

Abstract
This article describes the development of microwave-assisted oxyanionic 5-exo-dig cyclization-Claisen rearrangement sequence as a convenient “one-pot” route to a variety of seven-membered carbocyclic ring systems. This process was used as the key transformation for the construction of several natural products, including frondosins A, B, and C.
Keywords: Claisen rearrangement, sigmatropic rearrangement, 5-exo-dig cyclization, microwave-assisted synthesis
1. Introduction
Carbocyclic seven-membered rings are found as common structural units in a variety of polycyclic natural products, such as the phorbol esters,1 guanacastepenes,2 guianolides3 and the frondosins.4 Due to their medicinal relevance, numerous research groups are actively engaged in developing strategies for their construction by synthetic means. However, unlike five and six-membered carbocycles which are readily accessible through various cyclization reactions, methods allowing the construction of seven-membered rings are generally limited to processes other than direct intramolecular reactions. Among the most important of these are various cycloaddition strategies, such as the [5+2] and [4+3] reactions, which have allowed the synthesis of a number of cycloheptanoid natural products.5
2. Sequential 5-Exo Cyclization/Claisen Rearrangement Strategy
We have recently demonstrated that a variety of cycloheptanoid fused ring systems may be conveniently accessed through a known6 but largely ignored tandem sequence that involves a base-catalyzed intramolecular cyclization of appropriately substituted acetylenic alcohols (4-pentyn-1-ols), followed by in situ Claisen rearrangement of the intermediate 2-alkylidenetetrahydrofurans.7
The requisite allyl vinyl ether precursor in these reactions is produced as a transient species through a 5-exo-dig process, which involves the intramolecular addition of an alkoxide moiety to a proximal triple bond. Upon heating, this intermediate undergoes in situ [3,3] sigmatropic rearrangement, affording a cyclohept-4-enone derivative. In a typical case, the tandem cyclization-Claisen rearrangement sequence is effected simply by treatment of an appropriately substituted acetylenic alcohol, dissolved in phenetole, diphenyl ether or DMF with approximately 10 mol-% of MeLi, and heating the mixture to 100–210 °C. It is noteworthy that although the generation of various furanyl systems by transition metal catalyzed 5-exo-dig cyclizations of acetylenic alcohols has been amply documented,8 the formation of 2-alkylidenetetrahydrofurans by intramolecular addition of an alkoxide moiety to triple bonds is much less common.
Although the initial experiments were conducted using conventional heating, it was later discovered that the use of microwave irradiation in sealed vials is particularly well suited for these processes.7e As shown in Table 1, this strategy was successfully utilized for the generation of several mono-, di-, tri- and tetracyclic seven-membered ring systems. As a rule, the tandem cyclization/Claisen rearrangement reactions conducted under microwave irradiation are operationally simpler, they proceed faster and afford higher yields of expected products.
Table 1.
Synthesis of seven-membered carbocyclic structures by sequential 5-exo cyclization/Claisen rearrangement strategy.
| Entry | 4-Alkyn-1-ol | Conditionsa | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 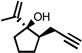 |
150 °C 12 minb |
 |
72 |
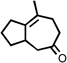
| 2 | 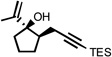 |
200 °C 15 minc |
 |
74 |
| 3 | 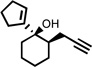 |
200 °C 4 minb |
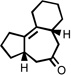 |
92 |
| 4 | 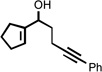 |
210 °C 45 minb |
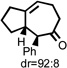 |
77 |
| 5 | 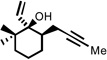 |
200 °C 15 minc |
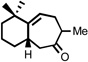 |
77 |
| 6 | 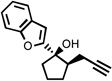 |
200 °C 30 minc |
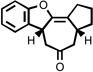 |
63 |
| 7 | 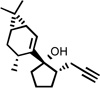 |
185 °C 60 mind |
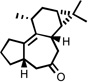 |
76 |
Reactions were performed using a CEM microwave (unless otherwise noted) oven in the presence of 10–15 mol-% MeLi.
phenetole was used as the solvent.
DMF was used as the solvent.
Reaction was done using conventional heating (oil bath).
3. Application to Natural Product Synthesis: the Frondosins
Frondosins A–E comprise five related novel sesquiterpene hydroquinone derivatives recently isolated from the Micronesian marine sponge Dysidea frondosa.9 All members of the frondosin family (A–E) are antagonists of interleukin-8 (IL-8) and inhibitors of protein kinase C (PKC) in the low micromolar range.10 In addition to being involved in cellular inflammatory events,11 IL-8 is now known to also play an important role in tumor progression and metastasis in several human cancers,12 including lung cancers.12b It is has also been reported that IL-8, along with growth-regulated oncogene alpha, is involved in chemoattraction, neovascularization and stimulation of HIV-1 replication both in T-lymphocytes and macrophages.13 Importantly, it was recently demonstrated that compounds which inhibit the actions of IL-8 also inhibit HIV-1 replication.14
The unique structural features of the frondosins coupled with their therapeutic potential as lead compounds for the development of novel anti-inflammatory, anti-tumor and anti-HIV agents has sparked considerable interest in their total synthesis by several research groups. To date, these efforts have resulted in several published total syntheses of frondosin B,15a-f,16,17 one of frondosin C18 and a recent total synthesis of (+)-frondosin A by Trost et al.19
3.1 Frondosin C
Among the tetracyclic members of the frondosin family (frondosin B-E), frondosin C is unique in that its ring core is entirely carbocyclic.9 As depicted in Scheme 1, assembly of the tetracyclic skeleton of frondosin C was achieved in a straightforward fashion via the tandem cyclization-Claisen rearrangement strategy described above.18 Thus, ketone 2 was produced in 80% yield as a 2.7:1 mixture of diastereomers when the tertiary alcohol 1 was exposed to catalytic MeLi and microwave irradiation at 210 °C. Subsequent DDQ oxidation allowed installation of the requisite diene functionality in the B ring, and the resulting product 3 could be manipulated further in a short sequence of steps to yield the indene derivative 4. Finally, hypervalent iodine oxidation of 4 afforded the target (±)-frondosin C along with its epimer 8-epi-frondosin C, which were readily separated by standard column chromatography.18
Scheme 1.

Total synthesis of (±)-frondosin C and (±)-8-epi-frondosin C.
Reagents and conditions: (a) 10 mol-% MeLi, 210 °C (µwave), phenetole, 40 min, 80%, (b) DDQ, DCM, 0 °C, 89%, (c) NaBH4, MeOH, 0 °C, 97%, (d) i. MsCl, TEA, DCM, ii. TsNHNH2, TEA, 70% (2 steps), (d) TBAF, THF, rt, 96%, (e) BTIB, MeCN/H2O, 0°C, 10 min.
3.2 Frondosin B
The bicyclic 6-7 core of frondosin B was readily constructed in a “one-pot” procedure from secondary alcohol 5 as depicted in Scheme 2.16 The resulting ketone 6 was subsequently methylated under kinetic conditions, affording compound 7 in excellent yield. The key step in the remainder of the sequence was the Lewis acid induced cyclization of hydroquinone 8 which provided the entire tetracyclic scaffold of frondosin B. Subsequent isomerization of the trisubstituted double bond was achieved by refluxing 9 in benzene in the presence of catalytic TsOH.16
Scheme 2.

Total synthesis of (±)-frondosin B.
Reagents and conditions: (a) 10 mol-% MeLi, 210 °C (µwave), phenetole, 45 min, 80%, (b) i. LHMDS, 89%, THF, −78 °C, ii. MeI −60 °C, 92% (c) CAN, MeCN/H2O, rt, 96% (d) i. H2, Pd/C, 5 min, (e) BF3•OEt2, DCM, 0 °C, 95% (2 steps), (f) TsOH, benzene, reflux, 5 h, 70%.
3.3 Frondosin A
Following the successful total synthesis of frondosin B, it was envisioned that ketone 7 might also serve as a useful precursor to frondosin A, potentially requiring only a few additional steps to complete the target natural product, namely: (1) isomerization of the trisubstituted double bond, (2) epimerization at C8, (3) installation of the requisite exocyclic double bond via standard methenylation chemistry, and (4) aromatic O-demethylation.
Although most of the planned steps were relatively straightforward, the C5-C6 double bond in 7 was surprisingly resistant to isomerization under a variety of conditions attempted.20 However, the desired transformation could be achieved indirectly, using the Saegusa oxidation21 as the key step as shown in Scheme 3. This sequence initially resulted in the formation of two regioisomeric dienes 10 and 11; however, it was discovered that the undesired isomer 10 could be efficiently and quantitatively converted to the less conjugated isomer 11 – probably via the fully conjugated enol tautomer 10a22 – on simple heating in refluxing ethyl acetate.
Scheme 3.

Synthesis of 8-epi-Frondosin A dimethyl ether.
Reagents and conditions: (a) i. TMSOTf, NEt3, DCM, ii. Pd(OAc)2, CH3CN, 75%, (c) H2, Pd/C, 95%, (d) Mg, TiCl4, DCM/THF, 73%.
Brief exposure of diene 11 to catalytic hydrogenation conditions provided ketone 12 in 95% yield and subsequent olefination of the hindered ketone according to the method of Yan et al.23 provided the epimer of frondosin A 13 as its dimethyl ether derivative in 51.5% overall yield from compound 7.
Additionally, ketone 12 could also be isomerized under basic conditions (KHMDS) to install the requisite cis substitution pattern at the B ring, characteristic of naturally-occurring frondosin A. Thus, the synthesis of (±)-frondosin A dimethyl ether 15 was achieved in 70% isolated yield from 14 (Scheme 4). Given that compound 15 is the same advanced intermediate that Trost et al.19 recently converted to frondosin A via a one-pot oxidation-reduction protocol, this constitutes a formal total synthesis of racemic frondosin A. 20
Scheme 4.

Formal synthesis of (±)-frondosin A.
Reagents and conditions: (a) i. KHMDS, rt, 30 min, ii. AcOH, (b) Mg, TiCl4, DCM/THF, 70%.
4. Application to the Synthesis of Optically Active Systems
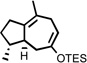
We have recently demonstrated that the tandem 5-exo cyclization/Claisen rearrangement sequence may be readily extended to the synthesis of optically active cycloheptenone derivatives.17 This methodology involves the use of nonracemic allylic alcohol starting materials (derived from the corresponding enones through asymmetric reduction) and relies on the ability of a single stereogenic center built within the incipient tetrahydrohydrofuranyl intermediate to confer stereochemical information to the final cycloheptenone product having up to two new stereocenters. Ultimately, the stereochemical integrity of the reaction is the result of a preferred chair-like six-membered transition state for the Claisen rearrangement, where the two prochiral centers of the 2-alkylidene tetrahydrofuran intermediate occupy a pseudo diequatorial relationship.7h
The feasibility of this sequence was tested using a number of prochiral α,β-unsaturated ketones which were efficiently reduced to optically active allylic alcohols using the well-established CBS methodology.24 Importantly, the resulting nonracemic alcohols were subsequently converted to optically active cycloheptenone derivatives with almost complete transfer of chirality via the cycloisomerization/Claisen rearrangement process (Figure 3).17
Figure 3.

General strategy for the generation of optically active cycloheptenone derivatives.
4.1 Total Synthesis of (–)-Frondosin B
As a direct application of the asymmetric methodology discussed above, optically active (–)-frondosin B was prepared in a straightforward seven-step sequence starting with enone 16 as shown in Scheme 5.17 Thus, 16 was treated with catalytic (–)-CBS reagent and excess catechol borane to afford the requisite nonracemic allylic alcohol 17 in 68% yield and 98% ee. Subsequent exposure to catalytic MeLi and microwave irradiation, followed by methylation of the resulting bicyclic ketone provided 18 in excellent chemical yield and ee. Further manipulation of this intermediate according to the sequence already described for the racemic analogue (Scheme 2) gave (–)-frondosin B, which possesses the opposite absolute stereochemistry compared to the naturally occurring frondosin B.
Scheme 5.

Total synthesis of (–)-frondosin B.
Reagents and conditions: (a) catechol borane, (–)-CBS reagent, toluene, −78 °C, 72h, 68%, 98% ee, (b) cat. MeLi, 210 °C (µwave), 1h, 79% (c) i. LHMDS, THF, −78 °C, ii. MeI, −60 °C, 85%, 97% ee, (d) CAN, MeCN/H2O, rt, 93% (e) i. H2, Pd/C, 5 min, (f) BF3•OEt2, DCM, 0 °C, 2% (2 steps), (g) cat. TsOH, benzene, reflux 5h, 68%.
In summary, sequential oxyanionic 5-exo-dig cyclization/Claisen rearrangement has been developed, allowing the straightforward synthesis of a number of interesting cycloheptanoid ring systems. The methodology is ideally suited for natural product synthesis and has been so far applied to the preparation of three meroterpenoid natural products frondosin A, B and C. In addition, an asymmetric variant of the process has been developed which may be readily employed to access optically active cycloheptenone ring systems.
Figure 1.

Representative natural products containing carbocyclic seven-membered rings.
Figure 2.

General strategy for the synthesis of polycyclic carbocyclic cycloheptanoid ring systems via tandem cyclization/Claisen rearrangement.
Acknowledgements
This research was supported by grants from the National Institutes of Health (NIGMS). T.V.O also gratefully acknowledges support from the Hans and Ella McCollum-Vahlteich ’21 endowment.
References
- 1.Evans FJ, editor. Naturally Occurring Phorbol Esters. Boca Raton, FL: CRC; 1986. [Google Scholar]
- 2.Bondi SM, Clardy J. J. Am. Chem. Soc. 2001;123:9900. doi: 10.1021/ja016176y. [DOI] [PubMed] [Google Scholar]
- 3.Wong H-F, Brown GD. J. Nat. Prod. 2002;65:481. doi: 10.1021/np0103113. [DOI] [PubMed] [Google Scholar]
- 4.Patil AD, Freyer AJ, Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047. [Google Scholar]
- 5.For a recent review, see Battiste MA, Pelphrey PM, Wright DL. Chem. Eur. J. 2006;12:3438. doi: 10.1002/chem.200501083.
- 6.Marvell EN, Titterington D. Tetrahedron Lett. 1980:2123. [Google Scholar]
- 7.(a) Ovaska TV, Roark JL, Shoemaker CM. Tetrahedron Lett. 1998;39:5705. [Google Scholar]; (b) Ovaska TV, Roses JB. Org. Lett. 2000;2:2361. doi: 10.1021/ol006139w. [DOI] [PubMed] [Google Scholar]; (c) Ovaska TV, Reisman SE, Flynn MA. Org. Lett. 2001;3:115. doi: 10.1021/ol006823a. [DOI] [PubMed] [Google Scholar]; (d) Ovaska TV, Ravi Kumar JS, Hulford CA, O’Sullivan MF, Reisman SE. Tetrahedron Lett. 2002;43:1939. [Google Scholar]; (e) McIntosh CE, Martinez I, Ovaska TV. Synlett. 2004:2579. [Google Scholar]; (f) Martinez I, Alford PE, Ovaska TV. Org. Lett. 2005;7:1133. doi: 10.1021/ol050144o. [DOI] [PubMed] [Google Scholar]; (g) Li X, Kyne RE, Ovaska TV. Org. Lett. 2006;8:5153. doi: 10.1021/ol0620848. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li X, Kyne RE, Ovaska TV. J. Org. Chem. 2007;72:6624. doi: 10.1021/jo0710432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For a recent review on transition metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes, see Alonso F, Beletskaya IP, Yus M. Chem. Rev. 2004;104:3079. doi: 10.1021/cr0201068.
- 9.Patil AD, Freyer AJ, Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047. [Google Scholar]
- 10.Hallock YF, Cardellina JH, II, Boyd MR. Nat. Prod. Lett. 1998;11:153. [Google Scholar]
- 11.(a) Seitz M, Dewald B, Gerber N, Baggiolini M. J. Clin. Invest. 1991;87:463. doi: 10.1172/JCI115018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller EJ, Cohen AB, Nagao D, Griffith RJ, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Am. Rev. Respir. Dis. 1992;146:247. doi: 10.1164/ajrccm/146.2.427. [DOI] [PubMed] [Google Scholar]
- 12.(a) Brat DJ, Bellail AC, Van Meir EG. Neuro-oncol. 2005;7:122. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu YM, Webster SJ, Flower D, Woll PJ. Br. J. Cancer. 2004;91:1970. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan A, Chen JJ, Yao PL, Yang PC. Front. Biosci. 2005:853. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 13.Lane BR, Liu J, Bock PJ, Schols D, Coffey MJ, Strieter RM, Polverini PJ, Markovitz DM. J. Virol. 2002;76:11570. doi: 10.1128/JVI.76.22.11570-11583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane BR, Lore K, Bock PJ, Andersson J, Coffey MJ, Strieter RM, Markovitz DM. J. Virol. 2001;75:8195. doi: 10.1128/JVI.75.17.8195-8202.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Inoue M, Frontier AJ, Danishefsky SJ. Angew. Chem., Int. Ed. 2000;39:761. [PubMed] [Google Scholar]; (b) Inoue M, Carson MW, Frontier AJ, Danishefsky SJ. J. Am. Chem. Soc. 2001;123:1878. doi: 10.1021/ja0021060. [DOI] [PubMed] [Google Scholar]; (c) Hughes CC, Trauner D. Angew. Chem., Int. Ed. 2002;41:1569. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (d) Hughes CC, Trauner D. Tetrahedron. 2004;60:9675. [Google Scholar]; (e) Kerr DJ, Willis AC, Flynn BL. Org. Lett. 2004;6:457. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]; (f) Reiter M, Torssell S, Lee S, MacMillan DWC. Chem. Sci. 2010;1:37. doi: 10.1039/C0SC00204F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Olson P, Davies HML. Org. Lett. 2008;10:573. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]; (h) Masters K-S, Flynn BL. J. Org. Chem. 2008;73:8081. doi: 10.1021/jo800682n. [DOI] [PubMed] [Google Scholar]; (i) Mehta G, Likhite NS. Tetrahedron Lett. 2008;49:7113. [Google Scholar]
- 16.Li X, Ovaska TV. Org. Lett. 2007;9:3837. doi: 10.1021/ol701633z. [DOI] [PubMed] [Google Scholar]
- 17.Ovaska TV, Sullivan JA, Ovaska SI, Winegrad JB, Fair JD. Org. Lett. 2009;11:2715. doi: 10.1021/ol900967j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Kyne RE, Ovaska TV. Tetrahedron. 2007;63:1899. [Google Scholar]
- 19.Trost BM, Hu Y, Horne DB. J. Am. Chem. Soc. 2007;129:11781. doi: 10.1021/ja073272b. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Keon AE, Sullivan JA, Ovaska TV. Org. Lett. 2008;10:3287. doi: 10.1021/ol8011343. [DOI] [PubMed] [Google Scholar]
- 21.Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011. [Google Scholar]
- 22.For related “deconjugative” processes of enone systems, see e.g. Rychnovsky SD, Mickus DE. J. Org. Chem. 1992;57:2732. ; (b) Rizvi SQA, Williams JR. J. Org. Chem. 1981;46:1127. [Google Scholar]; (c) Malhotra SK, Ringold HJ. J. Am. Chem. Soc. 1963;85:1538. [Google Scholar]
- 23.Yan T-H, Tsai C-C, Chien C-T, Cho C-C, Huang P-C. Org. Lett. 2004;6:4961. doi: 10.1021/ol0478887. [DOI] [PubMed] [Google Scholar]
- 24.For reviews, see: Corey EJ, Helal CJ. Angew. Chem., Int. Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. ; Byung TC. Tetrahedron. 2006;62:7621. [Google Scholar]