITC-Fluos (Roche, Penzberg, Germany) and propidium iodide (PI) (Sigma, St. Louis, MO) stainings, in which Annexin V bound to the exposed phosphatidylserine on the plasma membrane of the apoptotic cells.[11] Briefly, cells were rinsed with ice-cold PBS and then resuspended in 100 μL of binding buffer containing 140 mM NaCl, 2.5 mM CaCl2, 1.5 mM MgCl2 and 10 mM HEPES, pH 7.4. One-hundred microliters of Annexin V staining solution (1 μL of Annexin V stock solution being mixed with 100 μL of binding buffer) was added to the cells and incubated in the dark for 15 min at room temperature. The cells were washed with ice-cold PBS and resuspended in 600 μL of binding buffer. Immediately after mixing with 3 μL PI solution (50 μg/mL), the cells were analyzed by flow cytometry using a COULTER® EPICS® XL™ Flow Cytometer (Beckman Coulter, Inc., Fullerton, CA). The fluorescence of Annexin V and PI were monitored at emission wavelengths of 525 nm and 575 nm, respectively, with the excitation wavelength being at 488 nm for both probes. Approximately 10,000 cells were counted in each of the samples and data were statistically analyzed using an EXPO32 ADC analysis program. The percentage of early apoptotic cells was estimated by noting the Annexin V positive but PI negative cells, while the percentage of late apoptotic cells was estimated by noting the number of cells which were both Annexin V and PI positive. The total apoptotic cell death was calculated by summing up the percentages of early apoptosis and late apoptosis.
Statistical Analysis
Data were analyzed by one-way Analysis of Variance (ANOVA). Post-hoc multiple comparisons were performed using Least Significant Difference (LSD) test. P values <0.05 were regarded as statistically significant.
Results
(–)Sch B caused a time- (0-360 min) and dose- (7.5 and 15 μM) dependent increase in ROS production in H9c2 cardiomyocytes under the present experimental condition [Figure 2]. When the amount of ROS production was estimated by computing the area under the curve plotting the percent increase in fluorescence (with respect to the time-matched vehicle control) against time (up to 360 min), (–)Sch B at 7.5 and 15 μM were found to increase the ROS production by 24 and 32% (P<0.05), respectively, when compared with the vehicle control [Figure 3]. Pre- and co-incubations with ABT or DMTU almost completely abrogated the (–)Sch B-induced ROS production in H9c2 cardiomyocytes, with the inhibitory effect of ABT being more potent [Figure 3].
Figure 2.
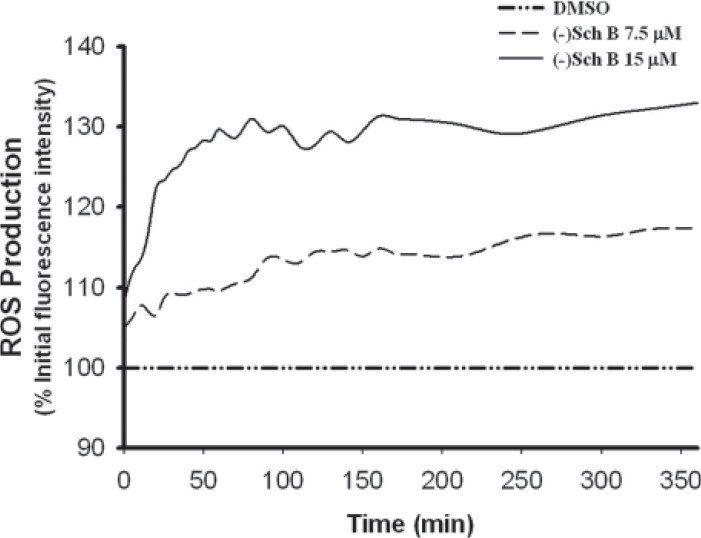
(–)Sch B-induced reactive oxygen species (ROS) production in H9c2 cardiomyocytes. (–)Sch B (7.5 or 15 μM) was incubated with H9c2 cardiomyocytes. Vehicle (i.e., DMSO) was added in untreated cells. Fluorescence intensity was monitored every minute for 360 minutes, as described in Materials and methods. Data, which were normalized with time-matched control, were expressed as a percentage of value at time zero. Values given are the mean of triplicate data
Figure 3.
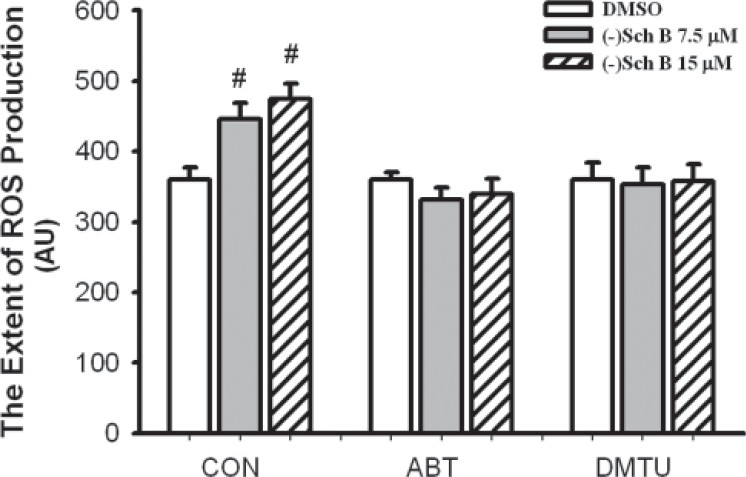
Effects of CYP inhibitor and antioxidant on (–)Sch B-induced ROS production in H9c2 cardiomyocytes. 1-aminobenzotriazol (ABT) and dimethylthiouracil (DMTU) were added at final concentrations of 10 mM and 20 mM, respectively, as described in Materials and methods. Respective vehicles were added to drug-untreated cells. Control (CON) group was not treated with ABT or antioxidant. The (–)Sch B (7.5 or 15 μM)-induced ROS production was monitored for 360 min. The extents of ROS production were estimated as described in Materials and methods. Values given are the mean±SD, with triplicate data. # Significantly different from the respective DMSO group (P<0.05)
Exposure of H9c2 cardiomyocytes to (–)Sch B (7.5 and 15 μM) for 6 h caused a dose-dependent increase (40 and 47%, P<0.05) in cellular GSH level at 16 h post-(–)Sch B exposure [Figure 4a]. Pre- and co-treatment with ABT or DMTU during the (–)Sch B exposure strongly suppressed the stimulation of cellular GSH level [Figure 4a]. Similar exposure of H9c2 cardiomyocytes to (–)Sch B-induced Hsp 25 and Hsp 70 production in a dose-dependent manner (12 and 25%, 33 and 42%, respectively, P<0.05) at 16 h post-(–)Sch B exposure [Figure 4b]. Pre- and co-treatment with ABT or DMTU during the (–)Sch B exposure completely abrogated the induction of Hsp 25, with the inhibition by ABT being more significantly [Figure 4b]. Similarly, the induction of Hsp 70 by (–)Sch B were also strongly suppressed by pre- and co-treatment with ABT or DMTU [Figure 4c].
Figure 4.
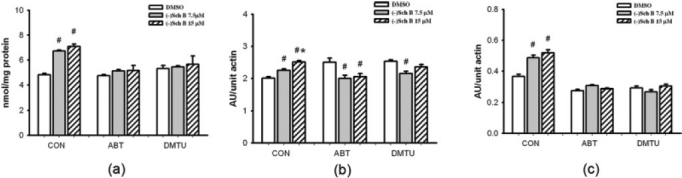
Effects of CYP inhibitor and antioxidant pre/co-treatment on (–)Sch B-induced increases in cellular-reduced glutathione (GSH) level and heat shock protein (Hsp) 25/70 production in H9c2 cardiomyocytes. Cells were pre/co-treated with ABT or DMTU, as described in Materials and methods. Cells were exposed to (–)Sch B (7.5 or 15 μM) for 6 h. Cellular GSH and Hsp 25/70 levels were measured at 16 h post-(–)Sch B exposure, as described in Materials and methods. values given are the mean±SD, with triplicate data. #Significantly from the respective DMSO group (P<0.05); *significantly different from the control (–)Sch B 7.5 μM group (P<0.05)
Exposure to (–)Sch B (15 μM) for 6 h, in the absence or presence of ABT or DMTU, did not affect the extent of apoptosis in cultured H9c2 cardiomyocytes [Figure 5]. Hypoxia/reoxygenation induced apoptosis in H9c2 cardiomyocytes, with the extent of apoptosis being increased by 1-fold (P<0.05), as compared to the non-hypoxia/reoxygenation control [Figure 5]. (–)Sch B strongly suppressed the hypoxia/reoxygenation-induced apoptosis at 16-h post-drug exposure, with the degree of protection being 48% (P<0.05). The cytoprotection afforded by (–)Sch B against hypoxia/reoxygenation-induced apoptosis was almost completely abrogated by pre- and co-treatment with ABT or DMTU during the 6-h exposure to (–)Sch B.
Figure 5.
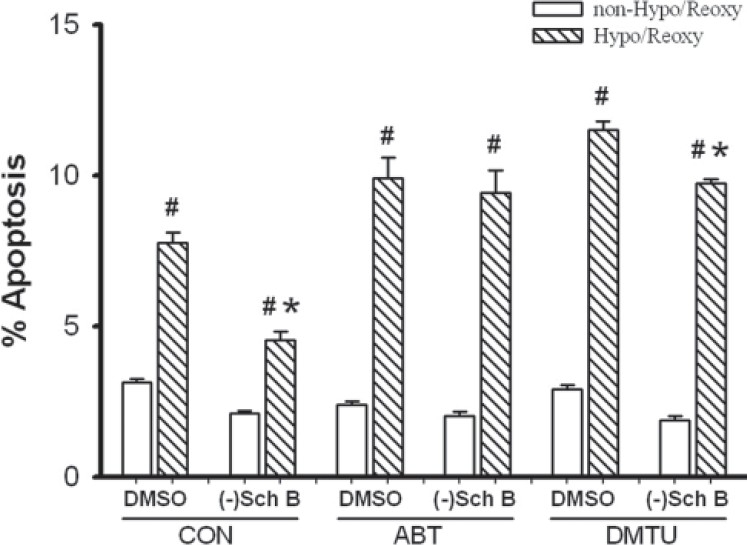
Effects of CYP inhibitor and antioxidant pre/co-treatment on (–)Sch B protection against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes. Cells were pre/co-treated with ABT or DMTU as described in Figure 4. They were exposed to (–)Sch B (15 mM) for 6 h followed by hypoxia/reoxygenation (Hypo/Reoxy) challenge (hypoxia for 2 h and reoxygenation for 16 h) at 16-h post-(–)Sch B exposure. The extent of apoptosis was measured as described in Materials and methods. values given are the mean±SD, with triplicate data. #Significantly differently from the respective non-Hypo/Reoxy group (P<0.05); *significantly different from the respective DMSO-treated Hypo/Reoxy group (P<0.05)
Discussion
A recent study in our laboratory has shown that CYP-catalyzed reaction is likely involved in the Sch B-induced glutathione antioxidant response in rat hearts.[7] Presumably, the protective response is triggered by ROS arising from the CYP-catalyzed metabolism of Sch B.[7] In the present study, (–)Sch B, an active stereoisomer of Sch B, was found to stimulate ROS production in H9c2 cardiomyocytes in situ, which could be completely suppressed by ABT or antioxidant.
The exposure of H9c2 cardiomyocytes to (–)Sch B for 6 h resulted in an optimal stimulation of cellular GSH level and Hsp 25/70 production at 16 h post-exposure. The (–)Sch B-induced glutathione antioxidant and heat shock responses were associated with the cytoprotection against hypoxia/reoxygenation-induced apoptosis. Both glutathione antioxidant/heat shock responses and cytoprotection against hypoxia/reoxygenation-induced apoptosis were abrogated by ABT or antioxidant pre/co-treatment during the 6-h (–)Sch B exposure. Taken together with the observation that the extent of (–)Sch B-induced ROS production was greatly reduced by ABT or DMTU treatment, our results strongly suggested the causal involvement of CYP-mediated ROS production in triggering cellular protective responses in H9c2 cardiomyocytes. In this regard, the cumulative amount of ROS generated within 6-h exposure (but not 3 h) to (–)Sch B culminated in optimal glutathione antioxidant and heat shock responses in H9c2 cardiomyocytes (data not shown). Conceivably, the sustained production of ROS from up to 6 h of CYP-catalyzed metabolism of (–)Sch B elicits a redox signaling, possibly through mitogen-activated protein kinases (MAPK) pathway, with resultant transcription activation of antioxidant/heat shock response element.[12–14] A recent study indicated that (–)Sch B could activate all three MAPK pathways, namely, extracellular signal-regulated protein kinase, p38 MAPK and C-Jun-NH2-terminal kinases, in H9c2 cardiomyocytes within 6 h of treatment.[15]
It is well established that oxidative stress is involved in the pathogenesis of ischemia/reperfusion injury,[16,17] and this pathological condition can be mimicked by subjecting cultured cardiomyocytes to hypoxia/reoxygenation challenge.[18–20] A burst of ROS production arising from the restoration of oxygen to the previous hypoxic cardiomyocytes increases intracellular oxidative stress, which can eventually contribute to necrotic and/or apoptotic cell death.[19] Consistently, our results indicated that a 2-h period of hypoxia followed by 16 h of reoxygenation caused significant apoptotic cell death in cultured H9c2 cardiomyocytes. Signs of cytotoxicity such as depletion of GSH and alteration of calcium homeostasis have been observed in hypoxia/reoxygenation-challenged cells.[6] The cytoprotection afforded by (–)Sch B against hypoxia/reoxygenation-induced apoptosis may mainly be related to the enhanced cellular glutathione status and the subsequent increased resistance of mitochondria to Ca2+-induced permeability transition.[6] As regards the role of Hsp 25 and Hsp 70 in cytoprotection, an earlier study in our laboratory has demonstrated that the cardioprotection afforded by Sch B against myocardial I/R injury is partly contributed by the induction of Hsp 25 and Hsp 70 in rats.[5]
Conclusions
Taken together, the results suggest that (–)Sch B can elicit cellular glutathione antioxidant and heat shock responses through the intermediacy of CYP-catalyzed ROS production, thereby protecting against oxidant-induced apoptosis in H9c2 cardiomyocytes.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Hancke JL, Burgos R, Ahumada F. Schisandra chinensis (Turcz) baill. Fitoterapia. 1999;70:451–71. [Google Scholar]
- 2.Yim TK, Ko KM. Schisandrin B protects against myocardial ischemia-reperfusion injury by enhancing myocardial glutathione antioxidant status. Mol Cell Biochem. 1999;196:151–6. [PubMed] [Google Scholar]
- 3.Ko KM, Yiu HY. Schisandrin B modulates the ischemia-reperfusion induced changes in non-enzymatic antioxidant levels in isolated-perfused rat hearts. Mol Cell Biochem. 2001;220:141–7. doi: 10.1023/a:1010979404447. [DOI] [PubMed] [Google Scholar]
- 4.Chiu PY, Ko KM. Time-dependent enhancement in mitochondrial glutathione status and ATP generation capacity by schisandrin B treatment decreases the susceptibility of rat hearts to ischemia-reperfusion injury. Biofactors. 2003;19:43–51. doi: 10.1002/biof.5520190106. [DOI] [PubMed] [Google Scholar]
- 5.Chiu PY, Ko KM. Schisandrin B protects myocardial ischemia-reperfusion injury partly by inducing Hsp25 and Hsp70 expression in rats. Mol Cell Biochem. 2004;266:139–44. doi: 10.1023/b:mcbi.0000049151.79238.30. [DOI] [PubMed] [Google Scholar]
- 6.Chiu PY, Luk KF, Leung HY, Ng KM, Ko KM. Schisandrin B stereoisomers protect against hypoxia/reoxygenation-induced apoptosis and inhibit associated changes in Ca2+-induced mitochondrial permeability transition and mitochondrial membrane potential in H9c2 cardiomyocytes. Life Sci. 2008;82:1092–101. doi: 10.1016/j.lfs.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Chen N, Ko KM. Schisandrin B-induced glutathione antioxidant response and cardioprotection are mediated by reactive oxidant species production in rat hearts. Biol Pharm Bull. 2010;33:825–9. doi: 10.1248/bpb.33.825. [DOI] [PubMed] [Google Scholar]
- 8.Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res. 1991;69:1476–686. doi: 10.1161/01.res.69.6.1476. [DOI] [PubMed] [Google Scholar]
- 9.Griffith OW. Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal Biochem. 1980;106:207–12. doi: 10.1016/0003-2697(80)90139-6. [DOI] [PubMed] [Google Scholar]
- 10.Ip SP, Che CT, Kong YC, Ko KM. Effects of schisandrin B pretreatment on tumor necrosis factor-alpha induced apoptosis and Hsp70 expression in mouse liver. Cell Stress Chaperones. 2001;6:44–8. doi: 10.1043/1355-8145(2001)006<0044:EOSBPO>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 12.Chen YC, Tsai SH, Shen SC, Lin JK, Lee WR. Alternative activation of extracellular signal-regulated protein kinases in curcumin and arsenite-induced HSP70 gene expression in human colorectal carcinoma cells. Eur J Cell Biol. 2001;80:213–21. doi: 10.1078/0171-9335-00158. [DOI] [PubMed] [Google Scholar]
- 13.Tang SY, Wang H, Zhang W, Halliwell B. Notopterygium forbesii boiss extract and its active constituents increase reactive species and heme oxygenase-1 in human fetal hepatocytes: Mechanisms of action. Chem Res Toxicol. 2008;21:414–23. doi: 10.1021/tx800301f. [DOI] [PubMed] [Google Scholar]
- 14.Jeong GS, Lee DS, Li B, Byun E, Kwon DY, Park H, et al. Protective effect of sauchinone by upregulating heme oxygenase-1 via the P38 MAPK and Nrf2/ARE pathways in HepG2 cells. Planta Med. 2010;76:41–7. doi: 10.1055/s-0029-1185906. [DOI] [PubMed] [Google Scholar]
- 15.Chiu PY, Chen N, Leong PK, Leung HY, Ko KM. Schisandrin B elicits a glutathione antioxidant response and protects against apoptosis via the redox-sensitive ERK/Nrf2 pathway in H9c2 cells. Mol Cell Biochem. 2011;350:237–50. doi: 10.1007/s11010-010-0703-3. [DOI] [PubMed] [Google Scholar]
- 16.Repine JE. Oxidant-antioxidant balance: Some observations from studies of ischemia-reperfusion in isolated perfused rat hearts. Am J Med. 1991;91:45–53. doi: 10.1016/0002-9343(91)90283-4. [DOI] [PubMed] [Google Scholar]
- 17.Kang YJ, Li G, Saari JT. Metallothionein inhibits ischemia-reperfusion injury in mouse heart. Am J Physiol. 1999;276:993–7. doi: 10.1152/ajpheart.1999.276.3.H993. [DOI] [PubMed] [Google Scholar]
- 18.Kim JS, Ohshima S, Pediaditakis P, Lemasters JJ. Nitric oxide protects rat hepatocytes against reperfusion injury mediated by the mitochondrial permeability transition. Hepatology. 2004;39:1533–43. doi: 10.1002/hep.20197. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda N, Mori T, Nakamura H, Shigekawa M. Mechanisms of reoxygenation-induced calcium overload in cardiac myocytes: Dependence on pHi. J Surg Res. 1995;59:712–8. doi: 10.1006/jsre.1995.1228. [DOI] [PubMed] [Google Scholar]
- 20.Strickler J, Jacobson KA, Liang BT. Direct preconditioning of cultured chick ventricular myocytes. Novel functions of cardiac adenosine A2a and A3 receptors. J Clin Invest. 1996;98:1773–9. doi: 10.1172/JCI118976. [DOI] [PMC free article] [PubMed] [Google Scholar]