

Abstract
Extracellular ATP stimulates proliferation of vascular smooth muscle cells (VSMC) through activation of G protein-coupled P2Y purinergic receptors. We have previously shown that ATP stimulates a transient activation of protein kinase A (PKA), which, together with the established mitogenic signaling of purinergic receptors, promotes proliferation of VSMC (Hogarth DK, Sandbo N, Taurin S, Kolenko V, Miano JM, Dulin NO. Am J Physiol Cell Physiol 287: C449–C456, 2004). We also have shown that PKA can phosphorylate β-catenin at two novel sites (Ser552 and Ser675) in vitro and in overexpression cell models (Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. J Biol Chem 281: 9971–9976, 2006). β-Catenin promotes cell proliferation by activation of a family of T-cell factor (TCF) transcription factors, which drive the transcription of genes implicated in cell cycle progression including cyclin D1. In the present study, using the phosphospecific antibodies against phospho-Ser552 or phospho-Ser675 sites of β-catenin, we show that ATP can stimulate PKA-dependent phosphorylation of endogenous β-catenin at both of these sites without affecting its expression levels in VSMC. This translates to a PKA-dependent stimulation of TCF transcriptional activity through an increased association of phosphorylated (by PKA) β-catenin with TCF-4. Using the PKA inhibitor PKI or dominant negative TCF-4 mutant, we show that ATP-induced cyclin D1 promoter activation, cyclin D1 protein expression, and proliferation of VSMC are all dependent on PKA and TCF activities. In conclusion, we show a novel mode of regulation of endogenous β-catenin through its phosphorylation by PKA, and we demonstrate the importance of this mechanism for ATP-induced proliferation of VSMC.
Keywords: purinergic, protein kinase A, cyclin D1
extracellular atp is released from sympathetic nerves, activated platelets, inflammatory cells, and endothelial cells to stimulate vascular smooth muscle cell (VSMC) contraction and proliferation (16). The levels of ATP rise dramatically during vascular injury, hypoxia, or inflammatory cell activation (9, 14). Under these conditions, ATP stimulates proliferation of VSMC and adventitial fibroblasts, which contributes to vascular hypertrophy during atherosclerosis, restenosis, or pulmonary hypertension (6, 12, 14). The mitogenic signaling of ATP in VSMC is mediated by P2Y purinergic receptors and includes activation of phospholipase C, release of inositol trisphosphate, and diacyl glycerol, Ca2+ mobilization, and activation of various protein kinases (Ca2+/calmodulin-dependent kinase, protein kinase C, and mitogen-activated protein kinases) (4, 12, 15, 20). We and others have previously shown that ATP stimulates a transient cAMP production and PKA activation, which is required for maximal DNA synthesis in response to ATP (19, 31). However, the molecular mechanism by which PKA may promote VSMC proliferation upon purinergic receptor stimulation remains elusive.
β-Catenin is a multifunctional protein that controls cell-cell adhesion and cell proliferation. In the latter function, β-catenin stimulates T-cell factor (TCF)/lymphoid enhancer factor transcription factors to induce transcription of a variety of growth-promoting genes, including c-myc (3) and cyclin D1 (29). In quiescent cells, β-catenin is maintained at low levels in the cytoplasm through phosphorylation by casein kinase-1 at Ser45 and by glycogen synthase kinase-3 (GSK-3) at Ser33/Ser37/Thr41 sites, respectively (23), and its subsequent ubiquitination and degradation by the proteosome (2, 7). Inhibition of GSK-3 through Wnt signaling results in a decrease in phosphorylation of β-catenin at Ser33/Ser37/Thr41 sites, its stabilization, and activation of TCF-dependent gene transcription (30). Mutations of β-catenin or of its regulatory proteins, resulting in the accumulation of β-catenin and the activation of TCF-dependent gene transcription, are frequently found in various types of cancers (5, 21). β-Catenin signaling is also implicated in VSMC proliferation in vitro and in vivo during vascular injury (22, 25).
We have recently discovered that PKA can phosphorylate β-catenin at Ser552 and Ser675 sites, and this phosphorylation by PKA promotes transcriptional activity of β-catenin in over-expression cell models (28). In the present study, we sought to examine whether ATP, through PKA, can stimulate phosphorylation of endogenous β-catenin at Ser552 and Ser675 sites and how this translates to ATP-induced proliferation of VSMC.
MATERIALS AND METHODS
Cell culture
The rat VSMC were isolated from Wistar-Kyoto rat aortas by enzymatic digestion and maintained as described previously (10). Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml streptomycin, 250 ng/ml amphotericin B, and 100 U/ml penicillin. Twenty-four hours before stimulation, the cells were serum deprived using Dulbecco's modified Eagle's medium containing 0.1% bovine serum albumin and 2 mM l-glutamine. Transient transfections were performed by using LipofectAMINE-PLUS reagent (Invitrogen) following the standard manufacturer's protocol. Adenovirus-meditated gene transduction was performed as described previously (19). This study was approved by the University of Chicago Biosafety and Animal Care and Use Committees.
Reagents
The cDNA for Flag-tagged β-catenin and its mutants were described previously (28). The cyclin D1 promoter (−1,745 base pairs) luciferase reporter was from Dr. Richard Pestell. The TCF/lymphoid enhancer factor luciferase reporter (TOP) and its negative control (FOP) plasmids were from Upstate Biotechnology. The dominant negative PKA plasmid (dnPKA) was described previously (8). The dominant negative TCF-4 plasmid (dnTCF-4) was from Dr. Tong-Chuan He. Adenovirus encoding protein kinase inhibitor PKI (Ad-PKI) was described previously (19). Adenovirus encoding the dominant negative TCF-4 mutant was from Vector Biolabs. Antibodies against β-catenin, phospho-S552-β-catenin, and phospho-S675-β-catenin were from Cell Signaling Technology. Antibodies against Flag and β-actin were from Sigma-Aldrich. Antibodies against Flag and β-actin were from Sigma Aldrich. antibodies against TCF-4 and cyclin D1 were from Santa Cruz Biotechnology. Antibodies against ERK1/2 were from Dr. Michael Dunn.
Immunoprecipitation and Western blot analysis
Cells were lysed in a buffer containing 150 mM NaCl, 20 mM TRIS (pH 7.5), 1 mM EDTA, 1 mM EGTA, 0.5% Triton X-100, protease inhibitors (1 mg/ml leupeptin, 1 mg/ml aprotinin, and 1 mM PMSF), and phosphatase inhibitors (1 mM NaF, 200 mM Na-orthovanadate). The lysates were cleared by centrifugation at 14,000 g for 10 min. Immunoprecipitation of Flag-tagged β-catenin proteins was performed using agarose-conjugated mouse anti-Flag antibodies (Sigma-Aldrich). The immunoprecipitation of endogenous β-catenin was performed using agarose-conjugated goat anti-β-catenin antibodies (Santa Cruz Biotechnology). Immunoprecipitation of TCF-4 was performed by incubating cleared cell lysates with 10 μg/ml rabbit polyclonal TCF-4 antibodies (Santa Cruz Biotechnology) at 4°C overnight, followed by incubation with protein A/protein G-conjugated agarose beads. The immune complexes were washed three times with 1 ml lysis buffer, boiled in Laemmli buffer, and subjected to polyacrylamide gel electrophoresis and Western blotting with the desired primary antibodies, followed by horseradish peroxidase-conjugated secondary antibodies, and developed by enhanced chemiluminescence reaction (Pierce). The digital chemiluminescence images were taken by a Luminescent Image Analyzer LAS-3000 (Fujifilm).
Nonradioactive in vitro assay for PKA activity
Following stimulation with desired agonists, the cells (grown in 12-well plates) were lysed in 0.1 ml/well lysis buffer containing 25 mM HEPES (pH 7.5), 0.5% NP-40, protease inhibitors (1 mg/ml leupeptin, 1 mg/ml aprotinin, and 1 mM PMSF), and phosphatase inhibitors (1 mM NaF, 200 mM Na-orthovanadate) (19). The lysates were cleared from insoluble material by centrifugation at 20,000 g for 10 min, and 5 μl cleared lysates were subjected to a kinase reaction with the fluorescence-labeled PKA substrate kemptide (Promega) following the manufacturer's protocol. The reaction was stopped by boiling the samples for 10 min. The phosphorylated kemptide was separated from the non-phosphorylated one by 0.8% agarose electrophoresis. The fluorescent images were taken by Luminescent Image Analyzer LAS-3000.
Luciferase reporter assay
Cells were transfected with desired luciferase reporter plasmid, thymidine kinase (TK)-driven renilla plasmid (transfection efficiency control), and cDNA encoding a gene of interest or an empty plasmid, serum starved overnight, followed by stimulation with 30 μM ATP for 12 h. The cells were washed twice with PBS, lysed in protein extraction reagent, and assayed for luciferase and renilla activity using the corresponding assay kits (Promega, Madison, WI). To account for differences in transfection efficiency, luciferase activity of each sample was normalized to renilla activity.
The [3H]thymidine uptake assay was performed as described previously (19). Serum-starved VSMC were stimulated with 30 μM ATP for 24 h. [3H]thymidine (1 μCi/ml) was added 6 h after cell stimulation for 18 h. The cells were then washed twice with ice-cold PBS, precipitated with 10% trichloroacetic acid (TCA) for 30 min, washed once with 5% TCA, and lysed in a solution containing 0.1% NaOH and 0.1% SDS for 15 min. The lysates were analyzed for radioactivity by scintillation spectrometry.
RESULTS
To examine whether ATP stimulates phosphorylation of β-catenin at Ser552 and Ser675 sites that are the PKA substrates in vitro (28), we used the phosphospecific antibodies generated against the corresponding phosphorylation sites of β-catenin. We first characterized the specificity of antibodies by assessing phosphorylation of overexpressed (in Cos-7 cells) wild-type (WT) β-catenin or its phosphorylation-deficient mutants. As shown in Fig. 1, both antibodies recognized WT-β-catenin after PKA stimulation by forskolin. The S552A mutation abolished the recognition by phospho-Ser552 antibodies, but not by phospho-Ser675 antibodies. The S675A mutation abolished the recognition by phospho-Ser675 antibodies, but not by phospho-Ser552 antibodies. The double S552A/S675A mutation abolished the recognition by either antibodies.
Fig. 1.

Specificity of phospho-S552 and phospho-S675 β-catenin antibodies. Cos-7 cells were transfected with Flag-tagged β-catenin or the corresponding mutants of β-catenin and stimulated with 10 μM forskolin (FSK) for 5 min. The proteins were immunoprecipitated (IP) with Flag antibodies followed by Western blotting (WB) with phospho-S552 or phospho-S675 β-catenin antibodies as indicated. The equal amounts of immunoprecipitated β-catenin or the corresponding mutants were confirmed by Western blotting with Flag antibodies. WT, wild-type.
Having confirmed the specificity of these antibodies, we then examined the phosphorylation of endogenous β-catenin in VSMC stimulated by ATP. As shown in Fig. 2, ATP stimulated phosphorylation of β-catenin at both Ser-552 (Fig. 2B) and Ser-675 (Fig. 2C) sites, which was abolished by PKA inhibition through adenovirus-mediated overexpression of PKI. The effectiveness of PKI was confirmed by the inhibition of PKA activity as assessed by in vitro PKA assay of cell lysates (Fig. 2A). The specificity of PKI was confirmed by showing that it had no effect on ATP-induced phosphorylation of ERK1/2, as detected by electrophoretic mobility shift assay of phosphorylated ERK1/2 (Fig. 2E). It is noteworthy that β-catenin was also significantly phosphorylated at Ser675 site at the basal state, but probably not by PKA, because PKI expression did not attenuate this basal phosphorylation (Fig. 2C).
Fig. 2.
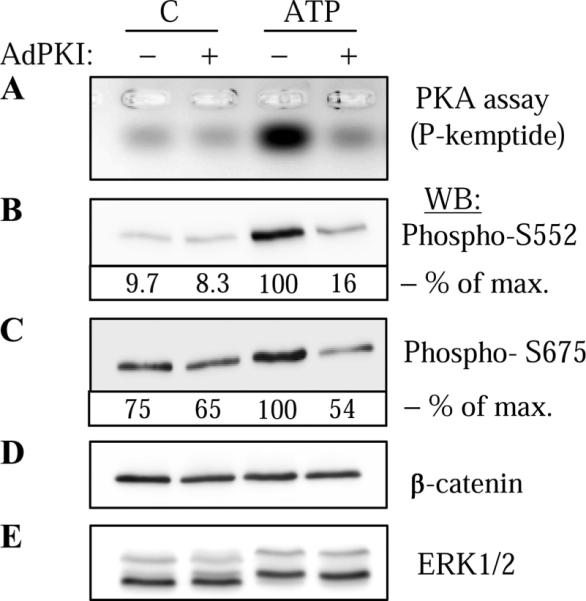
ATP-induced phosphorylation of endogenous β-catenin by PKA in vascular smooth muscle cells (VSMC). VSMC were transduced with control adenovirus (−) or adenovirus encoding PKA inhibitor PKI (Ad-PKI), stimulated with 30 μM ATP for 5 min and lysed. Cell lysates were analyzed for PKA activity (A) or were subjected to Western blotting with phospho-S552 (B) or phospho-S675 (C) β-catenin antibodies as indicated. The equal amounts of endogenous β-catenin were confirmed by Western blotting with β-catenin antibodies (D). For control purposes, ERK1/2 phosphorylation was assessed by electrophoretic mobility shift assay following Western blotting with ERK1/2 antibodies (E). The densitometry of selected blots is shown as % of maximal (max) response to ATP.
We then examined how the PKA-dependent phosphorylation of β-catenin by ATP affects its transcriptional activity in VSMC by using a luciferase reporter driven by TCF-binding elements. As shown in Fig. 3A, ATP stimulated a profound induction of TCF-luciferase activity. This increase was inhibited by overexpression of dominant negative PKA mutant (dnPKA), the efficiency and specificity of which were confirmed in our previous studies (8, 19). Given that the interaction between TCF transcription factors and β-catenin is critical for TCF-dependent gene transcription, we examined the effect of ATP (and the role of PKA) on β-catenin binding to the TCF-4 isoform that is expressed in VSMC. Figure 3B shows that β-catenin binding to TCF-4 was significantly increased on stimulation with ATP, as assessed by coimmunoprecipitation. This increased β-catenin/TCF-4 interaction was abolished by overexpression of PKI. Together, these data show that endogenous β-catenin is phosphorylated by PKA in VSMC stimulated by ATP and suggest that ATP-induced phosphorylation of β-catenin by PKA stimulates TCF-dependent gene transcription by promoting association of β-catenin with TCF-4.
Fig. 3.

ATP-induced, PKA-mediated activation of T-cell factor (TCF)-dependent gene transcription in VSMC. A: PKA-dependent activation of TCF reporter by ATP. VSMC were transfected with cDNA for TCF-luciferase (Luc) reporter (TOPflash) or the mutated control reporter (FOPflash) along with a renilla reporter driven by thymidine kinase promoter (TK-RL), and with an empty vector or cDNA for PKA dominant negative mutant (dnPKA). Following the stimulation of cells with 30 μM ATP for 12 h, luciferase activity was measured and normalized to a corresponding renilla activity. The TOP-Luc/TK-RL values were subtracted from the FOP-Luc/TK-RL values. Data represent means ± SD from a representative of three experiments performed in triplicate. *P < 0.01. B: PKA-dependent interaction between endogenous TCF-4 and β-catenin in response to ATP. VSMC were transduced with Ad-PKI, stimulated with ATP for 5 min, and lysed. Endogenous TCF-4 was immunoprecipitated from cell lysates, and the immune complexes or total cell lysates were examined by Western blotting with desired antibodies as indicated. The densitometry of a selected blot is shown as % of maximal response to ATP.
The β-catenin/TCF transcription complex drives the expression of several genes critical for cell cycle progression including cyclin D1. Figure 4A shows that ATP stimulates a profound induction of cyclin D1 promoter activity as assessed by luciferase reporter driven by – 1,745 cyclin D1 promoter. Overexpression of dominant negative mutant of TCF-4 (dnTCF-4) significantly attenuated ATP-induced cyclin D1 promoter activation (Fig. 4A). The efficiency of dnTCF-4 was confirmed by its inhibition of TCF-luciferase reporter activity (Fig. 4B). The specificity of dnTCF-4 was confirmed by showing no effect of dnTCF-4 on ATP-induced cAMP response element (CRE) reporter activity (Fig. 4C). Furthermore, the stimulation of cyclin D1 promoter by ATP was also significantly decreased by dnPKA (Fig. 4D). Together, these data suggest that activation of cyclin D1 promoter by ATP in VSMC is mediated, at least in part, by PKA and TCF-4.
Fig. 4.

ATP-induced activation of cyclin D1 promoter in VSMC is mediated by TCF-4 and PKA. VSMC were transfected with a control TK-RL DNA, a desired luciferase reporter for cyclin D1 promoter (A and D), TCF (Top/Fop) (B), or cAMP response element (CRE; C), along with the empty vector or the cDNAs for the TCF-4 dominant negative mutant (dnTCF-4; A–C), or dnPKA (D). Following the stimulation of cells with 30 μM ATP for 12 h, luciferase activity was measured and normalized to a corresponding renilla activity. Data represent means ± SD from a representative of three experiments performed in triplicate. *P < 0.01.
We next examined whether the above experiments translate functionally to the expression of the endogenous cyclin D1 protein and to proliferation of VSMC. As shown in Fig. 5A, ATP stimulated a time-dependent expression of cyclin D1 with the maximum at 12 h of exposure. Adenovirus-mediated transduction of either PKI (Fig. 5B), or dnTCF-4 (Fig. 5C) inhibited ATP-induced cyclin D1 expression without affecting the levels of housekeeping β-actin protein. Furthermore, having previously demonstrated that ATP-induced VSMC proliferation is partially mediated by PKA (19), and given the activation of β-catenin/TCF-4 signaling by PKA established in the present study and before (28), we examined the role of TCF-4 in ATP-induced proliferation of VSMC. As shown in Fig. 6, adenovirus-mediated transduction of dnTCF-4 significantly decreased DNA synthesis in response to ATP. Together, these results demonstrate the importance of PKA-mediated β-catenin/TCF-4 signaling in ATP-induced proliferation of VSMC.
Fig. 5.

ATP-induced activation of cyclin D1 protein expression in VSMC is mediated by TCF-4 and PKA. A: time course of cyclin D1 protein expression in response to 30 μM ATP as assessed by Western blotting with cyclin D1 antibodies. B and C: VSMC were transduced with control adenovirus or with Ad-PKI (B) or with Ad-dnTCF-4 (C). Cells were stimulated with 30 μM ATP for 12 h, and cell lysates were analyzed by Western blotting with antibodies against cyclin D1 and β-actin. The densitometry of selected blots is shown as % of maximal response to ATP.
Fig. 6.
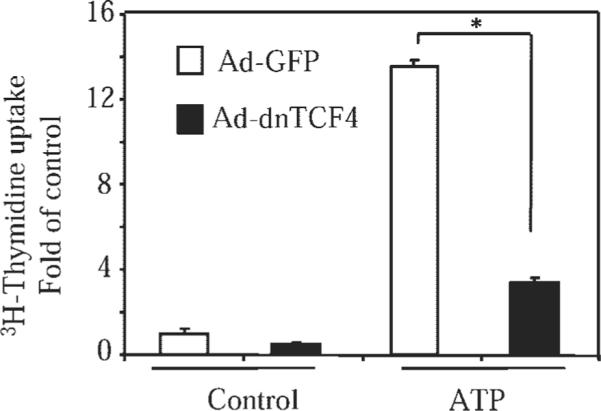
ATP-induced DNA synthesis in VSMC is mediated by TCF-4. VSMC were transduced with control adenovirus [AD-green fluorescent protein (GFP)] or adenovirus encoding dnTCF-4 (Ad-dnTCF-4), followed by stimulation with 30 μM ATP for 12 h. The [3H]thymidine uptake assay was then performed as described in MATERIALS AND METHODS. Data represent means ± SD from a representative of three experiments performed in triplicate. *P < 0.01.
DISCUSSION
The present study describes three major findings: 1) endogenous β-catenin can be phosphorylated by PKA at Ser552 and Ser675 in response to ATP in VSMC; 2) ATP-induced phosphorylation of endogenous β-catenin by PKA promotes its interaction with TCF-4, resulting in an increased transcriptional activity of TCF-4; and 3) ATP stimulates cyclin D1 expression and proliferation of VSMC in a manner dependent on PKA and TCF-4 activities. Even though the present study focused on VSMC and ATP as an agonist, to our knowledge, these three findings have not been previously reported for endogenous β-catenin regulation in other cells or by other stimuli.
It is accepted that the activity of β-catenin is controlled at the level of its stability through regulated proteolysis. As such, phosphorylation of β-catenin by GSK-3 at Ser33, Ser37, and Thr41 targets it for proteasomal degradation; whereas inhibition of GSK-3 through Wnt signaling results in accumulation of unphosphorylated β-catenin (7). Previous studies by others and us showed that GSK-3 activity can be inhibited through its phosphorylation by Akt (17) or by PKA (1, 13, 28). In our additional experiments, ATP stimulated a PKA-dependent phosphorylation of GSK-3α in VSMC (data not shown). However, this did not lead to accumulation of β-catenin, the levels of which were already easily detected in quiescent VSMC cells (Fig. 2D). Instead, we show for the first time that endogenous β-catenin can be directly activated (Fig. 3A) through its phosphorylation by PKA at Ser552 and Ser675 sites (Fig. 2). This represents an additional new mode of β-catenin regulation that was previously not appreciated. Furthermore, we provide a molecular mechanism for PKA-dependent activation of β-catenin by showing for the first time that phosphorylation of β-catenin by PKA promotes its association with TCF-4 in response to ATP (Fig. 3B).
β-Catenin and TCF-4 stimulate transcription of many genes implicated in cell cycle progression, including cyclin D1, c-myc, c-jun, and others (3, 11, 27, 29). In the present study, we show that cyclin D1 promoter activation and protein expression in response to ATP are dependent on both PKA and TCF-4 activities in VSMC (Figs. 4 and 5). It is noteworthy that cyclin D1 promoter also contains CREs known to be activated by PKA (18). However, even though ATP stimulated activation of artificial CRE reporter (Fig. 4C), the ATP-induced cyclin D1 promoter activation was not dependent on CRE, because mutation of CRE in cyclin D1 promoter did not affect its activation by ATP (data not shown). Thus we believe that in ATP responses, PKA activates cyclin D1 promoter not through CRE, but through β-catenin/TCF-4 axis.
Finally, we show for the first time that ATP-induced proliferation of VSMC is dependent on the activity of TCF-4 (Fig. 6). Proliferation of VSMC induced by other growth stimuli, such as serum, β-cellulin (epidermal growth factor receptor ligand), or platelet-derived growth factor, is also dependent on β-catenin/TCF-4 activity (22, 26, 32). In these cases, TCF-4 stimulation likely occurs through activation of Akt, inhibition of GSK-3, and stabilization of unphosphorylated (at GSK-3 sites) β-catenin (26). As discussed above, ATP stimulated TCF-4 through a different mechanism, i.e., through a direct phosphorylation of β-catenin by PKA (at sites distinct from those that are phosphorylated by GSK-3), resulting in increased association of β-catenin with TCF-4.
Regarding PKA, its role in cell proliferation depends on the cell type as well as on the stimulus. In VSMC, stimulation of PKA through β-adrenergic signaling inhibits proliferation, whereas PKA activation through purinergic or endothelin signaling promotes VSMC proliferation and hypertrophy, respectively (19, 28, 31). This agonist-specific role of PKA can be explained at least in part by 1) differential duration of PKA activation by these agonists [sustained PKA activation through β-adrenergic stimulation vs. transient PKA activation through purinergic or endothelin stimulation (8)]; and 2) inability of β-adrenergic stimuli to induce mitogenic signaling (MAP kinase, etc.), which is activated by purinergic or endothelin stimulation (19, 24). The transient nature of PKA activation by ATP is likely due to 1) relatively small cAMP increase in response to ATP compared with β-adrenergic stimulation; and 2) simultaneous activation of Ca2+/calmodulin-dependent phosphodiesterases by ATP, but not by β-adrenergic stimulation (data not shown). Thus we favor a notion that transient PKA activation works together with the established mitogenic signaling to promote VSMC proliferation in response to ATP. The present study describes one potential mechanism by which PKA may contribute to ATP-induced proliferation of VSMC through phosphorylation of β-catenin and activation of TCF-dependent gene transcription.
ACKNOWLEDGMENTS
We thank Dr. Richard Pestell, Dr. Tong-Chuan He, and Dr. Michael Dunn for providing the DNA and antibody reagents.
GRANTS This study was supported by National Heart, Lung, and Blood Institute Grant HL-071755 (to N. O. Dulin) and American Heart Association postdoctoral fellowship awards (to S. Taurin and N. Sandbo).
REFERENCES
- 1.Abbott KL, Loss JR, 2nd, Robida AM, Murphy TJ. Evidence that Galpha(q)-coupled receptor-induced interleukin-6 mRNA in vascular smooth muscle cells involves the nuclear factor of activated T cells. Mol Pharmacol. 2000;58:946–953. doi: 10.1124/mol.58.5.946. [DOI] [PubMed] [Google Scholar]
- 2.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-Catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu-Amer Y, Ross FP, McHugh KP, Livolsi A, Peyron JF, Teitelbaum SL. Tumor necrosis factor-alpha activation of nuclear transcription factor-kappaB in marrow macrophages is mediated by c-Src tyrosine phosphorylation of Ikappa Balpha. J Biol Chem. 1998;273:29417–29423. doi: 10.1074/jbc.273.45.29417. [DOI] [PubMed] [Google Scholar]
- 4.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14:3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 6.Burnstock G. Potential therapeutic targets in the rapidly expanding field of purinergic signalling. Clin Med. 2002;2:45–53. doi: 10.7861/clinmedicine.2-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnstock G. Purinergic signaling and vascular cell proliferation and death. Arterioscler Thromb Vasc Biol. 2002;22:364–373. doi: 10.1161/hq0302.105360. [DOI] [PubMed] [Google Scholar]
- 8.Davis A, Hogarth K, Fernandes D, Solway J, Niu J, Kolenko V, Browning D, Miano JM, Orlov SN, Dulin NO. Functional significance of protein kinase A activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell Signal. 2003;15:597–604. doi: 10.1016/s0898-6568(02)00148-1. [DOI] [PubMed] [Google Scholar]
- 9.Di Virgilio F, Solini A. P2 receptors: new potential players in atherosclerosis. Br J Pharmacol. 2002;135:831–842. doi: 10.1038/sj.bjp.0704524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dulin NO, Niu J, Browning DD, Ye RD, Voyno-Yasenetskaya T. Cyclic AMP-independent activation of protein kinase A by vasoactive peptides. J Biol Chem. 2001;276:20827–20830. doi: 10.1074/jbc.C100195200. [DOI] [PubMed] [Google Scholar]
- 11.Emala CW, Liu F, Hirshman CA. Giα but not Gqα is linked to activation of p21ras in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 1999;276:L564–L570. doi: 10.1152/ajplung.1999.276.4.L564. [DOI] [PubMed] [Google Scholar]
- 12.Erlinge D. Extracellular ATP: a growth factor for vascular smooth muscle cells. Gen Pharmacol. 1998;31:1–8. doi: 10.1016/s0306-3623(97)00420-5. [DOI] [PubMed] [Google Scholar]
- 13.Fang X, Yu SX, Lu Y, Bast RC, Jr, Woodgett JR, Mills GB. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci USA. 2000;97:11960–11965. doi: 10.1073/pnas.220413597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerasimovskaya EV, Ahmad S, White CW, Jones PL, Carpenter TC, Stenmark KR. Extracellular ATP is an autocrine/paracrine regulator of hypoxia-induced adventitial fibroblast growth. Signaling through extracellular signal-regulated kinase-1/2 and the Egr-1 transcription factor. J Biol Chem. 2002;277:44638–44650. doi: 10.1074/jbc.M203012200. [DOI] [PubMed] [Google Scholar]
- 15.Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA. PKC-δ and CaMKII-δ2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2004;286:C1281–C1289. doi: 10.1152/ajpcell.00202.2003. [DOI] [PubMed] [Google Scholar]
- 16.Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardt SE, Sadoshima J. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 18.Herber B, Truss M, Beato M, Muller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:1295–1304. [PubMed] [Google Scholar]
- 19.Hogarth DK, Sandbo N, Taurin S, Kolenko V, Miano JM, Dulin NO. Dual role of PKA in phenotypic modulation of vascular smooth muscle cells by extracellular ATP. Am J Physiol Cell Physiol. 2004;287:C449–C456. doi: 10.1152/ajpcell.00547.2003. [DOI] [PubMed] [Google Scholar]
- 20.Kunapuli SP, Daniel JL. P2 receptor subtypes in the cardiovascular system. Biochem J. 1998;336:513–523. doi: 10.1042/bj3360513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 22.Quasnichka H, Slater SC, Beeching CA, Boehm M, Sala-Newby GB, George SJ. Regulation of smooth muscle cell proliferation by betacatenin/T-cell factor signaling involves modulation of cyclin D1 and p21 expression. Circ Res. 2006;99:1329–1337. doi: 10.1161/01.RES.0000253533.65446.33. [DOI] [PubMed] [Google Scholar]
- 23.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- 24.Sandbo N, Taurin S, Yau DM, Kregel S, Mitchell R, Dulin NO. Downregulation of smooth muscle alpha-actin expression by bacterial lipopolysaccharide. Cardiovasc Res. 2007;74:262–269. doi: 10.1016/j.cardiores.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Seye CI, Kong Q, Erb L, Garrad RC, Krugh B, Wang M, Turner JT, Sturek M, Gonzalez FA, Weisman GA. Functional P2Y2 nucleotide receptors mediate uridine 5′-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation. 2002;106:2720–2726. doi: 10.1161/01.cir.0000038111.00518.35. [DOI] [PubMed] [Google Scholar]
- 26.Shin HS, Lee HJ, Nishida M, Lee MS, Tamura R, Yamashita S, Matsuzawa Y, Lee IK, Koh GY. Betacellulin and amphiregulin induce upregulation of cyclin D1 and DNA synthesis activity through differential signaling pathways in vascular smooth muscle cells. Circ Res. 2003;93:302–310. doi: 10.1161/01.RES.0000086803.64109.9E. [DOI] [PubMed] [Google Scholar]
- 27.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 29.Tetsu O, McCormick F. β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 30.Van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J Biol Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- 31.Wang DJ, Huang NN, Heppel LA. Extracellular ATP and ADP stimulate proliferation of porcine aortic smooth muscle cells. J Cell Physiol. 1992;153:221–233. doi: 10.1002/jcp.1041530202. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL. A role for the β-catenin/T-cell factor signaling cascade in vascular remodeling. Circ Res. 2002;90:340–347. doi: 10.1161/hh0302.104466. [DOI] [PubMed] [Google Scholar]