

Abstract
Background
The lymphatic system plays a key role in tissue fluid homeostasis and lymphatic dysfunction due to genetic defects or lymphatic vessel obstruction can cause lymphedema, disfiguring tissue swellings often associated with fibrosis and recurrent infections without available cures to date. In this study, retinoic acids (RAs) were determined to be a potent therapeutic agent that is immediately applicable to reduce secondary lymphedema.
Methods and Results
We report that RAs promote proliferation, migration and tube formation of cultured lymphatic endothelial cells (LECs) by activating FGF-receptor signaling. Moreover, RAs control the expression of cell-cycle checkpoint regulators such as p27Kip1, p57Kip2 and the aurora kinases through both an Akt-mediated non-genomic action and a transcription-dependent genomic action that is mediated by Prox1, a master regulator of lymphatic development. Moreover, 9-cisRA was found to activate in vivo lymphangiogenesis in animals based on mouse trachea, matrigel plug and cornea pocket assays. Finally, we demonstrate that 9-cisRA can provide a strong therapeutic efficacy in ameliorating the experimental mouse tail lymphedema by enhancing lymphatic vessel regeneration.
Conclusions
These in vitro and animal studies demonstrate that 9-cisRA potently activates lymphangiogenesis and promotes lymphatic regeneration in an experimental lymphedema model, presenting it as a promising novel therapeutic agent to treat human lymphedema patients.
Keywords: lymphangiogenesis, lymphedema, retinoic acids, lymphatic regeneration, therapy
Introduction
Lymphatic vessels are essential for tissue fluid homeostasis, lipid absorption and immune cell transport. The lymphatic system is known to be derived from the blood vascular system 1. During development, lymphatic endothelial cells (LECs) are differentiated from blood vascular endothelial cells (BECs) in the cardinal vein and the newly differentiated LECs migrate out to form the initial lymphatic vessel1, 2. The homeodomain transcription factor Prox1 plays an essential role in this LEC-differentiation process as the master regulator that suppresses the BEC-phenotype and induces LEC-fate 1.
Lymphedema, or tissue swelling, resulting from abnormal tissue fluid accumulation, can be caused by lymphatic malformation due to genetic defects or secondary lymphatic obstruction due to surgery or radiation, and presents a considerable physical and social burden as there is not treatment currently available. Recently, important progress has been reported in the development of treatments based on mesenchymal stem cells or growth factors coupled with lymph node transplantation 3, 4. However, extensive studies are still required before these experimental therapies will become available to treat human lymphedema patients.
Retinoic acids (RAs) are comprised of biologically active metabolites of vitamin A and are involved in a broad range of biological processes in vertebrate development by regulating genes important for cell proliferation, differentiation, apoptosis and metabolism 5, 6. RAs have been shown to arrest cell cycle progression and cell proliferation by upregulating key cell cycle inhibitors, such as p21Cip1/CDKN1A 7, p27Kip1/CDKN1B 8, p16INK4a/CDKN2A 9, p14INK4b/CDKN2B 10 and p19INK4D/CDKN2D 11 , and thus have been developed as therapeutic agents for various types of diseases, including breast cancer and leukemia12. RAs also modulate the interaction of endothelial cells with mural cells by suppression of Tie2 signaling 13, and RA-deficient mouse embryos display uncontrolled proliferation and decreased maturation of endothelium, resulting in defective vascular remodeling during embryogenesis 14. In contrast, the pro-angiogenic effect of RAs has also been documented and RAs stimulate transcription and translation of VEGF in non-endothelial cells. Recently, all-trans-RA (ATRA) has been shown to be important in the early steps of lymphatic development during embryogenesis 15. However, the effect of RAs in post-developmental lymphatic regeneration remains to be elucidated.
Here, we report that RAs stimulate proliferation, tube formation and migration of primary human LECs via the FGF signaling pathway. Moreover, 9-cisRA stimulates LEC-proliferation by regulating the cell-cycle related target genes, p27 kip1, p57kip2 and the aurora kinases through both genomic and non-genomic mechanisms. Consistent with these findings, in this study, various animal models demonstrate that 9-cisRA induces in vivo lymphangiogenesis and provides a therapeutic effect in an experimental lymphedema model. Together, the current findings provide support for the development of 9-cisRA as a therapeutic agent to treat human lymphedema patients.
Methods
Mouse Lymphedema Model
The protocols related to this mouse lymphedema model were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Southern California. We largely followed the previously established protocol for inducing experimental lymphedema in the tails of mice 16. Two surgeons performed a total of three independent experiments using two different mouse strains (C57BL/6J and BALB/c) purchased from the Jackson Laboratory (Bar Harbor, ME). Briefly, under a dissecting microscope, we removed a circumferential 2-mm-wide piece of skin located approximately 1 cm distal of the tail base and severed the deeper lymphatics running alongside the major blood vessels, with special attention not to damage blood vessels. From post-surgical day 2, we intraperitoneally (i.p.) injected 100 µL vehicle (10 µL of 100% ethanol and 90 µL of Sunflower seed oil (Sigma Aldrich)) into the control mouse group or 100 µL vehicle solution containing 9-cisRA (0.8 mg/kg) daily. The diameter of the proximal and distal sides of the surgical site in the tail was measured every other day. At the end of the experiments, mouse tails were surgically removed and processed for further immunohistochemical analyses.
Statistical Analysis
The outcome measures are expressed as the mean ± standard deviation per experimental condition, unless noted otherwise. Analysis of Variance (ANOVA) was used to detect the differences in outcome measures across the experimental and control groups for all in vitro and in vivo experiments. Pairwise comparisons of least-squares means between groups were adjusted using Dunnett’s or Tukey’s test whenever appropriate. A mixed linear model with the autoregressive covariance structure overtime was used to compare tail diameters over time by treatment and side of wounds. The analyses were performed using the SAS statistical package version 9.2 (SAS Institute Inc., Cary, North Carolina, USA). All reported P values were two-sided at a significance level of 0.05.
Supplemental Methods
Detailed information on the cell culture reagents and assays for cell proliferation, migration, tube formation, immunofluorescence, gene expression, luciferase, chromatin immunoprecipitation (ChIP), corneal pocket assay, mouse trachea and matrigel plug are available in the online-only Data Supplement.
Results
Retinoic acids promote the proliferation, migration and tube formation of primary human lymphatic endothelial cells
We investigated the effect of RAs on LEC-proliferation using various RA derivatives such as 9-cisRA, ATRA, TTNPB (pan-RAR ligand) and AM580 (RARα-specific ligand) 17, and found that all of these RA derivatives enhanced the proliferation of primary LECs (Figure 1A). We then chose 9-cisRA, which has been FDA-approved to treat Kaposi’s sarcoma, an endothelial tumor with a lymphatic endothelial phenotype, and found that it promotes LEC-proliferation in a dose-dependent manner (Figure 1B). The effect of 9-cisRA on LEC-migration was also studied by scratch assays on LECs that had been pre-treated with either vehicle or 9-cisRA. At 24 hours, the scratched area was fully recovered in the 9-cisRA-treated LECs, but not in the vehicle-treated control LECs (Figure 1C), indicating that 9-cisRA treatment significantly promoted the migration of primary LECs. Assays using the modified Boyden chamber that evaluates the directional migratory activity of LECs by 9-cisRA yielded results that are consistent with this finding (Figure 2D). In addition, 9-cisRA enhanced the tube-forming capability of LECs on the surface of the matrigel (Figure 1D). Taken together, these in vitro studies demonstrate that 9-cisRA significantly activates the proliferation, migration and tube formation of cultured primary human dermal LECs.
Figure 1.
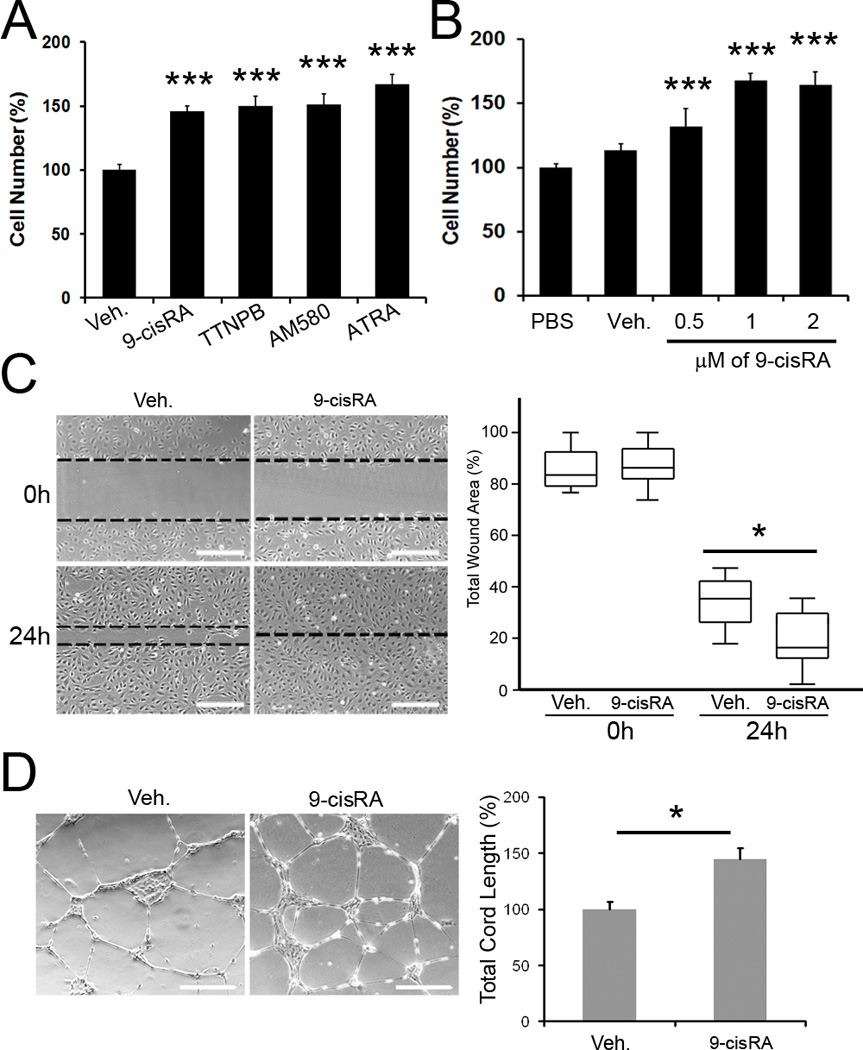
Retinoic acids activate proliferation, migration and tube formation of primary human LECs. (A) Activation of LEC-proliferation by various RA derivatives. Primary human LECs in a low serum media (1% FBS) were incubated with 1 µM of 9-cisRA, TTNPB, AM580, all-trans RA (ATRA) or vehicle (ethanol, 0.1%) alone. After 48 hours, the total cell number was estimated and displayed as a percent cell number against the vehicle alone (Veh)-treated control group. Bars represent the standard deviation (SD) of quadruplicates. Asterisks indicate p< 0.001 against the vehicle alone control. (B) Dose-dependent activation of LEC-proliferation in a low serum media (1% FBS) by 9-cisRA compared to phosphate-buffered saline (PBS) or vehicle alone (Veh) for 48 hours. Bars represent the standard deviation (SD) of quadruplicates. Asterisks indicate p< 0.001 against the vehicle alone control (Veh). (C) Effect of 9-cisRA on the migration of primary LECs. Monolayers of LECs were pre-treated with vehicle alone or vehicle containing 9-cisRA (1 µM) and scratched with a pipette tip to make a wound. After 24 hours, the total remaining wounded area was determined by image analysis and expressed as a percentage of the total wounded area in the box and whisker plot. Asterisk, p< 0.05. (D) Effect of 9-cisRA on tube formation of primary LECs. Cells were pre-treated with vehicle alone or vehicle with 9-cisRA (1 µM) for 24 hours and an equal number of cells was seeded onto the growth factor-depleted matrigel. After 24 hours, five representative images were taken and the total tube length per image was calculated and charted as a percentage of the vehicle-treated group. Asterisk, p< 0.05.
Figure 2.
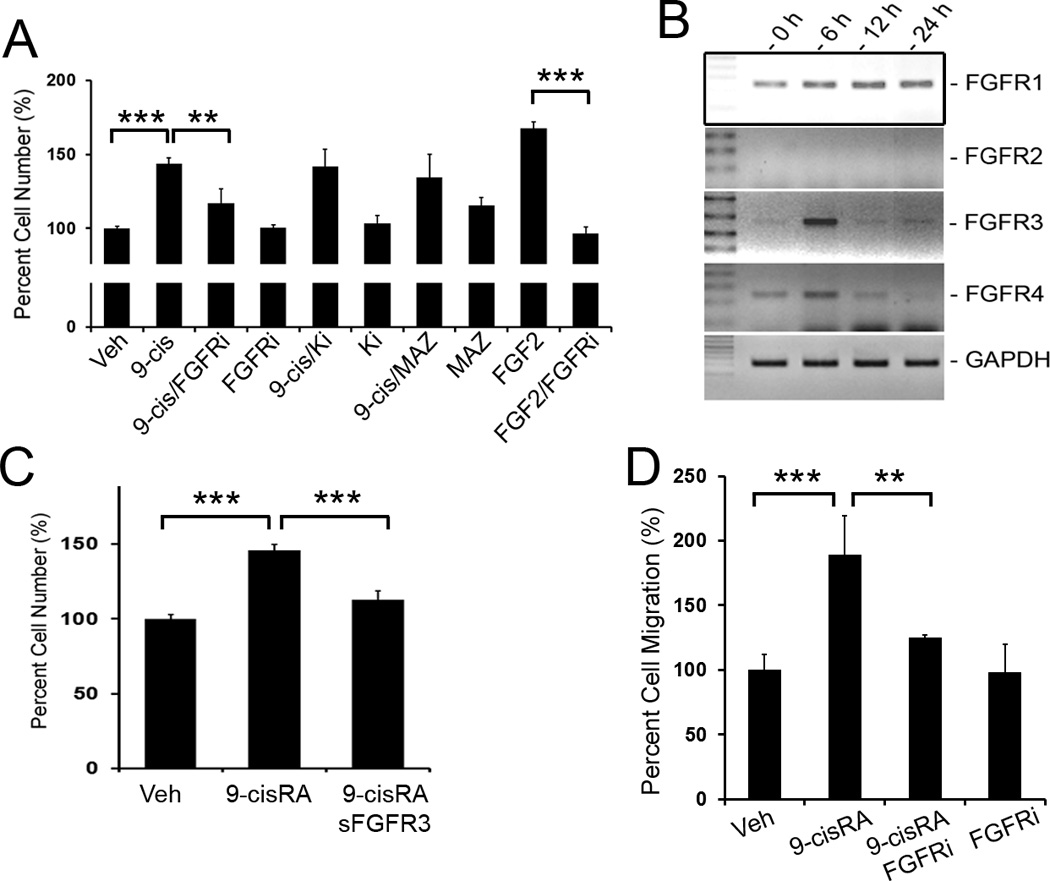
FGF pathway plays a key role in 9-cisRA-induced LEC-proliferation. (A) 9-cisRA-induced proliferation for 48 hours was significantly abrogated by a FGF-receptor inhibitor (FGFRi, PD173074, 50 nM) 23, but not by inhibitors for VEGFR-2 (Ki8751, 50 nM) 24 or VEGFR-3 (MAZ51, 50 nM) 25. Three asterisks, p< 0.001; two asterisks, p < 0.01. (B) Expression of four FGF receptors (FGFR1 to FGFR4) and GAPDH (internal control) in LECs treated with 9-cisRA (1 µM) for 0, 6, 12 and 24 hours was determined by semi-quantitative RT-PCR. None of these genes was regulated by vehicle alone treatment (data not shown). (C) Pre-incubation of LECs with a soluble FGFR3 recombinant protein (1 µg/ml) reduced the 9-cisRA-promoted LEC proliferation. Three asterisks, p< 0.001. (D) Chemotactic migration assay using modified Boyden chambers (Fluoroblok™). Primary LECs were allowed to migrate toward serum-free media containing vehicle alone, 9-cisRA (1 µM), FGFR inhibitor (FGFRi, 50 nM) or 9-cisRA (1 µM)/FGFR inhibitor (FGFRi, 50 nM) for 3 hours. The migrated cells were quantified and expressed as a percentage against the vehicle control. Three asterisks, p< 0.001; two asterisks, p< 0.01.
FGF signaling plays a key role in 9-cisRA-induced LEC proliferation and migration
We then set out to define the mechanism for the 9-cisRA-induced mitogenic and migration activities of LECs. Since VEGF and FGF are among the best characterized cytokines that activate proliferation of LECs 18–22, we investigated whether either or both of the two signaling pathways were involved in 9-cisRA-induced LEC proliferation by using specific inhibitors for each pathway. Primary LECs were incubated with chemical inhibitors against FGF receptors 23, VEGFR-2 24 and VEGFR-3 25 prior to 9-cisRA treatment. Interestingly, while the FGF receptor inhibitor resulted in a significant suppression of the 9-cisRA-induced LEC proliferation (Figure 2A), neither the VEGFR-2 nor VEGFR-3 inhibitor exhibited any suppression (Figure 2A). This finding prompted us to further evaluate the expression of FGF receptors in LECs upon 9-cisRA treatment. Semi-quantitative RT-PCR analyses revealed that the expression of FGFR-1 was largely unchanged and FGFR-2 was not detected in LECs (Figure 2B). In comparison, FGFR-3 and FGFR-4 were transiently upregulated by ~8 and ~2-fold, respectively, at 6-hours after 9-cisRA-treatment (Figure 2B). We have previously reported that FGFR-3 is prominently expressed in LECs over blood vessel endothelial cells (BECs) and plays an important role in FGF signaling in LECs 26. To define the essential role of FGFR3 in the 9-cisRA-induced activation of LEC-proliferation, primary LECs were pre-incubated with a soluble recombinant FGFR-3 protein prior to 9-cisRA treatment. Indeed, soluble FGFR-3 recombinant protein significantly suppressed the 9-cisRA-activated LEC proliferation (Figure 2C), suggesting that soluble FGFR-3 protein sequesters FGF ligands that mediate 9-cisRA-induced LEC proliferation. Consistent with this, the FGFR inhibitor suppressed the 9-cisRA-induced migratory activity of LECs based on a directional migration assay using a modified Boyden chamber (Figure 2D). Together, these data indicate that FGF/FGFR signaling significantly contributes to the 9-cisRA-induced proliferation and migration of primary LECs.
Regulation of p27Kip1, p57Kip2 and the aurora kinases A/B in LECs by 9-cisRA
Since RAs have been reported to induce growth arrest of many different types of cells, including immortalized B-cells 8, breast cancer cells 27 and neuronal cells 10 through an upregulation of CDK inhibitors, we studied the 9-cisRA regulation of various cell-cycle regulators in LECs. In contrast to previous studies, two major CDK inhibitors, p27Kip1 (CDKN1B) and p57Kip2 (CDKN1C), were strongly downregulated in 9-cisRA-treated LECs based on real-time RT-PCR (qRT-PCR), Western blot and immunofluorescence studies (Figure 3A–D). Importantly, it has been reported that p27Kip1 and p57Kip2 function askey cell-cycle brakes and that their inhibition is sufficient to induce the cells to enter the S-phase of the cell cycle 28. In addition, the Western blot analyses revealed that 9-cisRA treatment induced the expression of the aurora kinases A and B, which play an important role in the G2/M-phase of cell cycle progression 29 (Figure 3E). Taken together, the data demonstrate that 9-cisRA controls the expression of two major CDK inhibitors (p27Kip1 and p57Kip2) and two aurora kinases (AURKA and AURKB), which play critical functions in the G1/S and G2/M transition in LECs, respectively.
Figure 3.

Regulation of p27Kip1, p57Kip2 and aurora kinases by 9-cisRA in LECs. (A) qRT-PCR analyses show that the expression of CDKN1B/p27Kip1 and CDKN1C/p57Kip2 was significantly downregulated in 9-cisRA (1 µM) - treated LECs. Asterisk, p< 0.01 against 0 hour control; bars, the standard deviation (SD) of quadruplicates. Vehicle alone treatment did not alter the expression (data not shown). Western blot analyses also revealed 9-cisRA-mediated downregulation of the protein expression of p27Kip1 (B) and p57Kip2 (C). (D). Immunofluorescence staining assays demonstrated the downregulation of p27Kip1 and p57Kip2 after 18-hour of 9-cisRA (1 µM) treatment. Bars, 20 µm. (E) Protein expression of the aurora kinases A and B were transiently upregulated at 12 hours in LECs treated with 9-cisRA (1 µM).
Genomic and non-genomic mechanisms for the 9-cisRA-mediated regulation of p27Kip1, p57Kip2 and aurora kinases in LECs
It is known that RAs exert their functions through two modes of action; a conventional, genomic mechanism that relies on nuclear hormone receptor-mediated transcription, and a more rapid form of non-genomic action that does not require gene transcription 12. Recent studies have identified a novel, non-genomic effect of RAs in cell cycle regulation through PI3 kinase 12. We thus investigated the possibility of p27Kip1 downregulation by the PI3K/Akt pathway resulting from 9-cisRA, since Akt activation is the major signal for downregulation of p27Kip1, but not p57Kip230. Indeed, we detected rapid phosphorylation of Akt by 9-cisRA within 10 minutes, whereas vehicle treatment did not show any sign of Akt activation (Figure 4A). Moreover, we found that, from the first hour of 9-cisRA treatment, p27Kip1 became phosphorylated at its serine 10 residue (Figure 4B), which is known to be important for degradation of the protein 31. Consistent with this, pre-treatment of LECs with an Akt inhibitor (LY294002) effectively blocked the 9-cisRA-induced LEC proliferation and migration (Figure 4C and 4D). These data indicate that Akt-mediated p27Kip1 downregulation and phosphorylation may play a key role in the 9-cisRA-induced non-genomic effect on LEC-proliferation and migration.
Figure 4.

Mechanism for 9-cisRA-mediated regulation of p27Kip1, p57Kip2 and the aurora kinases in LECs. (A) Non-genomic action of 9-cisRA on the rapid activation of Akt in LECs. The Akt protein is phosphorylated in LECs within 10 minutes of 9-cisRA (1 µM) treatment. (B) Phosphorylation of p27Kip1 at serine 10 (pp27(S10)) occurs from the first hour of 9-cisRA treatment of LECs. Expression of the whole p27Kip1 protein was also shown. (C) Essential role of the Akt-mediated non-genomic action of 9-cisRA in LEC proliferation. LECs were pretreated with the PI3K signal inhibitor LY294002 (LY 50 nM (+), 100 nM (++)) for 30 minutes before 9-cisRA treatment (1 µM) and allowed to grow for 48 hours before determination of the relative cell numbers. Note that the inhibition of the PI3K/Akt signal blocked 9-cisRA-induced LEC proliferation. (D) Similarly, LECs were pretreated with LY294002 (LY, 100 nM (++)) for 30 minutes before 9-cisRA treatment (1 µM) and allowed to migrate in the modified Boyden chamber for 3 hours before measurement of the relative number of migrated cells. Note the essential role of the Akt-mediated non-genomic action of 9-cisRA in LEC migration. (E) qRT-PCR assays showed that knockdown of Prox1 in LECs resulted in downregulation of p57Kip2, but not p27Kip1. (F) Adenoviral overexpression of Prox1 (AdProx1) in HUVECs for 48 hours strongly induced p57Kip2, but not p27Kip1, as determined by qRT-PCR. AdCTR, control adenovirus. (G) ChIP assays for Prox1 in LECs performed against the p57Kip2 promoter using two independent PCR primer sets showed a physical association of the Prox1 protein with the p57Kip2 promoter. (H) Luciferase-reporter assays measuring the response of the different length proximal promoters of p57Kip2 to Prox1-mediated activation. The reporter constructs were named by the position of its 5’ and 3’ ends of the promoter fragments relative to the transcriptional start site (+1) of p57Kip2. (I) Prox1 ChIP was performed against the p57Kip2 promoter from LECs treated with vehicle (Veh) or 9-cisRA (1 µM) for 0.5 or 4 hours. The expression level of Prox1 protein was also determined in LECs treated with 9-cisRA for 2 hours. (J) Western blot assay showing that overexpression of Prox1 in the presence of 9-cisRA did not induce the expression of p57Kip2. (K) Prox1 ChIP revealed that, upon treatment of LECs with 9-cisRA (1 µM, 4 hours), Prox1 dissociated from the promoters of the aurora kinase kinases A (AURKA) and B (AURKB) and Survivin. Prox1 did not bind to the promoters of CCNB1 and CCNB2. For panels C, D, E, F and H, data are shown as the mean ± standard deviation (SD) and asterisks present p< 0.05.
On the other hand, the expression of p57Kip2 has been shown to be regulated by Prox1, the master regulator for lymphatic development32. In support of this, when we inhibited the expression of Prox1 with a small interfering RNA (siRNA) in primary LECs, only p57Kip2, but not p27Kip1, was significantly downregulated (Figure 4E). Moreover, when Prox1 was overexpressed by adenovirus in human umbilical venous endothelial cells (HUVECs), a cell type that does not express the lymphatic marker Prox1, p57Kip2, but not p27Kip1, was significantly upregulated (Figure 4F). Importantly, chromatin immunoprecipitation (ChIP) assay revealed that Prox1 was physically associated with the p57Kip2 promoter (Figure 4G). We performed p57Kip2 promoter-reporter assays and found that Prox1 strongly activated thep57Kip2 promoter (Figure 4H). These data indicate that the Prox1 protein binds to the p57Kip2 promoter and directly activates its transcription in LECs. Notably, Western blot studies revealed that the expression of the p57Kip2 protein was strongly downregulated within the first 1~2 hours of 9-cisRA treatment (Figure 3C). ChIP assay revealed that while the Prox1 protein did bind to the p57Kip2 promoter in the vehicle-treated LECs, 9-cisRA treatment induced a rapid dissociation of the Prox1 protein from the p57Kip2 promoter within 0.5-hours, without affecting the Prox1 protein expression level (Figure 4I). This finding indicates that 9-cisRA treatment immediately decreased the DNA-binding affinity of Prox1 toward the p57Kip2 promoter. Moreover, because Prox1 overexpression induced p57Kip2 expression in the absence of 9-cisRA, but did not induce in the presence of 9-cisRA (Figure 4J), we conclude that 9-cisRA regulates p57Kip2 expression predominantly by modulating Prox1 binding to its promoter in LECs. Taken together, these data demonstrate that 9-cisRA inhibits Prox1 binding to the p57Kip2 promoter and thus downregulates p57Kip2, resulting in the cell cycle progression of LECs.
We searched for additional cell cycle-related genes regulated by Prox1 and studied the transcriptional profiles of various cell cycle-related genes after Prox1 knockdown in LECs. While knockdown of Prox1 alone yielded only moderate changes in cell-cycle genes, simultaneous knockdown of both Prox1 and RXRα, a nuclear receptor of 9-cisRA, synergistically upregulated several important cell cycle-related genes, such as the aurora kinases A and B, survivin, cyclin B1 and B2, KI67 nuclear antigen, as well as the NEK2 and PBK kinases (Table 1), indicating that Prox1 and RXRα may cooperatively repress the expression of these cell cycle-related genes. Moreover, ChIP studies showed that Prox1 also binds to the promoters of aurora kinases A/B and survivin, and that treatment of 9-cisRA caused a rapid dissociation of Prox1 from their promoters within 4 hours (Figure 4K). Taken together, these studies demonstrate that 9-cisRA activates cell cycle progression through both genomic and non-genomic actions and that Prox1, the master regulator for lymphatic differentiation, is intricately involved in the genomic effect of 9-cisRA on cell cycle progression.
Table 1.
Fold changes in the expression of various cell cycle-related genes upon knockdown of PROX1 and/or RXRα for 24 hours in lymphatic endothelial cells
| Probe ID | Gene | iC | iP | iR | iP/iR | Gene Title | Gene Function |
|---|---|---|---|---|---|---|---|
| 208079_s_at | AURKA | 1.0 | 1.7 | 1.2 | 3.6 | Aurora kinase A | Cell cycle kinase |
| 209464_at | AURKB | 1.0 | 1.4 | 1.1 | 3.0 | Aurora kinase B | Cell cycle kinase |
| 202095_s_at | BIRC5 | 1.0 | 1.5 | 1.1 | 3.5 | Survivin | Anti-apoptosis |
| 214710_s_at | CCNB1 | 1.0 | 1.8 | 1.2 | 3.6 | Cyclin B1 | G2/M-specific cyclin |
| 202705_at | CCNB2 | 1.0 | 1.4 | 1.0 | 3.2 | Cyclin B2 | G2/M-specific cyclin |
| 212021_s_at | MKI67 | 1.0 | 1.4 | 1.2 | 3.1 | Proliferation-related antigen | Cell proliferation |
| 204641_at | NEK2 | 1.0 | 1.6 | 1.1 | 3.6 | NIMA (never in mitosis gene a)-related kinase 2 | Mitotic regulation |
| 219148_at | PBK | 1.0 | 1.9 | 1.1 | 4.3 | PDZ binding kinase | Mitotic kinase |
iC, control siRNA; iP, Prox1 siRNA; iR, RXRα siRNA; Probe ID, probe identification number of Human Genome U133 Plus 2.0 Array from Affymetrix
9-cisRA enhances in vivo lymphangiogenesis
In order to extend the in vitro findings, we set out to evaluate the pro-lymphangiogenic potential of 9-cisRA and performed various in vivo lymphangiogenesis assays using the mouse trachea, matrigel plugs and the cornea. For the mouse trachea assay, vehicle solution mixed with or without 9-cisRA was dropped into mouse nostrils daily and trachea specimens were collected after five days and stained for the lymphatic vessel marker, LYVE-1 (Figure 5A–E). We found that, compared to the vehicle control, 9-cisRA significantly activated lymphangiogenesis, determined by the total length of the capillary lymphatic vessels on the luminal side of the trachealis muscle, where lymphatic vessels are otherwise only sparsely present (Figure 5A vs. 5B). Moreover, whereas the vehicle-treated lymphatic vessels were round-ended, 9-cisRA-treated lymphatic vessels displayed lymphatic tipped cells with jagged filopodia (Figure 5C vs. 5D). This assay indicates that 9-cisRA activated lymphatic vessels to sprout and then invade into the stroma of the trachea, a typical feature of active lymphangiogenesis.
Figure 5.

Promotion of in vivo lymphangiogenesis by 9-cisRA. (A–E) Mouse trachea assay: Lymphangiogenesis was activated in 9-cisRA-treated trachea (B, D) (5 mice) compared to vehicle-treated trachea (A, C) (5 mice). Low magnification images (A, B) of the tracheal mucosa are shown (Bars, 400 µm). Enlarged images of the boxed areas in panels A and B show that the lymphatic vessels in the vehicle-treated trachea are round-ended (arrowheads in C), whereas the lymphatics in the 9-cisRA-treated trachea have jagged ends (arrows in D), indicating active lymphatic sprouting. (E) Total length of the lymphatic vessels was measured on the luminal side of the trachealis muscle, which is largely devoid of lymphatic vessels. (F–H) Mouse subcutaneous matrigel plug assay: Matrigel premixed with vehicle alone (F) or 9-cisRA (10 µM) (G) was subcutaneously injected (two matrigel plugs per mouse, 5 mice each group) and harvested after two weeks for immunohistochemistry (IHC) analyses using an anti-podoplanin antibody. The relative lymphatic vessel area was determined in the matrigel using the NIH ImageJ software (H). Bar in (G), 50 µm. (I–K) 9-cisRA stimulates lymphangiogenesis in the mouse cornea. Representative LYVE-1 (red) immunofluorescent images are shown to demonstrate lymphatic vessel ingrowth into the cornea implanted with vehicle pellet (I) or 9-cisRA pellet (J) after 14 days. Dotted line: demarcation between the cornea and conjunctiva. Original Magnification: X100. (K) A significant difference was observed in the lymphatic coverage area between the vehicle (10 mice) and 9-cisRA (13 mice) groups. Error bars represent S.E.M. (L, M) Subcutaneous matrigel plug assay using the previously reported lymphatic-specific GFP mice 33 to visualize the newly formed lymphatic networks: Matrigel premixed with vehicle alone (L) or vehicle containing 9-cisRA (10 µM) (M) was injected into the skin of the lymphatic-specific GFP mice (two matrigel plugs per mouse, 5 mice each group) and isolated after two weeks. Lymphatic vessels within the matrigels were directly visualized under a fluorescent stereoscope.
We also performed a matrigel plug assay by subcutaneously implanting growth factor-depleted matrigel that had been pre-mixed with vehicle or 9-cisRA. After two weeks, matrigel plugs were harvested, paraffin-embedded and analyzed for lymphatic capillaries using an anti-podoplanin antibody (Figure 5F vs. 5G). We found that the 9-cisRA-containing matrigel plugs recruited roughly 2-fold more podoplanin-positive lymphatic vessels than vehicle alone matrigel plugs. Moreover, activation of lymphangiogenesis by 9-cisRA was also assessed by mouse cornea micropocket assay. A slow-release pellet containing vehicle or 9-cisRA was implanted into the cornea and newly formed lymphatic vessels were quantified after 14-days by LYVE-1 staining (Figure 5I vs. 5J). This assay demonstrates that 9-cisRA induced lymphatic vessel growth four times more than vehicle alone. In addition, we made use of the lymphatic-specific GFP transgenic (Prox1-GFP) mice that we recently reported 33 in order to directly visualize the macroscopic view of lymphatic in growth into matrigel implants containing 9-cisRA. Consistent with the results from the assays above, 9-cisRA stimulated a significantly higher lymphangiogenic activity than vehicle alone in matrigel implants (Figure 5L vs. 5M). Taken all together, the trachea, matrigel plug and cornea micropocket assays consistently demonstrate that 9-cisRA can strongly activate in vivo lymphangiogenesis.
Therapeutic efficacy of 9-cisRA in experimental mouse tail lymphedema
We next investigated the therapeutic effect of 9-cisRA by using the mouse tail lymphedema model that has been utilized as a model of human secondary lymphedema 16. We induced the experimental lymphedema by surgically obstructing the dermal and deep lymphatic vessels in both female and male mouse tails, and daily i.p. administered 9-cisRA or vehicle alone starting from post procedure day 2. For 3~4 weeks, we measured the diameters of the tails on the proximal and distal sides of the incisions. Lymphedema was induced in two mouse strains, C57BL/6J (Figure 6) and BALB/c (Supplemental Figure 1) by two different researchers and the outcome was consistent. From both mouse strains, we observed that 9-cisRA treatment significantly reduced the secondary tail lymphedema. Specifically, in female mice of both strains, lymphedema had similarly developed in the vehicle and 9-cisRA-treated groups in the first week post-surgery. However, the lymphedema in the vehicle-treated mice continuously developed in the second week and reached to the peak in the third week, before tapering off in the fourth week (Figure 6A and 6B). In a clear comparison, the lymphedema of the 9-cisRA-treated mice began to resolve from the second week on and returned largely to the base-line level at the end of the fourth week, demonstrating a potent therapeutic effect of 9-cisRA in this mouse experimental lymphedema model. In male mice of both strains, however, whereas the lymphedema in the vehicle-treated male mice peaked at the first week and maintained throughout the second half of the experiment, the 9-cisRA-treatment clearly decreased the formation of the lymphedema and also accelerated the resolution of the tissue swelling in the tail (Figure 6D and 6E). This distinct pattern of lymphedema formation and resolution between the genders and the therapeutic effect of 9-cisRA were also detected with BALB/c mice (Supplemental Figure 1). We then harvested the tails from the two groups and performed immunohistochemical analyses of the lymphatic vessels (Figure 6C and 6F). Importantly, lymphatic regeneration was much more prominent in the 9-cisRA-treated group than the vehicle-treated group, indicating that the 9-cisRA-promoted amelioration of lymphedema is associated with increased lymphatic regeneration. Quantitative analyses of the lymphatic vessels are shown in Supplemental Figure 6. In order to investigate whether 9-cisRA promotes skin wound healing, which may help to indirectly resolve the lymphedema, we evaluated the effect of 9-cisRA on wound healing in the skin and found that daily i.p. treatment of 9-cisRA did not accelerate or promote wound healing (Supplemental Figure 2). Taken together, these findings demonstrate that 9-cisRA effectively promotes lymphatic regeneration in experimental mouse lymphedema model and is clinically applicable to the treatment of post-surgical lymphedema in humans.
Figure 6.

Therapeutic effect of 9-cisRA on secondary lymphedema and lymphatic vessel regeneration. Experimental lymphedema was surgically induced in the tail of C57BL/6J mice and vehicle alone (ethanol/seed oil) or vehicle with 9-cisRA (0.8 mg/kg) was i.p. injected daily from day 2. Images of secondary lymphedema development in female (A–C) and male (D–F) mice treated with vehicle (Veh) or 9-cisRA (9cRA) for 32 days. The diameter of the tail on the distal (white arrowheads) and proximal sides of the wounds was measured every other day and charted for the female (B) and male (E) mice. Six females and 5 males were used for the vehicle-treated group and 7 females and 6 males were for the 9-cisRA-treated group. Cross, p< 0.05; asterisk, p< 0.01. (C, F) After 32 days, prominent regeneration of LYVE-1-positive lymphatic vessels was detected in the 9-cisRA-treated mice, compared to the vehicle-treated group (Veh), in both genders. For statistical analyses, a mixed linear model with the autoregressive covariance structure overtime was used to compare tail diameters over time by treatment and distal side of the wounds. Statistically significant difference (p< 0.001) was observed for the distal side of the wounds in females from days 12 to 32 and in males from days 6 to 32. No statistical difference was found for the proximal side of the wounds between the two groups in both genders. Similar data was obtained from an experiment using BALB/c mice (Supplemental Figure 1).
Discussion
RAs have been shown to induce cell cycle arrest in various cell types and are in clinical use as therapeutic agents 12. In particular, 9-cisRA has been shown to have an anti-angiogenic effect and received FDA-approval for the topical treatment of cutaneous lesions of Kaposi’s sarcoma, an endothelial cancer 12. Unexpectedly, however, our initial study to understand the effect of RAs on the function of Prox1 in human primary LECs led us to fortuitously uncover a previously unidentified role of RAs in post-developmental lymphatic regeneration. Subsequent investigation revealed that RAs can potently induce lymphangiogenesis in vitro and in vivo, and also demonstrated therapeutic efficacy in an experimental lymphedema model. Consistent with this, these findings are supported by a recent study showing that ATRA promotes the early steps of lymphatic differentiation and development 15. In strong agreement, we found that ATRA potently induced LEC-proliferation in vitro (Figure 1) and lymphatic vessel growth in the mouse cornea and matrigel plug assays (Supplemental Figure 3). In contrast, 9-cisRA neither activated proliferation of human dermal BECs (Supplemental Figure 4), nor promoted blood vessel growth, when implanted in the mouse cornea (data not shown). Since 9-cisRA and ATRA can bind and activate both RXR and RAR 5, we hypothesize that the lymphangiogenic activity of RA may be mediated by either or both of these RA nuclear receptors, and perhaps also by other RA-binding proteins, such as RBP, CRBP and CRABP.
In this study, we dissected the molecular mechanisms underlying the pro-lymphangiogenic effect of 9-cisRA and found that 9-cisRA exerts its effects through both genomic and non-genomic actions. The transcription-dependent genomic action of 9-cisRA was found to be mediated through the regulation of PROX1 gene expression and Prox1 protein activity. The data clearly showed that 9-cisRA rapidly dissociates the Prox1 protein from the promoter of the cell-cycle inhibitor p57Kip2, which leads to the downregulation of p57Kip2. 9-cisRA was also found to upregulate the aurora kinases A and B, serine/threonine kinases essential for cell cycle progression through the G2/M phase, by a genomic effect which involves the dissociation of the Prox1 protein from the promoter. Moreover, siRNA-mediated knockdown of Prox1 and RXRα in LECs resulted in a cooperative upregulation of multiple cell cycle-regulated genes. Therefore, Prox1 plays a key role in mediating the genomic action of 9-cisRA so as to stimulate LEC proliferation. On the other hand, downregulation and phosphorylation of p27Kip1 by 9-cisRA indicate that the non-genomic effect of 9-cisRA for LEC-proliferation may be mediated by Akt activation, since Akt activation is the major signal for repressing the expression of p27Kip1 30 . The blocking of Akt activation prevented 9-cisRA-induced LEC proliferation and migration (Figure 4C and 4D). Interestingly, the mRNA level of anothercell cycle regulator p21Cip1 was found to be transiently increased by 9-cisRA treatment (Supplemental Figure 5). Taken together, we conclude that enhanced LEC proliferation and migration by 9-cisRA is a combined outcome of both genomic and non-genomic actions. To the best of our knowledge, this is the first report of both the effect of 9-cisRA on lymphangiogenesis and its therapeutic efficacy in secondary lymphedema. Although several pro-lymphangiogenic factors have previously been implicated in the treatment of lymphedema 34, 9-cisRA is a metabolite of vitamin A that has been extensively characterized for its biochemical, physiological, pharmacological and toxicological properties, and thus can be immediately used to treat human patients. In fact, 9-cisRA and other RAs are already being prescribed for various human diseases. Therefore, our present study places 9-cisRA as one of the most promising therapeutic agents against human lymphedema, a disease without any currently available effective treatment.
Conclusions
We have shown 9-cisRA to be a potent lymphangiogenic factor that stimulates proliferation, migration and tube formation of cultured LECs and lymphatic vessel growth in various animal models. Furthermore, 9-cisRA treatment significantly accelerated the resolution of surgically-induced mouse tail lymphedema. Therefore, this evidence provides strong support for further development of 9-cisRA as a therapeutic agent to treat human lymphedema.
Supplementary Material
Acknowledgments
Funding Sources: This study was supported by the Marches of Dimes Foundation (Y.H), American Cancer Society (Y.H), NIH/National Institute of Child Health and Human Development (Y.H), American Heart Association (Y.H) and NIH/National Eye Institute (L.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: None
References
- 1.Wigle JT, Oliver G. Prox1 function is required for the development of the murine lymphatic system. Cell. 1999;98:769–778. doi: 10.1016/s0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- 2.Wigle JT, Harvey N, Detmar M, Lagutina I, Grosveld G, Gunn MD, Jackson DG, Oliver G. An essential role for prox1 in the induction of the lymphatic endothelial cell phenotype. EMBO J. 2002;21:1505–1513. doi: 10.1093/emboj/21.7.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lahteenvuo M, Honkonen K, Tervala T, Tammela T, Suominen E, Lahteenvuo J, Kholova I, Alitalo K, Yla-Herttuala S, Saaristo A. Growth factor therapy and autologous lymph node transfer in lymphedema. Circulation. 2011;123:613–620. doi: 10.1161/CIRCULATIONAHA.110.965384. [DOI] [PubMed] [Google Scholar]
- 4.Tammela T, Saaristo A, Holopainen T, Lyytikka J, Kotronen A, Pitkonen M, Abo-Ramadan U, Yla-Herttuala S, Petrova TV, Alitalo K. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat Med. 2007;13:1458–1466. doi: 10.1038/nm1689. [DOI] [PubMed] [Google Scholar]
- 5.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid x receptors: X-ploring their (patho)physiological functions. Cell Death Differ. 2004;11 Suppl 2:S126–S143. doi: 10.1038/sj.cdd.4401533. [DOI] [PubMed] [Google Scholar]
- 6.Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell. 2008;134:921–931. doi: 10.1016/j.cell.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jing Y, Xia L, Waxman S. Targeted removal of pml-raralpha protein is required prior to inhibition of histone deacetylase for overcoming all-trans retinoic acid differentiation resistance in acute promyelocytic leukemia. Blood. 2002;100:1008–1013. doi: 10.1182/blood.v100.3.1008. [DOI] [PubMed] [Google Scholar]
- 8.Zancai P, Cariati R, Rizzo S, Boiocchi M, Dolcetti R. Retinoic acid-mediated growth arrest of ebv-immortalized b lymphocytes is associated with multiple changes in g1 regulatory proteins: P27kip1 up-regulation is a relevant early event. Oncogene. 1998;17:1827–1836. doi: 10.1038/sj.onc.1202089. [DOI] [PubMed] [Google Scholar]
- 9.Wainwright LJ, Lasorella A, Iavarone A. Distinct mechanisms of cell cycle arrest control the decision between differentiation and senescence in human neuroblastoma cells. Proc Natl Acad Sci U S A. 2001;98:9396–9400. doi: 10.1073/pnas.161288698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tokumoto YM, Tang DG, Raff MC. Two molecularly distinct intracellular pathways to oligodendrocyte differentiation: Role of a p53 family protein. EMBO J. 2001;20:5261–5268. doi: 10.1093/emboj/20.18.5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu TX, Zhang JW, Tao J, Zhang RB, Zhang QH, Zhao CJ, Tong JH, Lanotte M, Waxman S, Chen SJ, Mao M, Hu GX, Zhu L, Chen Z. Gene expression networks underlying retinoic acid-induced differentiation of acute promyelocytic leukemia cells. Blood. 2000;96:1496–1504. [PubMed] [Google Scholar]
- 12.Bushue N, Wan YJ. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285–1298. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki Y, Komi Y, Ashino H, Yamashita J, Inoue J, Yoshiki A, Eichmann A, Amanuma H, Kojima S. Retinoic acid controls blood vessel formation by modulating endothelial and mural cell interaction via suppression of tie2 signaling in vascular progenitor cells. Blood. 2004;104:166–169. doi: 10.1182/blood-2003-09-3293. [DOI] [PubMed] [Google Scholar]
- 14.Lai L, Bohnsack BL, Niederreither K, Hirschi KK. Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development. 2003;130:6465–6474. doi: 10.1242/dev.00887. [DOI] [PubMed] [Google Scholar]
- 15.Marino D, Dabouras V, Brandli AW, Detmar M. A role for all-trans-retinoic acid in the early steps of lymphatic vasculature development. J Vasc Res. 2010;48:236–251. doi: 10.1159/000320620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutkowski JM, Moya M, Johannes J, Goldman J, Swartz MA. Secondary lymphedema in the mouse tail: Lymphatic hyperplasia, vegf-c upregulation, and the protective role of mmp-9. Microvasc Res. 2006;72:161–171. doi: 10.1016/j.mvr.2006.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaetano C, Catalano A, Illi B, Felici A, Minucci S, Palumbo R, Facchiano F, Mangoni A, Mancarella S, Muhlhauser J, Capogrossi MC. Retinoids induce fibroblast growth factor-2 production in endothelial cells via retinoic acid receptor alpha activation and stimulate angiogenesis in vitro and in vivo. Circ Res. 2001;88:E38–E47. doi: 10.1161/01.res.88.4.e38. [DOI] [PubMed] [Google Scholar]
- 18.Hong YK, Shin JW, Detmar M. Development of the lymphatic vascular system: A mystery unravels. Dev Dyn. 2004;231:462–473. doi: 10.1002/dvdy.20179. [DOI] [PubMed] [Google Scholar]
- 19.Oliver G, Alitalo K. The lymphatic vasculature: Recent progress and paradigms. Annu Rev Cell Dev Biol. 2005;21:457–483. doi: 10.1146/annurev.cellbio.21.012704.132338. [DOI] [PubMed] [Google Scholar]
- 20.Helotera H, Alitalo K. The vegf family, the inside story. Cell. 2007;130:591–592. doi: 10.1016/j.cell.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Makinen T, Alitalo K. Lymphangiogenesis in development and disease. Novartis Found Symp. 2007;283:87–98. doi: 10.1002/9780470319413.ch8. discussion 98-105, 238-141. [DOI] [PubMed] [Google Scholar]
- 22.Karpanen T, Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. 2008;3:367–397. doi: 10.1146/annurev.pathmechdis.3.121806.151515. [DOI] [PubMed] [Google Scholar]
- 23.Panek RL, Lu GH, Dahring TK, Batley BL, Connolly C, Hamby JM, Brown KJ. In vitro biological characterization and antiangiogenic effects of pd 166866, a selective inhibitor of the fgf-1 receptor tyrosine kinase. Journal of Pharmacology and Experimental Therapeutics. 1998;286:569–577. [PubMed] [Google Scholar]
- 24.Kubo K, Shimizu T, Ohyama S, Murooka H, Iwai A, Nakamura K, Hasegawa K, Kobayashi Y, Takahashi N, Takahashi K, Kato S, Izawa T, Isoe T. Novel potent orally active selective vegfr-2 tyrosine kinase inhibitors: Synthesis, structure-activity relationships, and antitumor activities of n-phenyl-n'-{4-(4-quinolyloxy)phenyl}ureas. J Med Chem. 2005;48:1359–1366. doi: 10.1021/jm030427r. [DOI] [PubMed] [Google Scholar]
- 25.Kirkin V, Thiele W, Baumann P, Mazitschek R, Rohde K, Fellbrich G, Weich H, Waltenberger J, Giannis A, Sleeman JP. Maz51, an indolinone that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo. Int J Cancer. 2004;112:986–993. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- 26.Shin JW, Min M, Larrieu-Lahargue F, Canron X, Kunstfeld R, Nguyen L, Henderson JE, Bikfalvi A, Detmar M, Hong YK. Prox1 promotes lineage-specific expression of fibroblast growth factor (fgf) receptor-3 in lymphatic endothelium: A role for fgf signaling in lymphangiogenesis. Mol Biol Cell. 2006;17:576–584. doi: 10.1091/mbc.E05-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dow R, Hendley J, Pirkmaier A, Musgrove EA, Germain D. Retinoic acid-mediated growth arrest requires ubiquitylation and degradation of the f-box protein skp2. J Biol Chem. 2001;276:45945–45951. doi: 10.1074/jbc.M103593200. [DOI] [PubMed] [Google Scholar]
- 28.Di Stefano V, Giacca M, Capogrossi MC, Crescenzi M, Martelli F. Knock-down of cyclin-dependent kinase inhibitors induces cardiomyocyte re-entry in the cell cycle. J Biol Chem. 2011;286:8644–8654. doi: 10.1074/jbc.M110.184549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salaun P, Rannou Y, Prigent C. Cdk1, plks, auroras, and neks: The mitotic bodyguards. Adv Exp Med Biol. 2008;617:41–56. doi: 10.1007/978-0-387-69080-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirza AM, Gysin S, Malek N, Nakayama K, Roberts JM, McMahon M. Cooperative regulation of the cell division cycle by the protein kinases raf and akt. Mol Cell Biol. 2004;24:10868–10881. doi: 10.1128/MCB.24.24.10868-10881.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, Meloche S. P27 cytoplasmic localization is regulated by phosphorylation on ser10 and is not a prerequisite for its proteolysis. EMBO J. 2001;20:6672–6682. doi: 10.1093/emboj/20.23.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrova TV, Makinen T, Makela TP, Saarela J, Virtanen I, Ferrell RE, Finegold DN, Kerjaschki D, Yla-Herttuala S, Alitalo K. Lymphatic endothelial reprogramming of vascular endothelial cells by the prox-1 homeobox transcription factor. EMBO J. 2002;21:4593–4599. doi: 10.1093/emboj/cdf470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B, Hong YK. Visualization of lymphatic vessels by prox1-promoter directed gfp reporter in a bacterial artificial chromosome-based transgenic mouse. Blood. 2011;117:362–365. doi: 10.1182/blood-2010-07-298562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.