

Abstract
The glycoprotein hormone receptors are G protein-coupled receptors containing a large extracellular domain fused to a prototypical serpentine domain. cis-activation occurs when binding of hormone to the extracellular domain stabilizes the serpentine domain in an active conformation. Studies by others suggested that these receptors can also signal by trans-activation, where hormone binding to one receptor protomer activates the serpentine domain of an associated protomer, as documented by the partial rescue of hormone-dependent signaling when a binding defective mutant is coexpressed with a signaling defective mutant. However, our characterizations of several LH receptor (LHR) mutants used in previous studies differ markedly from those originally reported. Also, when examining a pair of LHR mutants previously shown to functionally rescue in vitro as well as in vivo, in addition to finding that the properties of the individual mutants differ significantly from those originally described, we determined that when this pair of mutants was coexpressed in vitro, quantitative analyses did not indicate functional rescue. Additional data are presented that provide a plausible alternate explanation for the apparent in vivo trans-activation that was reported. Finally, using LHR mutants that we have documented to be expressed at the cell surface but to lack human chorionic gonadotropin binding activity or to be severely impaired in their ability to activate Gs, we did not observe functional rescue of human chorionic gonadotropin-stimulated cAMP when the mutants were coexpressed, even though bioluminescence resonance energy transfer analyses confirmed that the coexpressed mutants formed dimers. Taken altogether, our data substantively question the concept of functional rescue between LHR mutants.
The LH receptor (LHR) is a family A G protein-coupled receptor (GPCR) that is most closely related to the other members of the glycoprotein hormone receptor family, the FSH receptor (FSHR) and TSH receptor family. These receptors contain the prototypical serpentine domain containing seven-transmembrane helices attached to a large extracellular domain (ECD or ectodomain). The ECD is composed of a series of leucine-rich repeats that are connected to the serpentine domain via a cysteine-rich hinge region. Earlier studies demonstrated high human (h) chorionic gonadotropin (CG) affinity binding to the LHR ECD when expressed on its own (1, 2), and subsequent studies have shown the hormone-binding domain (HB) of the glycoprotein hormone receptors to be defined by the leucine-rich repeats (3, 4). By mechanisms not yet fully understood, the binding of agonist to the HB stabilizes the serpentine domain in an active conformation, permitting intracellular signaling through G proteins.
As with many other GPCR, the LHR has been shown to self-associate into dimers and oligomers (referred to henceforth as “dimers”) (5–7). LHR dimers can be detected under basal conditions by both coimmunoprecipitation and bioluminescence resonance energy transfer (BRET) analyses and the propensity for LHR dimerization does not appear to be affected by the activation state of the receptor (6). These findings, in addition to the observation that LHR dimers are detected in the endoplasmic reticulum as well as plasma membrane (6), suggest that LHR dimerization occurs early in the biosynthetic pathway and is most likely an obligate and constitutive process.
Earlier in vitro functional rescue studies had also suggested that the human (h) LHR may dimerize and suggested a unique means by which the receptors could signal via trans-activation (8–10). In these studies, cells were cotransfected with an hLHR mutant described as signaling inactive and an hLHR mutant described as binding inactive. When coexpressed, a modest degree of hormone-stimulated cAMP production was observed, suggesting functional rescue via trans-activation such that the hormone-occupied HB of the signaling-inactive mutant partially activated the serpentine domain of the binding-inactive mutant. More recently, it was reported that this phenomenon could also be observed in vitro between murine (m) LHR mutants as well as in vivo using mouse models (11). In an Lhcgr knockout background (LuRKO), BAC transgenic mice were generated that expressed either a reported binding-inactive mLHR or a reported signaling-inactive mLHR. Crossing of the mice yielded heterozygotes coexpressing both mutant mLHR. Whereas the male mice expressing either mutant alone exhibited infertility and hypogonadism, those coexpressing both mutant mLHR were fertile and exhibited a somewhat normal phenotype (11). These findings have been interpreted as demonstrating that LHR dimerization and trans-activation were phenomena that could occur in vivo.
However, as described by the studies presented herein, our findings seriously question whether functional rescue between LHR mutants occurs.
Results
Functional rescue between LHR mutants has been defined as the observation of the partial restoration of hCG-stimulated cAMP production when a mutant defective in hormone binding activity is coexpressed with a mutant defective in signaling activity (8–11). The initial goal of this study was to determine whether such receptor pairs could be shown to physically interact with each other by BRET analyses. Therefore, we first sought to replicate previously reported in vitro studies indicating functional rescue between hLHR mutants (8–10). These earlier studies had been performed in human embryonic kidney (HEK)293 cells, a model system that has been well validated to recapitulate wild-type (wt) and mutant hLHR receptor expression and signaling through Gs observed in gonadal cells (12–25). Therefore, we similarly used 293 cells for the present studies. We initially selected hLHR(K605R) [termed “hLHR(K583R)” in original reports (8–10]1 as a signaling-inactive mutant. HEK293 cells expressing this mutant were originally described to display normal hCG binding affinity but lack a detectable hCG-stimulated increase in cAMP production (8–10). To verify the functional properties of the mutant, we similarly examined 293 cells transfected with hLHR(K605R). Because hormone-stimulated cAMP production is dependent on cell surface receptor densities (26, 27), we analyzed the signaling properties of cells transfected under conditions that yielded the same cell surface receptor densities of wt or mutant receptor as determined by flow cytometry and by [125I]hCG binding to intact cells. As shown in Fig. 1A, we observed that K605R cells were indeed attenuated in their response to a maximally stimulating concentration of hCG. However, the mutant maintained a significant capacity for signaling because the maximal response of the K605R cells was approximately 30% that of the hLHR(wt) cells. Even when the cell surface density of K605R was decreased to about 50% of the wt receptor, there was still an appreciable hCG-stimulated cAMP response. Because K605R retains significant signaling capacity, we chose instead to use an hLHR mutant, hLHR(D405N,Y546F), which we have previously shown to be severely signaling impaired despite normal cell surface expression and hCG binding activity (28). The greater attenuation of signaling by this mutant is confirmed by the data shown in Fig. 1B. By comparing the data shown in panels A and B, which were obtained in the same experiment, it can also be determined that at the same cell surface receptor densities there is a much greater hCG-stimulated cAMP response by K605R than by D405N,Y546F. Therefore, hLHR(D405N,Y546F) was used for subsequent experiments testing potential functional rescue.
Fig. 1.

The previously reported signaling inactive hLHR(K605R) mutant exhibits only a partially attenuated signaling phenotype. A, HEK293 cells were transiently transfected to yield the same or lower cell surface densities of myc-hLHR(K605R) as compared with myc-hLHR(wt) as determined by flow cytometry using nonpermeabilized cells and [125I]hCG binding to intact cells, and basal and hCG-stimulated cAMP were measured. B, HEK293 cells were transiently transfected to yield the same cell surface density of myc-hLHR(D405N,Y546F) as myc-hLHR(wt) as determined by [125I]hCG binding to intact cells, and basal and hCG-stimulated cAMP were measured. The data in panels A and B were generated in the same experiment, which is representative of at least three independent experiments. Controls were generated using cells transfected with empty pcDNA3.1 (EV). Data shown are the mean ± sem of triplicate determinations. RLU, Relative light units.
As a binding-defective hLHR mutant, we initially selected I80A. This mutant (termed “I55A” in original reports) was reported to be expressed at the cell surface, but to lack detectable hCG-binding activity (8–10). To confirm this, we examined 293 cells transiently transfected with maximal plasmid concentrations encoding myc-hLHR(wt) or myc-hLHR(I80A). As shown in Table 1, a low but detectable level of [125I]hCG binding was observed in cells expressing I80A, and flow cytometry analyses indicated that hLHR(I80A) was expressed at much reduced levels at the cell surface. Consistent with this, confocal microscopy reveals very few cells that express hLHR(I80A) on the cell surface as compared with cells expressing hLHR(wt) (Fig. 2). Both the flow cytometry data (Table 1) and microscopy data (Fig. 2) indicate that the majority of hLHR(I80A) is retained intracellularly. To determine whether the decreased numbers of cell surface hLHR(I80A) were functional, we examined hCG-stimulated cAMP in transfected cells (Fig. 3). The cell surface density of hLHR(wt) was matched to that of hLHR(I80A) based on maximal [125I]hCG binding activity. Under these conditions, the I80A cells were observed to respond to hCG with increased cAMP production with a greater efficacy than that of cells expressing wt receptor. Competition binding assays were then performed and the mean ± sem of the inhibition constant (Ki) values determined from six independent experiments were 37.5 ± 2.5 and 20.0 ± 2.9 ng/ml hCG for the wt and I80A receptors, respectively, demonstrating that the I80A mutant binds hCG with high affinity. Taken altogether, our data indicate that, in contrast to what was originally reported (8–10), hLHR(I80A) is trafficking impaired, with little receptor at the cell surface. However, the I80A receptor that is at the cell surface binds hCG with high affinity, and it responds to hCG with increased cAMP production. Therefore, our data indicate that hLHR(I80A) is not an appropriate binding-defective mutant for examining potential functional rescue.
Table 1.
Reduced maximal hCG binding activity to 293 cells expressing hLHR(I80A) is associated with reduced cell surface expression of the mutant
| Transfection | Cell surface [125I]hCG bound (ng/106 cells) | Flow cytometry on nonpermeabilized cells (RLU) | Flow cytometry on permeabilized cells (RLU) |
|---|---|---|---|
| myc-hLHR(wt) | 10.8 | 8821 | 34,041 |
| myc-hLHR(I80A) | 0.6 | 1915 | 15,524 |
HEK293 cells were transiently transfected with maximal concentrations of plasmids encoding myc-hLHR(wt) or myc hLHR(I80A). Intact cells were assayed for [125I]hCG binding using a saturating concentration of [125I]hCG. Permeabilized and nonpermeabilized cells were assayed by flow cytometry for receptor expression using an antibody to the myc epitope tag. Results are shown from one experiment representative of at least three independent experiments. RLU, Relative light units.
Fig. 2.

hLHR(I80A) is expressed very poorly at the cell surface and mostly retained intracellularly. HEK293 cells were transiently transfected with the indicated constructs, and the receptors were visualized by immunofluorescence in cells that had been nonpermeabilized (top) or permeabilized (bottom). The microscope settings in all the panels were identical.
Fig. 3.
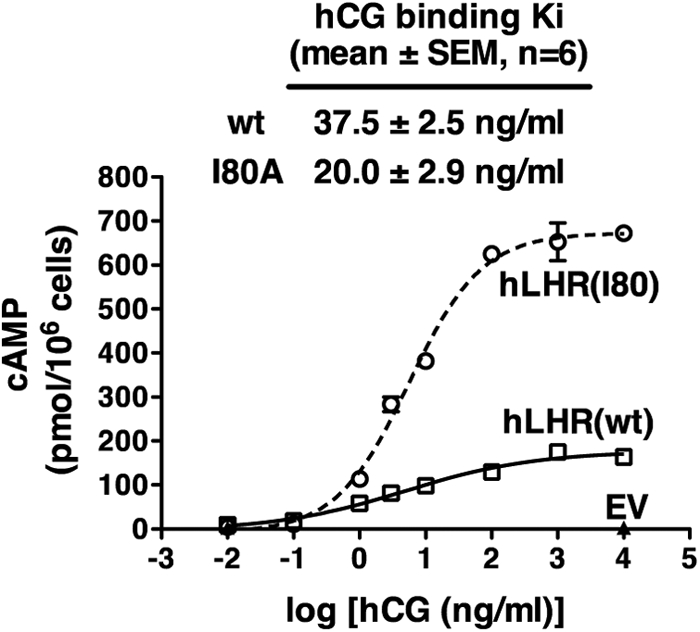
Cells expressing the previously reported binding- defective hLHR(I80A) respond to hCG with increased cAMP production. HEK293 cells were transiently transfected with a maximal concentration of plasmid encoding myc-hLHR(I80A) and a submaximal concentration of plasmid encoding myc-hLHR(wt). In the cAMP experiment shown, wt and I80A cells bound 0.7 and 0.6 ng [125I]hCG/106 cells. Under these conditions, hCG-stimulated cAMP was measured. The cAMP data shown are the mean ± sem of triplicate determinations within one experiment representative of two independent experiments. In separate experiments, the Ki values for hCG binding to cells expressing each of the constructs were determined by competition binding assays.
We therefore sought to identify an hLHR mutant that is expressed normally at the cell surface but lacks detectable hCG-binding activity. Toward this end, we created a chimeric receptor, designated hFL, which is composed of the ECD of the hFSHR fused to the serpentine domain of the hLHR. Flow cytometry to nonpermeabilized cells confirmed normal cell surface expression of the chimera. When analyzed in 293 cells expressing the same cell surface density of hLHR(wt), the hFL cells did not bind detectable levels of [125I]hCG and they responded to hCG with little or no cAMP (Fig. 4A). However, when matched to cells expressing the same density of cell surface hFSHR(wt), the hFL cells exhibited similar levels of [125I]hFSH binding and hFSH-stimulated cAMP (Fig. 4B). Thus, although hFL cannot bind hCG, the chimeric receptor is fully signaling competent. Therefore, hFL fulfills the criteria for an hCG binding-defective, but signaling-competent, hLHR mutant. In addition, we also chose to examine hLHR(I152T) as a binding-defective mutant. Identified by Qiao et al. (19) as a naturally occurring mutation in a 46, XY patient with a disorder of sexual differentiation, hLHR(I152T) in heterologous cells was reported to be expressed at the cell surface and to lack detectable hCG-binding activity. Using myc-hLHR(I152T), we similarly observed normal cell surface expression and little to no detectable [125I]hCG binding activity (Fig. 5), confirming its suitability as an hCG binding-defective LHR mutant with normal cell surface expression.
Fig. 4.

The hFL chimera does not bind or signal in response to hCG but binds and signals normally in response to hFSH. HEK293 cells were transiently transfected with EV, myc-hFL (a chimera composed of the ECD of the hFSHR fused to the serpentine domain of the hLHR), myc-hLHR(wt), or myc-hFSHR(wt), and cell surface receptor expression was quantified by flow cytometry using nonpermeabilized cells. A, hFL was compared with hLHR(wt) with respect to [125I]hCG binding and basal and hCG-stimulated cAMP under conditions of comparable cell surface receptor densities. B, hFL was compared with hFSHR(wt) with respect to [125I]hFSH binding and basal and hFSH-stimulated cAMP under conditions of comparable cell surface receptor densities. The data in panels A and B were generated in the same experiment, which is representative of at least three independent experiments. Data shown are the mean ± sem of triplicate determinations.
Fig. 5.

hLHR(I152T) is expressed at the cell surface but exhibits little to no hormone-binding activity. HEK293 cells were transiently transfected with increasing concentrations of plasmid encoding hLHR(wt) or hLHR(I152T). A, Flow cytometry on nonpermeabilized cells was performed to determine relative levels of cell surface receptor expresssion. B, The binding of [125I]hCG to intact cells was performed. Results shown are from one experiment representative of two independent experiments. RLU, Relative light units.
To determine whether the signaling-defective hLHR(D405N,Y546F), when coexpressed with the hCG binding-defective hFL or hLHR(I152T), would form heterodimers, quantitative BRET analyses were performed (Fig. 6). Saturable titration curves were obtained when D405N,Y546F was coexpressed with either hFL (Fig. 5A) or hLHR(I152T) (Fig. 5B). In contrast, BRET signals remained very low and did not yield hyperbolic curves when D405N,Y546F was coexpressed with HERG, a cell surface potassium ion channel that was included as a negative control. It is also important to note that the differences in the maximal BRET responses do not necessarily reflect differences in the extent of dimerization because the BRETmax is also dependent upon the relative conformations of the donor and acceptor to each other (29–31). Taken altogether, the data shown in Fig. 5 indicate that the signaling-defective hLHR(D405N,Y546F) can specifically dimerize with the hCG binding-defective hFL or hLHR(I80A).
Fig. 6.

BRET saturation curves confirm heterodimerization between the signaling impaired hLHR(D405N,Y546F) and either hFL or hLHR(I152T), which do not bind hCG. HEK293T cells were transiently transfected with a fixed concentration of the indicated Rluc fusion proteins and increasing concentrations of the indicated GFP2 fusion proteins under conditions in which the expression levels of Rluc fusion proteins within a given experiment were comparable. Energy transfer as measured by BRET2 was performed, with the data indicating the mean ± range of duplicate determinations within a given experiment. The data shown in panels A and B are from different experiments, each of which was repeated two (panel A) or three (panel B) times.
We then sought to examine potential functional rescue of hCG-stimulated signaling activity in dimers composed of an hLHR binding-defective mutant and an hLHR signaling-defective mutant. The experimental design entailed establishing HEK293 cells stably transfected with one of the mutants (mutant A) and transiently transfecting the stable cells with the other mutant (mutant B) such that the cell surface expression of mutant A would be approximately 10-fold greater than that of mutant B. Under these conditions, one would expect that most of mutant B would be heterodimerized with mutant A. We have previously found these conditions to be optimal for examining the potential effects of allosteric modulation between hLHR protomers within a receptor dimer (28). As shown in the top panel of Fig. 7, 293 cells stably transfected with empty vector (EV) or with the signaling defective hemagglutinin (HA)-hLHR(D405N,Y546F) were transiently transfected with the hCG binding-defective myc-hFL under conditions that generated closely matched levels of cell surface myc-hFL in the two cell lines. As additional controls, the stably transfected cells were also transiently transfected with EV. Low, but quantifiable, amounts of cAMP were produced in response to hCG in the EV cells transfected with hFL (yellow) and in the D405N, Y546F cells transfected with EV (red). Although detectable, these responses are extremely low (≤6%) when compared with hCG-stimulated cAMP produced by cells expressing hLHR(wt), which was more than 1000 pmol/106 cells (data not shown). Importantly, the hCG-stimulated cAMP response of the D405N,Y546F cells transfected with hFL (red and yellow) was no greater than the sum of hFL alone plus D405N,Y546F alone. Thus, the transfection of the binding defective hFL into cells stably expressing the signaling defective D405N,Y546F did not cause functional rescue between the associated receptors. In the bottom panel of Fig. 7, the two defective hLHR mutants were again tested for potential functional rescue, but this time the signaling defective HA-hLHR(D405N,Y546F) was transiently transfected into cells stably expressing EV or the hCG binding-defective myc-hFL. Again, conditions were chosen to closely match the cell surface expression levels of the signaling-defective mutant. Here too, the hCG-stimulated cAMP production in cells expressing both hFL and D405N,Y546F (red and yellow) was no greater than that of the sum of the response of cells expressing either mutant alone. Potential functional rescue between the signaling defective D405N,Y546F and the binding-defective hLHR(I152T) was also tested using the same experimental strategy (Fig. 8). Here too the hCG-stimulated response of cells coexpressing D405N,Y546F and I152T (red and yellow) was no greater than the sum of the responses of each mutant alone, again indicating a lack of functional rescue.
Fig. 7.

Absence of functional rescue between the signaling impaired hLHR(D405N,Y546F) and the hCG binding-impaired hFL. Top panel, Cells stably expressing EV or HA-hLHR(D405N,Y546) were transiently transfected with EV or myc-hFL as indicated to yield comparable cell surface expression of myc-hFL as determined by flow cytometry. Bottom panel, Cells stably expressing EV or myc-hFL were transiently transfected with HA-hLHR(D405N,Y546F) to yield comparable cell surface expression of HA-hLHR(D405N,Y546F) as determined by flow cytometry. Basal and hCG-stimulated cAMP were determined, in which the mean ± sem of triplicate determinations within a given experiment are shown. Experiments in the top and bottom panels were each performed at least three times. If functional rescue were occurring, the hCG-stimulated cAMP values shown by yellow and red squares would be expected to be greater than the sum of the yellow alone plus red alone. hCG-stimulated cAMP by cells expressing hLHR(wt) was more than 1000 pmol/106 cells.
Fig. 8.

Absence of functional rescue between the signaling- impaired hLHR(D405N,Y546F) and the hCG binding-impaired hLHR(I152T). Top panel, Cells stably expressing EV or HA-hLHR(D405N,Y546) were transiently transfected with EV or myc-hLHR(I152T) as indicated to yield comparable cell surface expression of myc-hLHR(I152T) as determined by flow cytometry. Bottom panel, Cells stably expressing EV or myc-hLHR(I152T) were transiently transfected with HA-hLHR(D405N,Y546F) to yield comparable cell surface expression of HA-hLHR(D405N,Y546F) as determined by flow cytometry. Basal and hCG-stimulated cAMP were determined, in which the mean ± sem of triplicate determinations within a given experiment are shown. Experiments in the top and bottom panels were each performed at least three times. If functional rescue were occurring, the hCG-stimulated cAMP values shown by yellow and red squares would be expected to be greater than the sum of the yellow alone plus red alone. hCG-stimulated cAMP by cells expressing hLHR(wt) was more than 1000 pmol/106 cells.
While our studies were nearing completion, a study was published describing both in vitro functional rescue in 293 cells as well as in vivo functional rescue in BAC transgenic mice between a particular set of defective mLHR mutants (11). The mLHR binding-defective mutant used in that study was described as C22A and, when expressed in 293 cells, it was shown to be expressed on the cell surface but to lack hormone-binding activity. The mLHR signaling-inactive mutant was described as containing a deletion of transmembrane helices 6 and 7 due to the deletion of amino acids Val553 to Ala689. This mutant, when expressed in 293 cells, was shown to be expressed at the cell surface, and cells expressing the mutant were shown to have normal hCG binding affinity but lack a detectable increase in cAMP in response to hCG. Coexpression of the two mutants in vitro in 293 cells indicated a partial functional rescue of hCG-stimulated cAMP production (11). Because the in vitro and in vivo findings of this study appeared to be in conflict with our results, suggesting a lack of functional rescue between hLHR mutants, we performed additional studies using these particular mLHR mutants.
Sequencing of the mLHR mutant, described as binding defective, confirmed that it contained a C22A substitution2 and the insertion of an HA epitope tag at the N terminus of the mature protein. Sequencing of the signaling-inactive mutant, which we will refer to henceforth as “mLHR(delTM6,7)” revealed that the 26-residue mLHR signal peptide was replaced with a different 30-residue signal peptide, which was followed by the appropriately placed FLAG epitope tag at the N terminus of the mature protein. We confirmed the deletion of the predicted third intracellular loop, transmembrane helix 6, the third extracellular loop, transmembrane helix 7, and most of the cytoplasmic tail. However, this resulted from the deletion of Val531 through Gln664 of the mLHR. In addition, we observed that the third nucleotide encoding Gln664 was not deleted, resulting in this large deletion to be followed by 18 residues different from the predicted final 11 residues of the wt mLHR. Furthermore, mLHR(delTM6,7) was found to have a substitution of residues 1–7 of the mature mLHR sequence. To determine whether these substituted residues at the N terminus would have any effect on mLHR function, we created a mutant in which only those substitutions were made. This N terminus mutant had normal cell surface expression and function (data not shown). Therefore, any alterations in properties of the mLHR(delTM6,7) can be attributed to the large deletion (and possibly the subsequent frameshift error) in the serpentine domain.
Using 293 cells stably transfected with either mLHR mutant or with the appropriately tagged wt mLHR, confocal microscopy was performed to visualize cell surface receptor (Fig. 8). In contrast to previously published data (11), we did not observe cell surface receptor expression in any cells expressing mLHR(delTM6,7), and the vast majority of cells expressing mLHR(C22A) did not exhibit any cell surface receptor expression. Occasionally, a well of cells indicated a lone cell showing cell surface expression of mLHR(C22A) (Fig. 9). The visualization of the two mutants in permeabilized cells indicated receptor expression, but at reduced levels (Fig. 9). In both stably and transiently transfected 293 cells, a similar lack of cell surface receptor expression for both mutants was also indicated by flow cytometry in nonpermeabilized cells. Permeabilized cells revealed intracellular expression of the mutants, albeit at reduced levels, as compared with wt (data not shown). Specific [125I]hCG binding to intact cells, when observed, was not reproducible and was generally too low (<0.1 ng [125I]hCG bound/106 cells) to be convincing.
Fig. 9.

mLHR(delTM6,7) and mLHR(C22A) are absent or nearly absent from the cell surface. Nonpermeabilized 293 cells were stably transfected with the indicated constructs, and the receptors were visualized by immunofluorescence. The microscope settings in the top panels were identical, and those in the bottom panel were identical. However, the settings between the top and bottom panels differed due to the different levels of expression of FLAG- vs. HA-tagged wt receptors.
Whether mLHR(C22A) also intrinsically lacks hormone-binding activity was difficult to address given its very poor cell surface expression. However, although we could not reliably detect hCG binding to intact cells expressing mLHR(C22A), we did observe detectable increases in hCG-stimulated cAMP in transiently transfected (Fig. 10A) or stably transfected (Fig. 11) 293 cells, albeit much reduced compared with cells transfected with mLHR(wt). To increase the total expression of mLHR(C22A) receptor protein, we expressed mLHR(C22A) in transiently transfected 293T cells. In the same experiments, basal cAMP, hCG-stimulated cAMP, and [125I]hCG binding were compared between 293 cells and 293T cells expressing mLHR(wt) or mLHR(C22A) (Fig. 10 and Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Cell surface [125I]-hCG binding was, as expected, increased in 293T cells as compared with 293 cells expressing mLHR(wt). Although [125I]hCG binding was detected in cells expressing mLHR(C22A) in one experiment and in this case it was higher in the 293T cells, binding to cells expressing this mutant could not be detected in the other two experiments (Supplemental Table 1). Similarly, the expression of mature mLHR(C22A) protein was too low to detect by Western blotting. However, quantitative RT-PCR confirmed approximately 60% greater levels of mLHR(C22A) mRNA in the 293T cells as compared with the 293 cells (Fig. 10B). As predicted, if the total levels of mLHR(C22A) were similarly greater in the 293T cells, leading to proportionally more mutant receptors on the cell surface, hCG provoked a greater fold increase in cAMP in 293T cells than the 293 cells expressing hLHR(C22A) (Fig. 10A and Supplemental Table 1). This is in contrast to 293 or 293T cells expressing mLHR(delTM6,7), where no detectable increases in cAMP were observed in response to hCG (data not shown), consistent with its lack of cell surface expression as well its lack of the critical intracellular regions necessary for coupling to Gs. As such, the clearly detectable hCG-stimulated cAMP production in mLHR(C22A) transfected cells demonstrates that this mutant must be capable of binding hCG, even if it may be with decreased affinity. Therefore, whereas previous studies showed mLHR (C22A) to be at the cell surface but lacking detectable hCG-binding activity (11), we observed mLHR(C22A) to be very poorly expressed at the cell surface and the extremely low levels of cell surface mutant receptor capable of binding and responding to hCG. Furthermore, mLHR (delTM6,7), originally described as being expressed normally at the cell surface and having normal hCG binding activity but no signaling activity (11), was shown in our studies to have no cell surface receptor expression due to intracellular retention (although it most likely would be signaling inactive if present at the cell surface).
Fig. 10.
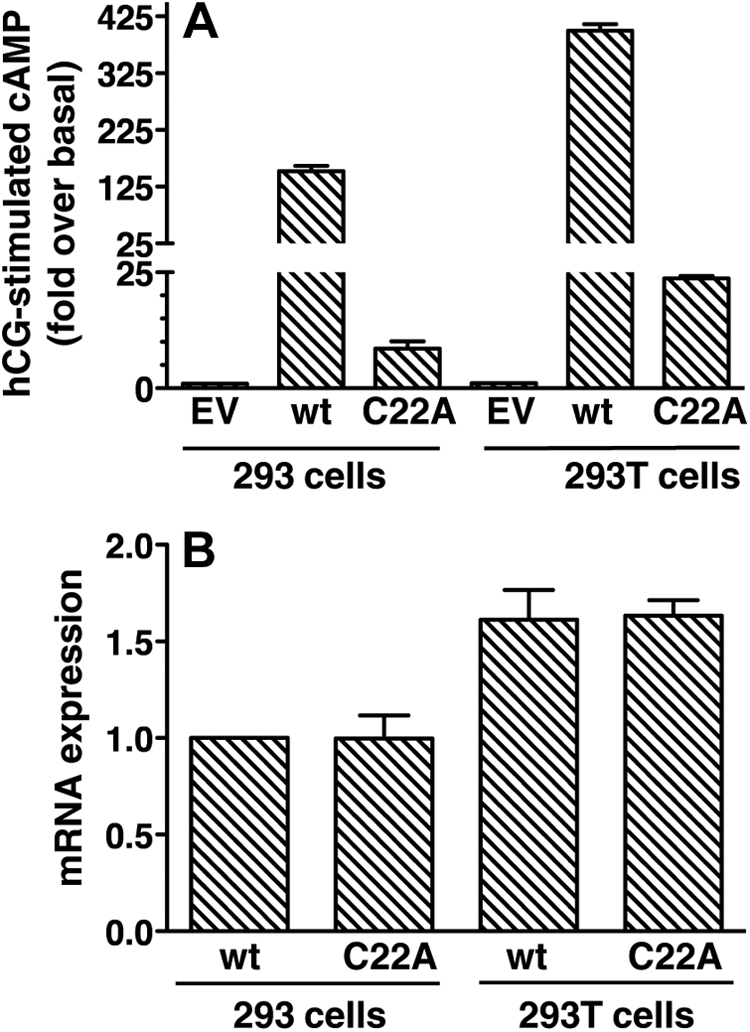
Expression of mLHR(C22A) in 293T cells results in higher levels of C22A mRNA and increased hCG-stimulated cAMP production. HEK293 cells and HEK293T cells were each transiently transfected with EV or plasmid encoding mLHR(wt) or mLHR(C22A) as indicated. Within the same experiment, basal and hCG-stimulated cAMP were measured to yield the fold stimulation (panel A), and quantitative RT-PCR was performed to yield the levels of mRNA relative to mLHR(wt) mRNA in 293 (panel B). Data shown are the mean ± sem of three independent experiments.
Fig. 11.

Absence of functional rescue between mLHR(C22A) and mLHR(delTM6,7). Top panel, Cells stably expressing EV or FLAG-mLHR(delTM6,7) were transiently transfected with EV or HA-mLHR(C22A) as indicated to yield comparable cell surface expression of HA-mLHR(C22A) as determined by flow cytometry. Bottom panel, Cells stably expressing EV or HA-mLHR(C22A) were transiently transfected with FLAG-hLHR(delTM6,7) to yield comparable cell surface expression of FLAG-hLHR(delTM6,7) as determined by flow cytometry. Basal and hCG-stimulated cAMP were determined, in which the mean ± sem of triplicate determinations within a given experiment are shown. The data in panels A and B are from the same experiment, which is representative of three independent experiments. If functional rescue were occurring, the hCG-stimulated cAMP values shown by yellow and red squares would be expected to be greater than the sum of the yellow alone plus red alone. hCG-stimulated cAMP by cells expressing hLHR(wt) was more than 1000 pmol/106 cells.
The coexpression of mLHR(C22A) with mLHR (delTM6,7) in vitro was previously shown to lead to functional rescue of hCG-stimulated cAMP (11). We independently examined this by transiently transfecting EV or mLHR(C22A) into cells stably expressing mLHR(delTM6,7) or EV or by transiently transfecting EV or mLHR(delTM6,7) into cells stably expressing mLHR(C22A) or EV (Fig. 11). Quantitative analyses of the data revealed no further increase in hCG-stimulated cAMP in the cells expressing both mutants as compared with either mutant alone, suggesting a lack of functional rescue. BRET analyses, however, indicated that coexpressed mLHR(C22A) and mLHR (delTM6,7) mutants dimerize with each other (Fig. 12). Thus, although mLHR(delTM6,7) shows a strong and saturable BRET2 curve when it is coexpressed with mLHR(C22A), it does not do so when coexpressed with the plasma potassium channel HERG. Again, the differences in maximal BRET between the pair of mLHR(wt) receptors and pairs of the mLHR mutants do not necessarily reflect differences in the amount of dimerized receptors because the relative orientations of the energy donors and acceptors also contribute to the maximal BRET signal (30). Furthermore, as with all BRET analyses on whole cells, the BRET signal may emanate from anywhere in the cell. Therefore, the data in Fig. 12 do not distinguish between intracellularly located dimers of the mutants vs. dimers on the cell surface.
Fig. 12.

BRET saturation curves confirm heterodimerization between the mLHR(C22A) and mLHR(delTM7,8). HEK293T cells were transiently transfected with a fixed concentration of the indicated Rluc fusion proteins and increasing concentrations of the indicated GFP2 fusion proteins under conditions in which the expression levels of Rluc fusion proteins within a given experiment were comparable. Energy transfer as measure by BRET2 was performed, with the data indicating the mean ± range of duplicate determinations within a given experiment. Data shown are from one experiment representative of three independent experiments.
Discussion
Experimental approaches other than functional rescue have been used to demonstrate homodimerization of the glycoprotein hormone receptors (5–7, 32, 33). In addition, studies by others demonstrating functional rescue between coexpressed LHR binding-defective and signaling-defective mutants have been used as additional data in support of LHR dimerization (8–11). In contrast to previous studies, however, the data presented herein do not indicate functional rescue between LHR mutants, even when testing a pair of mutants used by others to demonstrate functional rescue and when demonstrating dimerization between the mutants.
One confounding issue that immediately became apparent was that we did not observe similar properties of several LHR mutants described in previous functional rescue studies. We examined two different “signaling defective” mutants, hLHR(K605R) and mLHR(delTM6,7)3, each previously described as being expressed normally at the cell surface, binding hCG normally, and lacking a detectable cAMP response to hCG (8–11). hLHR(K605R), although partially attenuated in its cAMP response to hCG, nonetheless exhibited much higher signaling capabilities (∼30% of the wt response) than originally described. Much greater discrepancies were observed with mLHR(delTM6,7). Whereas this mutant was initially shown to be expressed at the cell surface and to bind hCG (11), our findings in both transiently and stably transfected cells indicated that the mutant was not at all expressed at the cell surface and, therefore, hCG binding to transfected cells was not detected. Although mLHR (delTM6,7), if expressed at the cell surface, would be predicted to be signaling inactive because it lacks most of the serpentine domain beyond transmembrane helix 5, our data argue that the lack of hCG-provoked cAMP accumulation in cells expressing mLHR(delTM6,7) is dictated by the lack of cell surface expression of the mutant rather than its intrinsic lack of signaling capabilities.
We also examined some LHR mutants previously described as binding defective. These had been reported to be localized normally at the cell surface but to lack hCG-binding activity (8–11). In contrast to what had been reported (8–10), we observed hLHR(I80A)4 to have much reduced cell surface expression compared with hLHR(wt). Our data further showed that the low levels of hLHR(I80A) at the cell surface did indeed bind hCG with high affinity. Examining the binding defective hLHR(C44A)5 (9) by flow cytometry, we did not observe cell surface expression of this mutant (data not shown). Our data suggest that the lack of hCG binding to cells expressing these two binding-defective hLHR mutants is due to the lack of trafficking of the mutants to the cell surface as a result of their misfolding and intracellular retention. Finally, we also examined the properties of the murine homolog of hLHR(C44A), which is the mLHR(C22A) binding-defective mutant used in a recent study demonstrating functional rescue both in vitro and in vivo (11). Similar to the original description of hLHR(C44A) (9), the mLHR(C22A) mutant was reported to be expressed normally at the cell surface but to lack detectable hCG binding activity and, therefore, to be unresponsive to hCG with respect to cAMP production (11). However, in our experiments mLHR(C22A) was observed to be very poorly expressed at the cell surface as determined by flow cytometry using stably or transiently transfected 293 cells and transiently transfected 293T cells. Confocal microscopy using stably transfected 293 cells revealed that the vast majority of cells exhibited no cell surface receptor expression, with a few isolated cells showing cell surface expression. Importantly, despite the vastly reduced cell surface expression of mLHR(C22A) in most cells, pools of cells expressing this mutant responded to hCG with increased cAMP production (albeit at much lower levels than wt receptor due to the low levels of cell surface mutant receptor). These data demonstrate that mLHR(C22A) must be capable of binding hCG. However, the overall cell surface expression was too low to permit quantification of the binding affinity and, therefore, we cannot rule out the possibility that the binding affinity may also be decreased. We reasoned that if we could increase the total amount of mLHR(C22A) expression, then even if the same very low percentage of total receptor were trafficked to the cell surface, there would be greater cell surface mLHR(C22A). Indeed, increasing the total expression of mLHR(C22A) by transfecting 293T cells, we observed a greater fold stimulation of cAMP production provoked by hCG in the 293T cells as compared with 293 cells. Therefore, other conditions that would increase the total expression of mLHR(C22A) would be expected to similarly result in increased cell responsiveness to LH or hCG.
Due to the discrepancies between the expression and functional properties originally reported and those we observed for the above mutants, we examined potential functional rescue between LHR mutants of our own design or choosing. We experimentally confirmed them to be expressed normally at the cell surface, but to exhibit little or no hCG binding activity (i.e. binding defective), or to bind hormone normally, but be severely impaired in hCG-provoked cAMP (i.e. signaling defective). Quantitative analyses of the data revealed no functional rescue. The lack of functional rescue in these latter cases cannot be attributed to a lack of receptor dimerization because the mutants were demonstrated to physically associate with each other by quantitative BRET analyses. However, it had been reported by others who described functional rescue between LHR mutants that not all pairs of LHR mutants could yield functional rescue (8). Therefore, we also examined functional rescue using a pair of LHR mutants previously shown to yield functional rescue when coexpressed in vitro (11), the mLHR(C22A) binding-defective and mLHR(TM6,7) signaling-defective mutants discussed above, which we had found to have phenotypes different from those originally described. However, even using these two mutants, we did not observe functional rescue despite BRET analyses showing their dimerization with each other.
Therefore, there are many discrepancies between previously published data and our data regarding properties of several LHR mutants used in functional rescue studies as well as in the presence or absence of functional rescue when testing coexpressed LHR mutants in vitro. We cannot account for the differences observed with respect to the properties of the individual LHR mutants. In both previous studies and in ours, the in vitro properties of the mutants were analyzed in 293 cells, which recapitulate the cell surface expression and activity toward Gs that is observed in gonadal cells (12–25). With respect to the presence or absence of functional rescue, it is important to note that our study design included negative controls of cells transfected with empty vector in place of one of the LHR mutants. This is important because there are always small increases in hCG-stimulated cAMP in cells expressing each mutant alone, and these levels must be taken into account when the two mutants are examined together. Furthermore, only those experiments in which the cell surface expression levels of a receptor were very tightly matched were used for analyses. Especially at low levels of receptor expression, very small changes in LHR cell surface expression lead to extremely large alterations in the maximal response to hCG (Refs. 26 and 27 and unpublished data). These factors may contribute to apparent discrepancies between other reports and ours with respect to in vitro functional rescue.
The mLHR(C22A) binding-defective and mLHR(TM6,7) signaling-defective mutants were also used in a recent in vivo study using BAC transgenic mice suggesting functional rescue between dimerized LHR mutants (11). Male mice homozygous for the binding-defective mLHR(C22A) or signaling-defective mLHR(delTM6,7) on an Lhcgr knockout (LuRKO) background exhibited hypogonadism and infertility, whereas offspring from crosses between the mice that coexpressed both mutants on the LuRKO background had a nearly normal reproductive phenotype (11). How might these compelling observations be reconciled with our observations that these two mutants, when coexpressed in vitro, do not yield functional rescue, that both mutants are primarily retained intracellularly, and that the small amount of mLHR(C22A) capable of responding to hCG? If the mLHR(C22A)/mLHR(delTM6,7) BAC transgenic mice expressed greater amounts of mLHR(C22A) protein than the BAC transgenic mice expressing only mLHR(C22A), the former could have sufficient (albeit still very reduced) levels of cell surface mLHR(C22A) to permit gonadal development and responsiveness. That a small increase in the cell surface expression of mLHR(C22A) would be sufficient to rescue the reproductive phenotype of the mLHR(C22A)/mLHR(delTM6,7) BAC transgenic mice is supported by studies on naturally occurring activating mutations in humans of the LHCCR. Thus, in males with activating mutations of the LHCCR, pubertal levels of testosterone (causing testotoxicosis or gonadotropin-independent precocious puberty) are secreted even though the activating mutations cause only a small increase in basal cAMP as compared with hormone-stimulated wt receptor (34, 35). For example, hLHR(D578G), the first and most common activating LHCGR mutation associated with testotoxicosis identified in the US population, causes only a 3-fold increase in basal cAMP (36). Therefore, only a small increase in the cell surface expression of the mostly intracellularly retained mLHR(C22A) would be predicted to be necessary to result in the secretion of testosterone levels capable of inducing and maintaining a fairly normal reproductive phenotype in the male mLHR(C22A)/mLHR(delTM6,7) BAC transgenic mice. BAC transgenic mice have the limitation that the founders are generally mosaic and the offspring may have variable transgene copy numbers with possible gene amplification (37). Therefore, the apparent rescue of the functional phenotype in the mLHR(C22A)/mLHR(delTM6,7) BAC transgenic mice is most likely due to an increased copy number of the mLHR(C22A) transgene in these mice relative to the mLHR(C22A) homozygous mice. We also considered the possibility that the coexpression of mLHR(C22A) with mLHR(delTM6,7) might increase the cell surface expression of mLHR(C22A), although this would be unlikely because mLHR(delTM6,7) itself is profoundly intracellularly retained. Examining this by flow cytometry, we did not observe greater cell surface expression of mLHR(C22A) when mLHR(delTM6,7) was present (data not shown).
Taken altogether, our studies seriously question the concept that functional rescue can occur between mutant LHR protomers within a dimer. We suggest that the apparent functional rescue observed in the in vivo studies may be explained by possible differences in mLHR(C22A) copy number and therefore protein expression between the particular mLHR(C22A) BAC transgenic mice and the crossed mLHR(C22A)/mLHR(delTM6,7) BAC transgenic mice used for phenotypic analyses (11). Therefore, although our studies and those of others using approaches other than functional rescue have demonstrated glycoprotein hormone receptor dimerization when the receptors are expressed as recombinant proteins in heterologous cells (5–7, 32, 33), we maintain that caution should be exercised when using data from functional rescue experiments as suggestive of glycoprotein hormone receptor dimerization.
Data from a multitude of studies in recent years support the general concept of GPCR associating into homo- and heterodimers and, in most cases, higher ordered oligomers as well (reviewed in Refs. 38–41). The precise mechanisms by which a GPCR dimer activates the G protein and how the composition of the receptor protomers within the dimer may affect signaling, however, are not yet fully understood. Several studies suggest an asymmetry between receptor protomers within a receptor dimer with respect to the binding to and activation of the G protein (42–46). Although the data presented herein argue against functional rescue resulting from trans-activation between dimerized mutant LHR protomers, our data do not rule out the possibility that dimerized LHR protomers allosterically influence each other. Indeed, it has been reported using other GPCR that trans-activation, as defined by an allosteric modulation of signaling between the protomers of a receptor dimer, occurs although the effects described may vary between receptor systems (47–49). With respect to the LHR, it has been shown that the coexpression of the signaling impaired hLHR(D405N,Y546F) with wt or constitutively active hLHR causes a small degree of attenuation of signaling through the wt or constitutively active receptor in a manner consistent with heterodimerization (28). Further support for an allosteric communication between protomers within a glycoprotein hormone receptor dimer come from studies using hormone desorption assays (6, 7, 33).
In summary our studies, if not refuting, substantively question and challenge earlier studies suggesting functional rescue between a hormone binding-defective LHR and a signaling-defective LHR, and we maintain that previous observations suggesting functional rescue may be explained by other confounding factors. Therefore, our findings argue against a model by which the agonist-occupied ECD of one glycoprotein receptor protomer within a dimer may activate the serpentine domain of the other protomer. We suggest, however, that the dimerization of the gonadotropin receptors allows for more subtle allosteric communication between the protomers that may serve to contribute to the regulation of signaling.
Materials and Methods
Plasmids and hormones
The cDNA for wt hLHR and mLHR were kindly given to us by Ares Advanced Technology (Ares-Serono Group, Randolph, MA) and Dr. Thomas Gudermann (University of Munich, Munich, Germany), respectively. For expression and functional studies, the cDNA were inserted into pcDNA3.1(neo) (Invitrogen, Carlsbad, CA), and mutations were introduced using standard methodologies. Where indicated, an epitope tag was placed at the N terminus of the mature protein. This modification did not affect LHR cell surface expression, binding, or signaling properties. The pcDNA3.1 plasmids encoding myc-mLHR(C22A) and FLAG-mLHR(delTM6,7) were kindly given to us by Dr. Ilpo Huhtaniemi (Imperial College of London, London, UK). For BRET studies, the cDNA were subcloned into pRluc or pGFP2 vectors (PerkinElmer, Waltham, MA), which insert Renilla luciferase (Rluc) or GFP2, respectively, at the C terminus of the protein. HERG-GFP2 was a generous gift from Dr. Alvin Shrier (Department of Physiology, McGill University, Montreal, Canada). Plasmids were prepared using Qiagen maxiprep kits (QIAGEN, Valencia, CA), and sequences were confirmed by automated DNA sequencing (performed by the DNA Core Facility of the University of Iowa Carver College of Medicine). Pregnant mare serum gonadotropin and highly purified recombinant hCG and FSH were purchased from Dr. A. Parlow and the National Institute of Diabetes and Digestive and Kidney Diseases National Hormone and Pituitary Program. Purified hCG and FSH were iodinated as previously described (50). A crude preparation of hCG, used only for the determination of nonspecific binding in hCG binding assays, was purchased from Sigma (St. Louis, MO).
Cells and transfections
HEK293 and 293T cells were obtained from the American Type Tissue Collection (Manassas, VA) and were maintained, plated for experiments, and transiently transfected as previously described (6). When transfecting cells with varying concentrations of plasmid encoding receptor cDNA, the total amount of plasmid was kept constant using EV. Stably transfected cells were established by transfecting cells with vectors containing a neo-selectable marker and, after removing the transfection cocktail, culturing the cells in growth media supplemented with 700 μg/ml G418 (selection media). Cells were grown in selection media for at least 2 wk before use in experiments and were maintained in selection media.
[125I]hCG binding assays
Maximal cell surface hCG-binding capacity was determined as previously described using intact cells incubated with [125I]hCG (500 ng/ml final concentration) with or without an excess of unlabeled crude hCG (50 IU/ml final concentration) (27). Determination of the Ki for competition of [125I]hCG by unlabeled hCG was performed by incubating intact cells with a subsaturating concentration of [125I]hCG (10 ng/ml final concentration) and increasing concentrations of the same preparation of highly purified recombinant, unlabeled, hCG. Data were analyzed using GraphPad Prism software (La Jolla, CA).
[125I]hFSH binding assays
Maximal cell surface hFSH binding capacity was determined as previously described using intact cells incubated with [125I]hFSH (300 ng/ml final concentration) with or without an excess of unlabeled pregnant mare serum gonadotropin (220 IU/ml final concentration) (27).
Flow cytometry to evaluate cell surface receptor expression
On the day of the experiment, HEK293 cells were washed once with filtered PBS for immunohistochemistry (PBS-IH, 137 mm NaCl, 2.7 mm KCl, 1.4 mm KH2PO4, 4.3 mm Na2HPO4, pH 7.4). Cells were then detached by washing with PBS-IH, collected by gentle centrifugation, and resuspended in PBS-IH. For detection of HA epitope tags, they were incubated 1 h at 4C in the presence or absence HA.11 monoclonal anti-HA antibody from Covance Laboratories, Inc. (Princeton, NJ) diluted 1:500 with PBS-IH containing 0.5% BSA (PBS-IH/BSA), washed with PBS-IH/BSA, and then incubated 1 h at 4 C with fluorescein isothiocyanate-conjugated goat antimouse IgG (1:350) from Sigma in PBS-IH/BSA. For detection of myc epitope tags, the primary antibody used was 9E10 monoclonal anti-myc antibody (1:20) from Santa Cruz (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Cells were washed with PBS-IH/BSA, resuspended in 1 ml PBS-IH/BSA, and filtered through a 70-μm BD Falcon cell strainer (BD Biosciences, Palo Alto, CA) into a clean test tube on ice. A BD DiVa fluorescence-activated cell sorter with a 488-nm laser was then used to quantify cell surface expression in 10,000 cells from each experimental group. Control gating was set using cells transfected with EV and stained with antibody as described. Arbitrary fluorescence units describing total cell surface fluorescence were determined as the product of the percent cells gated and the geometric mean fluorescence of the sample minus the respective product in the control (EV) group.
Determination of intracellular cAMP production
Intracellular cAMP in response to maximally stimulatory concentration of hCG (100 ng/ml final concentration) or hFSH (300 ng/ml) was measured as described previously (27).
BRET2 assays
HEK293T cells were transiently cotransfected with a fixed concentration of Rluc fusion proteins and increasing concentrations of GFP2 fusion proteins, where within a given experiment the total amount of plasmid transfected was made constant by the addition of EV and the concentrations of plasmids encoding the Rluc fusion proteins were adjusted so that, after substrate addition, bioluminescence values of the Rluc fusion proteins expressed alone were similar. The cells were collected and BRET2 ratios determined as previously described (6, 28, 33). The data were plotted using GraphPad Prism (La Jolla, CA).
Confocal imaging of receptors
HEK293 cells were washed, fixed, and treated with or without 1% Triton for permeabilization as previously described (51). Cells expressing FLAG-mLHR receptor were incubated with anti-FLAG M2 monoclonal antibody (Sigma product F3165) at a final concentration of 1:200, and cells expressing HA-mLHR receptor were incubated with anti-HA monoclonal antibody (Covance product MMS-101R) at a final concentration of 1:200 for 1 h at room temperature (RT). After washing, cells were incubated with fluorescein isothiocyanate-conjugated anti-mouse IgG (Sigma product F0257) at a final dilution of 1:500 for 1 h at RT. Cells were visualized by confocal microscopy as previously described (51).
Quantitative analyses of mLHR mRNA expression
HEK293 and HEK293T cells were each transiently transfected with plasmids encoding mLHR(wt) or mLHR(C22A) as described above. On the day of the experiment, total RNA was extracted using PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA). RNA (1 μg) was digested with deoxyribonuclease I for 15 min at RT and reverse transcribed with oligo-dT and SuperScript III Reverse Transcriptase (Invitrogen). RNA levels were measured by quantitative RT-PCR using IQ SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA) (52). The primers for Lhcgr and GADPH were the same as those reported by others (53, 54).
Supplementary Material
Acknowledgments
We thank Drs. Francesco DeMayo (Baylor College of Medicine) and Robert Kesterson (University of Alabama School of Medicine) for valuable discussions regarding mouse BAC transgenics and Dr. Mario Ascoli (The University of Iowa College of Medicine) for critical reading of the manuscript.
This work was supported by National Institutes of Health Grants HD022196 and DK068614.
Disclosure Summary: M.Z., R.G., and D.L.S. have nothing to declare.
Because the hLHR has a signal peptide and the N terminus of the mature protein has not been experimentally determined, the numbering system we have used in this and other reports starts with the initiation methionine. In contrast, the original reports by others initiated numbering with the presumed N terminus of the mature protein.
Because the signal peptide length was altered in the mLHR(delTM6,7) mutant, the numbering of residues of both mLHR mutants herein is based on the beginning of the predicted mature mLHR protein.
As numbered from the first residue of the signal peptide. Termed hLHR(I55A) in the original reports (8–10).
As numbered from the first residue of the signal peptide. Termed hLHR(C22A) in the original report (9).
- BRET
- Bioluminescence resonance energy transfer
- CG
- chorionic gonadotropin
- ECD
- extracellular domain
- EV
- empty vector
- FSHR
- FSH receptor
- GFP
- green fluorescent protein
- GPCR
- G protein-coupled receptor
- HA
- hemagglutinin
- HB
- hormone-binding domain
- HEK
- human embryonic kidney
- IH
- immunohistochemistry
- LHR
- LH receptor
- RT
- room temperature
- wt
- wild type.
References
- 1. Xie YB, Wang H, Segaloff DL. 1990. Extracellular domain of lutropin/choriogonadotropin receptor expressed in transfected cells binds choriogonadotropin with high affinity. J Biol Chem 265:21411–21414 [PubMed] [Google Scholar]
- 2. Davis D, Liu X, Segaloff DL. 1995. Identification of the sites of N-linked glycosylation on the follicle-stimulating hormone (FSH) receptor and assessment of their role in FSH receptor function. Mol Endocrinol 9:159–170 [DOI] [PubMed] [Google Scholar]
- 3. Thomas D, Rozell TG, Liu X, Segaloff DL. 1996. Mutational analyses of the extracellular domain of the full-length lutropin/choriogonadotropin receptor suggest leucine-rich repeats 1–6 are involved in hormone binding. Mol Endocrinol 10:760–768 [DOI] [PubMed] [Google Scholar]
- 4. Fan QR, Hendrickson WA. 2005. Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433:269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tao YX, Johnson NB, Segaloff DL. 2004. Constitutive and agonist-dependent self-association of the cell surface human lutropin receptor. J Biol Chem 279:5904–5914 [DOI] [PubMed] [Google Scholar]
- 6. Guan R, Feng X, Wu X, Zhang M, Zhang X, Hébert TE, Segaloff DL. 2009. Bioluminescence resonance energy transfer studies reveal constitutive dimerization of the human lutropin receptor and a lack of correlation between receptor activation and the propensity for dimerization. J Biol Chem 284:7483–7494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Urizar E, Montanelli L, Loy T, Bonomi M, Swillens S, Gales C, Bouvier M, Smits G, Vassart G, Costagliola S. 2005. Glycoprotein hormone receptors: link between receptor homodimerization and negative cooperativity. EMBO J 24:1954–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji I, Lee C, Song Y, Conn PM, Ji TH. 2002. Cis- and trans-activation of hormone receptors: the LH receptor. Mol Endocrinol 16:1299–1308 [DOI] [PubMed] [Google Scholar]
- 9. Lee C, Ji I, Ryu K, Song Y, Conn PM, Ji TH. 2002. Two defective heterozygous luteinizing hormone receptors can rescue hormone action. J Biol Chem 277:15795–15800 [DOI] [PubMed] [Google Scholar]
- 10. Jeoung M, Lee C, Ji I, Ji TH. 2007. Trans-activation, cis-activation and signal selection of gonadotropin receptors. Mol Cell Endocrinol 260–262:137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rivero-Müller A, Chou YY, Ji I, Lajic S, Hanyaloglu AC, Jonas K, Rahman N, Ji TH, Huhtaniemi I. 2010. Rescue of defective G protein-coupled receptor function in vivo by intermolecular cooperation. Proc Natl Acad Sci USA 107:2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laue L, Chan WY, Hsueh AJ, Kudo M, Hsu SY, Wu SM, Blomberg L, Cutler GB., Jr 1995. Genetic heterogeneity of constitutively activating mutations of the human luteinizing hormone receptor in familial male-limited precocious puberty. Proc Natl Acad Sci USA 92:1906–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laue L, Wu SM, Kudo M, Hsueh AJ, Cutler GB, Jr, Jelly DH, Diamond FB, Chan WY. 1996. Heterogeneity of activating mutations of the human luteinizing hormone receptor in male-limited precocious puberty. Biochem Mol Med 58:192–198 [DOI] [PubMed] [Google Scholar]
- 14. Kremer H, Martens JW, van Reen M, Verhoef-Post M, Wit JM, Otten BJ, Drop SL, Delemarre-van de Waal HA, Pombo-Arias M, De Luca F, Potau N, Buckler JM, Jansen M, Parks JS, Latif HA, Moll GW, Epping W, Saggese G, Mariman EC, Themmen AP, Brunner HG. 1999. A limited repertoire of mutations of the luteinizing hormone (LH) receptor gene in familial and sporadic patients with male LH-independent precocious puberty. J Clin Endocrinol Metab 84:1136–1140 [DOI] [PubMed] [Google Scholar]
- 15. Latronico AC, Abell AN, Arnhold IJ, Liu X, Lins TS, Brito VN, Billerbeck AE, Segaloff DL, Mendonca BB. 1998. A unique constitutively activating mutation in the third transmembrane helix of the luteinizing hormone receptor causes sporadic male gonadotropin independent precocious puberty. J Clin Endocrinol Metab 83:2435–2440 [DOI] [PubMed] [Google Scholar]
- 16. Latronico AC, Chai Y, Arnhold IJ, Liu X, Mendonca BB, Segaloff DL. 1998. A homozygous microdeletion in helix 7 of the luteinizing hormone receptor associated with familial testicular and ovarian resistance is due to both decreased cell surface expresssion and impaired effector activation by the cell surface receptor. Mol Endocrinol 12:442–450 [DOI] [PubMed] [Google Scholar]
- 17. Hirakawa T, Galet C, Ascoli M. 2002. MA-10 cells transfected with the human lutropin/choriogonadotropin receptor (hLHR): a novel experimental paradigm to study the functional properties of the hLHR. Endocrinology 143:1026–1035 [DOI] [PubMed] [Google Scholar]
- 18. Hirakawa T, Ascoli M. 2003. A constitutively active somatic mutation of the human lutropin receptor found in Leydig cell tumors activates the same families of G proteins as germ line mutations associated with Leydig cell hyperplasia. Endocrinology 144:3872–3878 [DOI] [PubMed] [Google Scholar]
- 19. Qiao J, Han B, Liu BL, Chen X, Ru Y, Cheng KX, Chen FG, Zhao SX, Liang J, Lu YL, Tang JF, Wu YX, Wu WL, Chen JL, Chen MD, Song HD. 2009. A splice site mutation combined with a novel missense mutation of LHCGR cause male pseudohermaphroditism. Hum Mutat 30:E855–E865 [DOI] [PubMed] [Google Scholar]
- 20. Richter-Unruh A, Martens JW, Verhoef-Post M, Wessels HT, Kors WA, Sinnecker GH, Boehmer A, Drop SL, Toledo SP, Brunner HG, Themmen AP. 2002. Leydig cell hypoplasia: cases with new mutations, new polymorphisms and cases without mutations in the luteinizing hormone receptor gene. Clin Endocrinol (Oxf) 56:103–112 [DOI] [PubMed] [Google Scholar]
- 21. Wu SM, Hallermeier KM, Laue L, Brain C, Berry AC, Grant DB, Griffin JE, Wilson JD, Cutler GB, Jr, Chan WY. 1998. Inactivation of the luteinizing hormone/chorionic gonadotropin receptor by an insertional mutation in Leydig cell hypoplasia. Mol Endocrinol 12:1651–1660 [DOI] [PubMed] [Google Scholar]
- 22. Martens JW, Verhoef-Post M, Abelin N, Ezabella M, Toledo SP, Brunner HG, Themmen AP. 1998. A homozygous mutation of the luteinizing hormone receptor causes partial Leydig cell hypoplasia: correlation between receptor activity and phenotype. Mol Endocrinol 12:775–784 [DOI] [PubMed] [Google Scholar]
- 23. Toledo SP, Brunner HG, Kraaij R, Post M, Dahia PL, Hayashida CY, Kremer H, Themmen AP. 1996. An inactivating mutation of the luteinizing hormone receptor causes amenorrhea in a 46, XX female. J Clin Endocrinol Metab 81:3850–3854 [DOI] [PubMed] [Google Scholar]
- 24. Kremer H, Kraaij R, Toledo SP, Post M, Fridman JB, Hayashida CY, van Reen M, Milgrom E, Ropers HH, Mariman E, Themmen APN, Brunner HG. 1995. Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat Genet 9:160–164 [DOI] [PubMed] [Google Scholar]
- 25. Laue L, Wu SM, Kudo M, Hsueh AJ, Cutler GB, Jr, Griffin JE, Wilson JD, Brain C, Berry AC, Grant DB, Chan WY. 1995. A nonsense mutation of the human luteinizing hormone receptor gene in Leydig cell hypoplasia. Hum Mol Genet 4:1429–1433 [DOI] [PubMed] [Google Scholar]
- 26. Zhang M, Mizrachi D, Fanelli F, Segaloff DL. 2005. The formation of a salt bridge between helices 3 and 6 is responsible for the constitutive activity and lack of hormone responsiveness of the naturally occurring L457R mutation of the human lutropin receptor. J Biol Chem 280:26169–26176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang M, Tao YX, Ryan GL, Feng X, Fanelli F, Segaloff DL. 2007. Intrinsic differences in the response of the human lutropin receptor versus the human follitropin receptor to activating mutations. J Biol Chem 282:25527–25539 [DOI] [PubMed] [Google Scholar]
- 28. Zhang M, Feng X, Guan R, Hébert TE, Segaloff DL. 2009. A cell surface inactive mutant of the human lutropin receptor (hLHR) attenuates signaling of wild-type or constitutively active receptors via heterodimerization. Cell Signal 21:1663–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hébert TE, Galés C, Rebois RV. 2006. Detecting and imaging protein-protein interactions during G protein-mediated signal transduction in vivo and in situ by using fluorescence-based techniques. Cell Biochem Biophys 45:85–109 [DOI] [PubMed] [Google Scholar]
- 30. Mercier JF, Salahpour A, Angers S, Breit A, Bouvier M. 2002. Quantitative assessment of β 1- and β 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J Biol Chem 277:44925–44931 [DOI] [PubMed] [Google Scholar]
- 31. Pfleger KD, Eidne KA. 2006. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET). Nat Methods 3:165–174 [DOI] [PubMed] [Google Scholar]
- 32. Thomas RM, Nechamen CA, Mazurkiewicz JE, Muda M, Palmer S, Dias JA. 2007. Follicle-stimulating hormone receptor forms oligomers and shows evidence of carboxyl-terminal proteolytic processing. Endocrinology 148:1987–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guan R, Wu X, Feng X, Zhang M, Hébert TE, Segaloff DL. 2010. Structural determinants underlying constitutive dimerization of unoccupied human follitropin receptors. Cell Signal 22:247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Latronico AC, Segaloff DL. 1999. Naturally occurring mutations of the luteinizing hormone receptor: Lessons learned about reproductive physiology and G protein-coupled receptors. Am J Hum Genet 65:949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Themmen APN, Huhtaniemi IT. 2000. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev 21:551–583 [DOI] [PubMed] [Google Scholar]
- 36. Shenker A, Laue L, Kosugi S, Merendino JJ, Jr, Minegishi T, Cutler GB., Jr 1993. A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature 365:652–654 [DOI] [PubMed] [Google Scholar]
- 37. Chandler KJ, Chandler RL, Broeckelmann EM, Hou Y, Southard-Smith EM, Mortlock DP. 2007. Relevance of BAC transgene copy number in mice: transgene copy number variation across multiple transgenic lines and correlations with transgene integrity and expression. Mamm Genome 18:693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Milligan G. 2008. A day in the life of a G protein-coupled receptor: the contribution to function of G protein-coupled receptor dimerization. Br J Pharmacol 153(Suppl 1):S216–S229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Terrillon S, Bouvier M. 2004. Roles of G-protein-coupled receptor dimerization. EMBO Rep 5:30–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dalrymple MB, Pfleger KD, Eidne KA. 2008. G protein-coupled receptor dimers: functional consequences, disease states and drug targets. Pharmacol Ther 118:359–371 [DOI] [PubMed] [Google Scholar]
- 41. Palczewski K. 2010. Oligomeric forms of G protein-coupled receptors (GPCRs). Trends Biochem Sci 35:595–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Damian M, Martin A, Mesnier D, Pin JP, Banères JL. 2006. Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J 25:5693–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Damian M, Mary S, Martin A, Pin JP, Banères JL. 2008. G protein activation by the leukotriene B4 receptor dimer. Evidence for an absence of trans-activation. J Biol Chem 283:21084–21092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jastrzebska B, Fotiadis D, Jang GF, Stenkamp RE, Engel A, Palczewski K. 2006. Functional and structural characterization of rhodopsin oligomers. J Biol Chem 281:11917–11922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Park PS, Lodowski DT, Palczewski K. 2008. Activation of G protein-coupled receptors: beyond two-state models and tertiary conformational changes. Annu Rev Pharmacol Toxicol 48:107–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prézeau L, Pin JP, Blahos J. 2005. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J 24:499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. 2009. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol 5:688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, Parmentier M, Springael JY. 2009. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem 284:31270–31279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Monnier C, Tu H, Bourrier E, Vol C, Lamarque L, Trinquet E, Pin JP, Rondard P. 2011. Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J 30:32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ascoli M, Puett D. 1978. Gonadotropin binding and stimulation of steroidogenesis in Leydig tumor cells. Proc Natl Acad Sci USA 75:99–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mizrachi D, Segaloff DL. 2004. Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol Endocrinol 18:1768–1777 [DOI] [PubMed] [Google Scholar]
- 52. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔ C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 53. Schmittgen TD, Zakrajsek BA. 2000. Effect of experimental treatment on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J Biochem Biophys Methods 46:69–81 [DOI] [PubMed] [Google Scholar]
- 54. Yamashita S, Tai P, Charron J, Ko C, Ascoli M. 2011. The Leydig cell MEK/ERK pathway is critical for maintaining a functional population of adult Leydig cells and for fertility. Mol Endocrinol 25:1211–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.