

Abstract
Iron is critical in multiple aspects of CNS development, but its role in neurodevelopment—the ability of iron deficiency to alter normal development—is difficult to dissociate from the effects of anemia. We developed a novel dietary restriction model in the rat that allows us to study the effects of iron deficiency in the absence of severe anemia. Using a combination of auditory brainstem response analyses (ABR) and electron microscopy, we identified an unexpected impact of nonanemic iron deficiency on axonal diameter and neurofilament regulation in the auditory nerve. These changes are associated with altered ABR latency during development. In contrast to models of severe iron deficiency with anemia, we did not find consistent or prolonged defects in myelination. Our data demonstrate that iron deficiency in the absence of anemia disrupts normal development of the auditory nerve and results in altered conduction velocity.
Introduction
Development of the CNS can be disrupted by gestational deficiency in multiple micronutrients, with iron deficiency being the most prevalent of such problems. An estimated 25% of pregnancies worldwide are thought to involve iron deficiency (ID) anemia (IDA), the most severe form of iron deficiency (Stoltzfus, 2003). This is increasingly recognized as a serious concern for CNS development, as early gestational IDA is associated with long-lasting cognitive abnormalities that affect language learning, behavior, general nerve conductivity (Algarín et al., 2003; Beard, 2003; Lozoff et al., 2008), altered motor function, and coordination (Shafir et al., 2008). Many of the impairments cannot be reversed with iron supplementation, suggesting a critical window of vulnerability exists during early development (Kwik-Uribe et al., 2000; Lozoff et al., 2000).
In contrast to the many studies on IDA, far less is known about the impact on CNS development of moderate ID, a condition where tissue iron concentrations are decreased but not severely enough to induce anemia. Although an estimated 50% of anemia cases are due to IDA, the prevalence of ID is in fact three to five times greater than IDA (Cameron and Neufeld, 2011). Importantly, emerging studies suggest that ID without anemia can have negative consequences on behavioral and neurocognitive outcomes (Kwik-Uribe et al., 2000; McCann and Ames, 2007) and it is thus imperative to develop approaches that allow further study of the impact of ID on neurodevelopment in the absence of anemia.
We have established a model of nonanemic maternal ID during gestation that generates offspring that are ID but show no signs of severe anemia. ID offspring show reduced levels of brain iron and significant age-specific alterations in conduction velocity as measured by auditory brainstem responses (ABRs), a noninvasive test used in clinical settings. As the latency response of wave I of the ABR is a readout of signal transduction along the auditory nerve and is influenced by the degree of myelination and by axonal size, we conducted a morphological analysis of the auditory nerve itself, which has never been investigated in ID. In contrast to studies in IDA where changes in ABR latency had been interpreted as a reflection of myelination defects, we found only transient and minor changes in expression of myelin proteins MBP and PLP in the auditory nerve without changes to other myelin components (MAG and MOG). A detailed ultrastructural analysis of myelination also revealed no disruption in myelin thickness or compaction. We did, however, find a significant change in axon diameter distribution with inappropriately large axons early in development and abnormally small axons at more mature stages. Together, these results suggest that iron plays an important role in proper axonal maturation and reveals a novel pathology caused by ID in the absence of anemia.
Materials and Methods
All protocols were approved by University Committee on Animal Resources at the University of Rochester. Eight-week old female Fisher rats (Charles River) were fed a control diet containing 240 μg/Fe/g (Harlan Teklad) for 1 week to acclimate. Animals were then divided into a control or iron-deficient (2–6 μg/Fe/g) diet group. Diet regimen began 2 weeks before mating and through the remainder of the experimental period.
Measurements of hematocrit levels and brain iron concentrations were performed as previously described (Mihaila et al., 2011). Control (Fe+) and iron deficient (Fe−) offspring underwent open-field ABR testing on P14, P21, and P40. ABR measurements and waveform analysis were performed as previously described (Polesskaya et al., 2010).
For EM analysis, animals were anesthetized, perfused with fixative (2% glutaraldehyde/4% PFA/0.1 m sodium cacodylate), and tissues were postfixed for 48 h. Cochleae were decalcified in 10% EDTA/PBS up to 3 d before sectioning and analysis.
For tissue harvest, animals were killed and auditory nerve (AN) samples were homogenized at 4°C in lysis buffer, subjected to SDS-PAGE, and probed for expression of ferritin (Cell Signaling Technology), myelin basic protein (Millipore), proteolipid protein (Novus Biologicals), protein zero (Abcam), phosphorylated neurofilament and neurofilament-M (Millipore), Caspr, MAG, MOG (Santa Cruz Biotechnology), and β-actin as loading control. Primary antibodies were applied overnight then incubated in HRP-conjugated secondary antibodies (Santa Cruz Biotechnology). Proteins were visualized via ECL-detecting reagents (GE Healthcare).
For oligodendrocyte counts, decalcified cochleae were cryosectioned longitudinally through the central modiolus and incubated in primary antibody CC1 rabbit polyclonal antibody (Abcam) followed by labeling with FITC and Texas Red-conjugated secondary antibodies (Santa Cruz Biotechnology). Cell counts were normalized to DAPI nuclei staining.
The number of animals per group analyzed is represented by n. Each control and ID group was composed of animals from at least three separate litters. Statistical analyses were performed with two-way ANOVA followed by Bonferroni posttest comparing ID to the control group. Variances of auditory axon size distribution were calculated per animal and then tested by two-way ANOVA between groups. A p value ≤0.05 was considered to be statistically significant.
Results
Offspring exposed to suboptimal iron levels during gestation show CNS iron depletion without developing anemia
We first observed that in utero nutritional ID did not result in postnatal anemia in the offspring (as detected by hematocrit values, red blood cell size, and hemoglobin content) but did cause significant reductions in tissue iron levels in the CNS. Dams were placed on an iron-restricted diet 2 weeks before mating and maintained on this diet throughout gestation to recapture the window of vulnerability (Mihaila et al., 2011). The dams maintained normal hematocrit values and produced normal sized, healthy looking pups with hematological values in the normal range at P1, P7, and P14 (Fig. 1A). We did observe a transient deviation in hematocrit value at P21.
Figure 1.
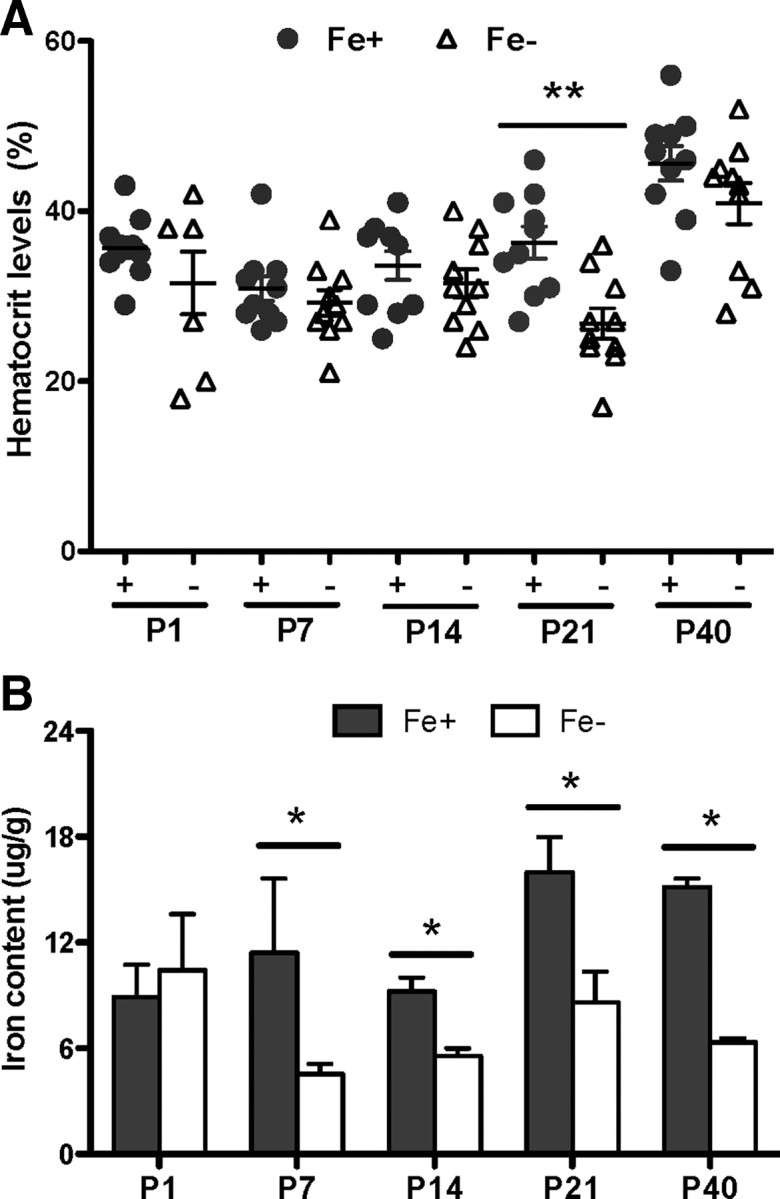
Gestational ID reduces postnatal brain iron concentration in the absence of severe anemia. A, Hematocrit levels from animals born to dams on Fe+ and Fe− diets. No significant differences were seen except at postnatal day 21. B, Iron content in whole brains measured by atomic absorption spectroscopy showed reductions in ID rat brains throughout postnatal development. Results are means ± SEM of pups from three separate litters. **p < 0.04, *p < 0.05 by two-way ANOVA followed by Bonferroni posttest.
Atomic absorption spectroscopic analysis on whole-brain tissue at different postnatal ages showed marked ID in the CNS of pups born to dams on an ID diet. Brain tissue from offspring in the ID group exhibited a significant reduction in iron content as early as P7, persisting until at least P40 and was only ∼50% of control at all ages (Fig. 1B).
CNS iron depletion in the offspring impairs auditory nerve function and alters myelin expression in an age-dependent manner
The ABR is a functional, noninvasive test that is widely used in humans to measure synchronous neural activity generated at several levels of the ascending auditory pathways, including the AN. The measurement of the ABR results in a wave diagram with wave I representing the neural activity along the AN. As the speed of impulse conduction is determined by axon diameter, myelination, and synapse integrity, insult-associated changes in conduction velocity are an indication of impairments in one or more of these parameters (Cankaya et al., 2003).
We found a significant decrease in ABR wave I latency in P14 ID pups compared with control animals, suggesting abnormally rapid signal conduction in the early AN (Fig. 2A). The ABR wave I latency decreased with age in both control and ID offspring and is characteristic of axonal maturation during this developmental window, but the absolute latency achieved at P40 was different in ID nerves. At P40, we found a significant increase in ABR wave I latency in ID animals, suggesting a slower signal conduction velocity in the AN (Fig. 2A). Consistent with our previous studies on offspring born to mothers with IDA (Mihaila et al., 2011), however, we did not detect any significant changes in ABR thresholds in ID, indicating the presence of intact hair cells. To confirm that the ABR impairments were associated with iron depletion in the target tissue, we determined the iron status in the ANs at all time points through analysis of the intracellular iron storage protein, ferritin (Fig. 2B). Ferritin levels were decreased in all ID samples, indicative of mobilization of intracellular iron stores, which occurs as a result of tissue ID (Chen et al., 1995).
Figure 2.

ID differentially impacts auditory nerve conduction at P14 and P40 with transient disruption of myelin proteins at P21. A, ID ABR peak 1 latencies (dashed line) are significantly faster at P14 and slower at P40 compared with controls (solid line). B, Quantified results of AN lysates probed for ferritin to establish tissue iron deficiency normalized to β-actin. C, Representative blots of AN samples probed for MBP, PLP, and P0 (protein zero). D, Myelin changes at P21 quantified; note significant reduction in mature MBP isoforms and PLP. E, Mature oligodendrocyte numbers, identified by CC1 positivity, do not change in controls or ID cochleae. Quantification presented as means ± SEM for n = 4 per group. *p < 0.05, **p < 0.03 by two-way ANOVA followed with Bonferroni posttest.
To determine whether the impaired ABR latencies at P14 and P40 were associated with changes in myelination, we examined expression of the myelin components myelin basic protein (MBP) and proteolipid protein (PLP) in the ANs from which ABR wave I responses were generated. There were no significant differences between ID and normal ANs at P14 and P40. We did, however, see differences in ID ANs at P21 with a specifically reduced expression of the 18.5 and 14 kDa MBP isoforms, which are associated with compact myelin generation in mature oligodendrocytes (Akiyama et al., 2002). The isoforms associated with the earlier stages of myelination (17 and 21.5 kDa) were not significantly different between ID and control ANs at this time point (Fig. 2C,D). The decrease in the mature MBP isoforms was not due to a decreased number of oligodendrocytes, as determined by CC1 immunoreactivity (Fig. 2E). Interestingly, decreased levels of mature MBP isoforms and PLP at P21 had no impact on the ABRs, such that ID animals approximated latencies in P21 controls (Fig. 2A).
Reduced iron levels in the auditory nerve alter axonal caliber without changes in myelin thickness
As we did not see a gross disruption of myelin protein expression and oligodendrocytes at P14 and P40 but still had altered latencies at these time points, we quantified axon diameter at P14, P21, and P40 by electron microscopy and found that normal maturational changes in axonal diameter were compromised in the ID offspring. The types of alterations were consistent with the abnormalities in the ABR seen at different ages (Fig. 3A,B). While the overall number of myelinated axons was not different in ID at any time point (Fig. 3I), control axons (Fig. 3B, gray) showed a developmental increase in diameter but ID axons (Fig. 3B, red) failed to significantly expand from P14 to P40. We also observed a significant variance in dispersion of axon diameters in ID ANs at time points when we detected an ABR defect. P14 ID nerves contained significantly larger axons than controls and displayed a broader distribution of axon diameters (Fig. 3C). When the ABR was normal, P21 ID axon diameters approximated those in control nerves with a similar size distribution (Fig. 3D). At P40, however, ID nerves lacked axons >3.5 μm and the broad distribution of axons characteristic for normal tissue was absent (Fig. 3E). Strikingly, the differences in axon diameters correlated precisely with the alterations in ABR latencies.
Figure 3.
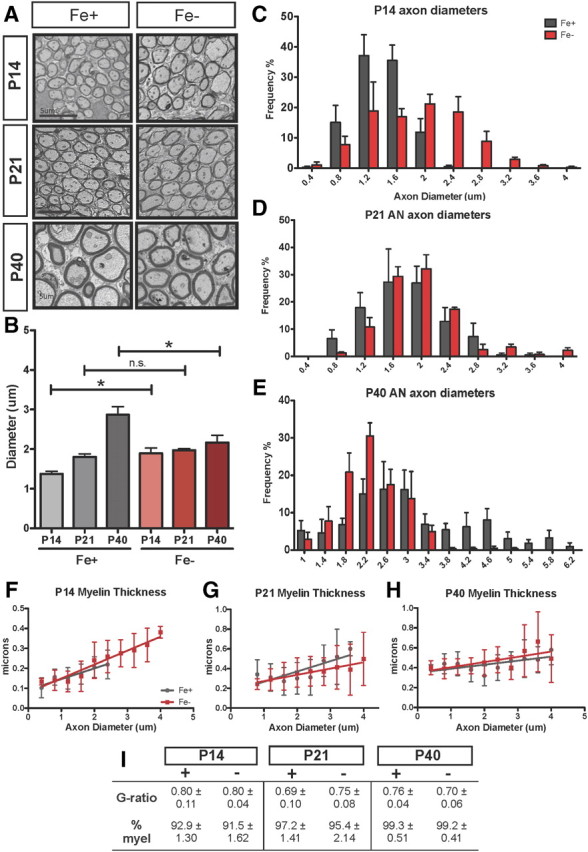
ID leads to larger axons at P14 but reduced diameter at P40 without changes to myelin thickness. A, Representative images of axons from iron-normal and ID auditory nerves. Axons were analyzed in five random images taken from EM cross-sections of Fe− and Fe+ auditory nerves. Scale bars, 5 μm. B, Quantification of mean axon diameters in Fe+ (gray) compared with Fe− (red) auditory nerves. *p < 0.03 at P14, *p < 0.05 at P40 by two-way ANOVA followed by Bonferroni posttest; n.s., not significant. C–E, Axon measurements at P14 (C), P21 (D), and P40 (E) were stratified by size frequency; note the substantial shift in Fe− axon diameters at P14 and the altered distribution at P40. Variance in Fe+ diameters increased significantly over time (**p < 0.001) but did not in Fe− animals (p = 0.37 by two-way ANOVA; data per age shown in C–E). F–H, Averaged myelin thickness plotted per respective axon diameter; quantified values are comparable in Fe+ and Fe− auditory nerves. I, G-ratio and percentage myelination (myel) at P14, P21, and P40. Results are means ± SEM of pups from at least three litters per group.
Although altered MBP and PLP expression at P21 suggested possible developmental defects in myelin maturation, we could not detect changes in myelin thickness via ultrastructural analysis of the ID nerve throughout development. As shown in Figure 3, F–H, the thickness of the myelin sheaths in axons of comparable sizes in ID and normal ANs were not significantly different at any time point. While we do not know whether the myelin thickness of large axons (>2.8 μm) in ID nerves at P14 absent in control tissue is appropriate, these axons are nevertheless myelinated and appear structurally normal. The ratio of axon to fiber diameter (G ratio) showed no difference between ID and control ANs (Fig. 3I).
Iron deficiency alters phosphorylation of neurofilament, independent of MAG signaling
Neurofilaments (NF) are major intrinsic factors in determining axonal diameter; the increased diameter of axons during postnatal development reflects an increase in axonal NF content as well as the proportion of phosphorylated NF (Pant and Veeranna, 1995). Phosphorylation of NF was only increased at P14 in ID nerves (Fig. 4A). In contrast, at P40, ID ANs had decreased phosphorylated NF levels, consistent with the overall decrease in axon diameter.
Figure 4.
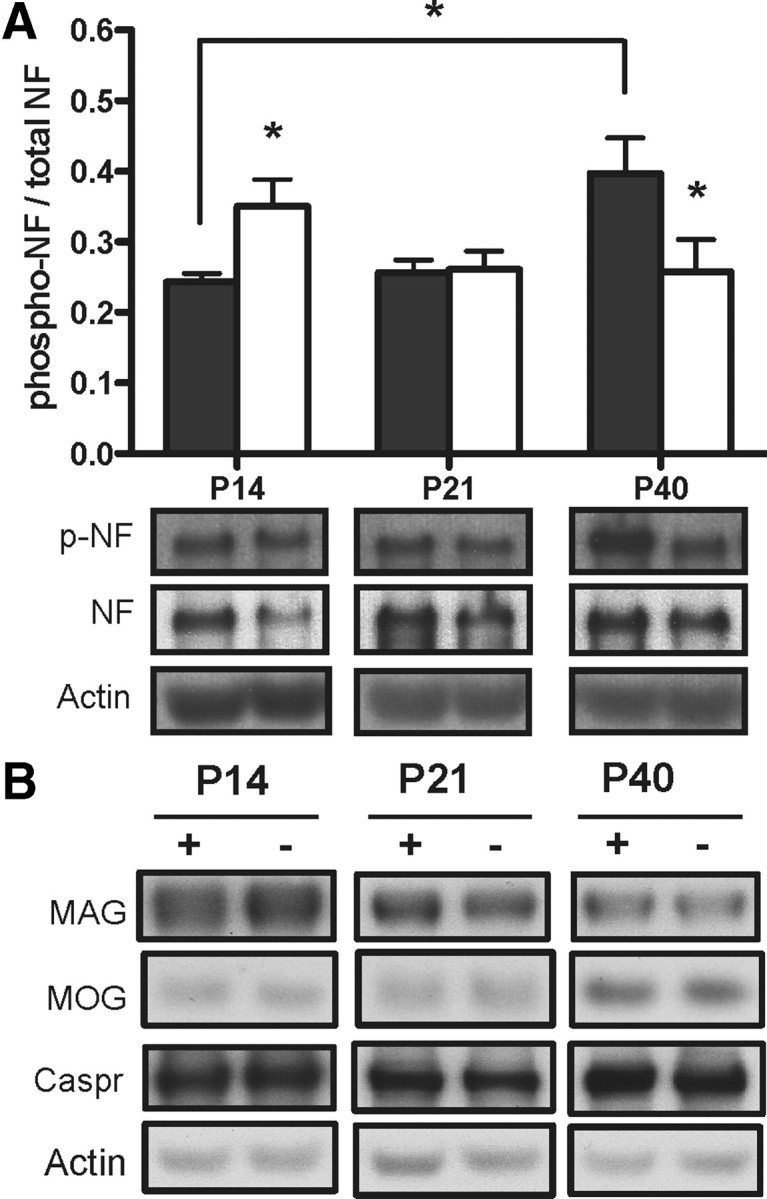
ID alters phosphorylation of neurofilament independent of MAG protein level. A, Quantified results of auditory nerve lysates probed for phosphorylated neurofilament M/H at P14, P21, and P40; note the increased phosphorylated NF at P14 compared with a reduction at P40. Representative blots of auditory nerve lysates probed for phosphorylated neurofilament (p-NF) and total neurofilament proteins. *p < 0.05 compared with respective time point control by two-way ANOVA followed by Bonferroni posttest. B, Representative blots of auditory nerve lysates probed for MAG, MOG, and Caspr with no altered expression. Results are means ± SEM of pups from at least three litters per group.
We next examined the expression of MAG, a myelin-associated glycoprotein that has been shown to modulate axonal caliber through phosphorylation of axonal NF proteins (Yin et al., 1998), MOG (which has been shown to be involved in axon–glia interactions), and the axonal glycoprotein Caspr (a contactin-associated protein important for myelin sheath attachment and impulse conduction). As shown in Figure 4B, none of these components showed any differences in expression in the ANs throughout development in iron deficiency.
Together, these results suggest that postnatal ABR defects associated with iron deficiency are not exclusively the result of a myelination defect. The disruptions to axon size and neural conduction in the absence of sufficient iron indicate that the cellular targets of iron deficiency may extend outside of the glial compartment.
Discussion
The motivation for this study was to understand the role of iron in neural development. The study of models of limited iron availability has provided important insight but such models are often associated with the induction of anemia, the most severe form of ID. To disentangle the consequences of ID from those of IDA, we tested various rat strains on a variety of ID diets and found that the Fisher rat on 2–6 μg/Fe/g represented a model in which dams never develop anemia while on the diet during pregnancy, ensuring that the growing embryos are not affected by hypoxia. The pups, which remained on the ID diet, were not significantly anemic either and manifested only a marginal transient decrease in hematocrit at P21. Critically, however, from P7 to P40, animals born to the ID dams had markedly lower brain iron levels and a decrease in iron storage capacity. This model thus provides an opportunity to determine the role of iron during development in the absence of anemia.
As expected, the offspring showed no gross abnormalities, e.g., normal weight and pallor. However, detailed analysis revealed a complex pattern of abnormal ABRs characterized by a decreased latency at P14 (i,e., faster impulse conduction), no impairment at P21, and increased latency at P40 (i.e., slower impulse conduction). ABR studies are used in humans as a noninvasive test to measure brain maturation defects (Roncagliolo et al., 1998; Amin et al., 2010). The interpretation that altered ABRs represent maturation defects is due to the correlation between conduction velocity, the increase in axonal diameter, and acquisition of myelin during development. A previous model of IDA showed increased latency of >0.2 ms ABRs (Mihaila et al., 2011), and increased latencies of >0.1 ms have been reported in human studies of IDA, where such changes are considered to represent significant functional impairments (Roncagliolo et al., 1998).
While the ABR analysis revealed a general impairment in brain function, which was specific to certain development ages, the challenge was to correlate this compound readout with a defined structural defect. To directly investigate the structural and functional relationship between ABR latency and anatomy, we isolated the distal auditory nerve, which produces wave 1 of the ABR. As wave 1 of the ABR is primarily mediated by the degree of myelination and axonal diameter, we reasoned that this analysis should provide insight into underlying causes of the defect we identified in ID animals. To our knowledge, no such study has previously been reported.
Our analysis of MBP and PLP expression in the AN revealed no significant change in the expression of early myelin proteins (MBP isoforms 21.5 and 17 kDa) at any time points. We observed no changes in the number of mature oligodendrocytes at any age. In addition, our measurements revealed no significant changes in myelin thickness or g-ratio. We did, however find a transient decrease in expression of the more mature MBP isoforms as well as PLP at P21, which coincides with the peak of myelination. This transient defect seemed to have no impact on the ABR, highlighting the multifactorial nature of the ABR.
The lack of a clear correlation of ABR changes with alterations in myelination suggested that other components contributed to the impaired ABR, and our studies revealed the unexpected finding of disruption in axonal maturation. For example, at P14 in control animals, the majority of axons were 0.5–1.5 μm in diameter, while at P40, axon sizes were spread across a wide range and reached >6 μm. In contrast, P14 ID axon distribution already reached up to 4 μm, which was maintained until P40 without further growth. Thus, the decreased latency at P14 was correlated with the presence of larger axons, while the increased latency at P40 was correlated with a relative scarcity of large axons in ID nerves. We cannot exclude the possibility that the change in axon size distribution was due to a selective loss of specific-sized axons in ID; however, this represents an unlikely explanation as no evidence was observed of axonal degeneration, excessive looping, or other defects associated with axonal degeneration (Edgar et al., 2009). While we did not observe a measurable change in myelin sheath thicknesses of comparably sized normal and ID axons, we did identify in ID axons significant changes in phosphorylated NF without changes to MAG, a known regulator of NF phosphorylation (Dashiell et al., 2002). ID ANs lacked the typical increase in NF phosphorylation, which is an important determinant of axonal caliber (Pant and Veeranna, 1995) and axonal transport rate in development (Shea et al., 2004). Our observation of normal Caspr levels in ID axons and lack of obvious impairment to overall node organization (data not shown) does not exclude but reduces concern for substantial node abnormality. While alternate explanations for the ABR defects certainly exist and include disruptions to unmeasured functions of oligodendrocytes, nodal formation, or synaptic function, the presented data strongly suggest that disrupted axonal size distribution plays a major factor in the ABR latency defects.
Impaired axonal size maturation in the ID AN raises the question of whether neuronal function is altered elsewhere, e.g., the target regions that are innervated by the potentially immature axons. The maturation of the AN is complex and not well understood but it has been shown in mice that AN axons enter the brain by E13–E14 and innervate the cochlear nuclei by E16, which is well before the onset of hearing (Carney and Silver, 1983). From E18 to approximately birth, axons enter the nuclear subdivisions, forming small side branches until appropriate targets are encountered and axons cease to grow. While still controversial, the AN is believed to grow into the brainstem before the migration of auditory neurons, which are then attracted to the AN axons and become postmitotic at day 10–14 (Rubel and Fritzsch, 2002). Hence, migration of most cochlear nucleus neurons coincides with the arrival of the AN axons, facilitating an interaction that is controlled by both intracellular and secreted molecules and is a critical part of proper brainstem development and synaptic maturation (Brumwell et al., 2000). As our tissue iron analysis suggests that the brain is not significantly iron deficient at the time of birth and only shows a reduction in iron content from P7 onwards, it seems likely that the origination and early innervations of AN axons would progress normally in ID and that defects will manifest at later stages, perhaps ultimately affecting the maturation of synaptic connections, which control processes like temporal processing. A paradigm for such disruption has been demonstrated through impaired dendritic arborization in the postnatal hippocampus under mild ID (Jorgenson et al., 2003). It will be of future interest to examine dendritic complexity in regions along the auditory pathway.
The data from our nutritional model demonstrate that in utero ID does not affect the pregnant dam and produces overall healthy, nonanemic offspring, but nonetheless is sufficient to cause both CNS iron depletion and readily detectable neurological impairments. Using ABR analysis as a noninvasive tool to assess neural function, combined with a detailed morphological analysis of the auditory nerve, we did not detect signs of hypomyelination, demyelination, a decrease in mature oligodendrocytes, or axonal degeneration, all of which have been observed in models of severe iron deficiency associated with anemia. In contrast, we report a novel phenotype that is characterized by the disruption of appropriate axonal maturation and continues to expand the cellular targets of iron deficiency outside of the glial compartment.
Footnotes
This work was supported by NIH Grants R01 HD059739 and R01 NS039511, and by the University of Rochester through grants from the Clinical and Translational Science Institute and the Environmental Health Science Center. We thank Ollivier Hyrien for outstanding assistance in the statistical evaluation of our data and Gayle Schneider, Kathy Barsz, and Kathryn Hutton for excellent technical assistance. We also would like to especially thank Camellia Mihaila for assistance with ABR data analysis; James Palis for his help with the blood analysis; and Karen Bentley, Core director of the EM facility, for her outstanding assistance with the EM analysis. We are also grateful to Mark Noble, Chris Pröschel, and all the members of our laboratory for stimulating discussion and insights.
The authors declare no competing financial interests.
References
- Akiyama K, Ichinose S, Omori A, Sakurai Y, Asou H. Study of expression of myelin basic proteins (MBPs) in developing rat brain using a novel antibody reacting with four major isoforms of MBP. J Neurosci Res. 2002;68:19–28. doi: 10.1002/jnr.10188. [DOI] [PubMed] [Google Scholar]
- Algarín C, Peirano P, Garrido M, Pizarro F, Lozoff B. Iron deficiency anemia in infancy: long-lasting effects on auditory and visual system functioning. Pediatr Res. 2003;53:217–223. doi: 10.1203/01.PDR.0000047657.23156.55. [DOI] [PubMed] [Google Scholar]
- Amin SB, Orlando M, Eddins A, MacDonald M, Monczynski C, Wang H. In utero iron status and auditory neural maturation in premature infants as evaluated by auditory brainstem response. J Pediatr. 2010;156:377–381. doi: 10.1016/j.jpeds.2009.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard J. Iron deficiency alters brain development and functioning. J Nutr. 2003;133:1468S–1472S. doi: 10.1093/jn/133.5.1468S. [DOI] [PubMed] [Google Scholar]
- Brumwell CL, Hossain WA, Morest DK, Bernd P. Role for basic fibroblast growth factor (FGF-2) in tyrosine kinase (TrkB) expression in the early development and innervation of the auditory receptor: in vitro and in situ studies. Exp Neurol. 2000;162:121–145. doi: 10.1006/exnr.2000.7317. [DOI] [PubMed] [Google Scholar]
- Cameron BM, Neufeld LM. Estimating the prevalence of iron deficiency in the first two years of life: technical and measurement issues. Nutr Rev. 2011;69(Suppl 1):S49. doi: 10.1111/j.1753-4887.2011.00433.x. [DOI] [PubMed] [Google Scholar]
- Cankaya H, Oner AF, Egeli E, Caksen H, Uner A, Akçay G. Auditory brainstem response in children with iron deficiency anemia. Acta Paediatr Taiwan. 2003;44:21–24. [PubMed] [Google Scholar]
- Carney PR, Silver J. Studies on cell migration and axon guidance in the developing distal auditory system of the mouse. J Comp Neurol. 1983;215:359–369. doi: 10.1002/cne.902150402. [DOI] [PubMed] [Google Scholar]
- Chen Q, Connor JR, Beard JL. Brain iron, transferrin and ferritin concentrations are altered in developing iron-deficient rats. J Nutr. 1995;125:1529–1535. doi: 10.1093/jn/125.6.1529. [DOI] [PubMed] [Google Scholar]
- Dashiell SM, Tanner SL, Pant HC, Quarles RH. Myelin-associated glycoprotein modulates expression and phosphorylation of neuronal cytoskeletal elements and their associated kinases. J Neurochem. 2002;81:1263–1272. doi: 10.1046/j.1471-4159.2002.00927.x. [DOI] [PubMed] [Google Scholar]
- Edgar JM, McLaughlin M, Werner HB, McCulloch MC, Barrie JA, Brown A, Faichney AB, Snaidero N, Nave KA, Griffiths IR. Early ultrastructural defects of axons and axon-glia junctions in mice lacking expression of Cnp1. Glia. 2009;57:1815–1824. doi: 10.1002/glia.20893. [DOI] [PubMed] [Google Scholar]
- Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci. 2003;25:412–420. doi: 10.1159/000075667. [DOI] [PubMed] [Google Scholar]
- Kwik-Uribe CL, Golub MS, Keen CL. Chronic marginal iron intakes during early development in mice alter brain iron concentrations and behavior despite postnatal iron supplementation. J Nutr. 2000;130:2040–2048. doi: 10.1093/jn/130.8.2040. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Jimenez E, Hagen J, Mollen E, Wolf AW. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics. 2000;105:E51. doi: 10.1542/peds.105.4.e51. [DOI] [PubMed] [Google Scholar]
- Lozoff B, Clark KM, Jing Y, Armony-Sivan R, Angelilli ML, Jacobson SW. Dose-response relationships between iron deficiency with or without anemia and infant social-emotional behavior. J Pediatr. 2008;152:696–702. doi: 10.1016/j.jpeds.2007.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann JC, Ames BN. An overview of evidence for a causal relation between iron deficiency during development and deficits in cognitive or behavioral function. Am J Clin Nutr. 2007;85:931–945. doi: 10.1093/ajcn/85.4.931. [DOI] [PubMed] [Google Scholar]
- Mihaila C, Schramm J, Strathmann FG, Lee DL, Gelein RM, Luebke AE, Mayer-Pröschel M. Identifying a window of vulnerability during fetal development in a maternal iron restriction model. PLoS One. 2011;6:e17483. doi: 10.1371/journal.pone.0017483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant H, Veeranna C. Neurofilament phosphorylation. Biochem Cell Biol. 1995;73:575–592. doi: 10.1139/o95-063. [DOI] [PubMed] [Google Scholar]
- Polesskaya O, Cunningham LL, Francis SP, Luebke AE, Zhu X, Collins D, Vasilyeva ON, Sahler J, Desmet EA, Gelbard HA, Maggirwar SB, Walton JP, Frisina RD, Jr, Dewhurst S. Ablation of mixed lineage kinase 3 (Mlk3) does not inhibit ototoxicity induced by acoustic trauma or aminoglycoside exposure. Hear Res. 2010;270:21–27. doi: 10.1016/j.heares.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncagliolo M, Garrido M, Walter T, Peirano P, Lozoff B. Evidence of altered central nervous system development in infants with iron deficiency anemia at 6 mo: delayed maturation of auditory brainstem responses. Am J Clin Nutr. 1998;68:683–690. doi: 10.1093/ajcn/68.3.683. [DOI] [PubMed] [Google Scholar]
- Rubel EW, Fritzsch B. Auditory system development: primary auditory neurons and their targets. Annu Rev Neurosci. 2002;25:51–101. doi: 10.1146/annurev.neuro.25.112701.142849. [DOI] [PubMed] [Google Scholar]
- Shafir T, Angulo-Barroso R, Jing Y, Angelilli ML, Jacobson SW, Lozoff B. Iron deficiency and infant motor development. Early Hum Dev. 2008;84:479–485. doi: 10.1016/j.earlhumdev.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB, Yabe JT, Ortiz D, Pimenta A, Loomis P, Goldman RD, Amin N, Pant HC. Cdk5 regulates axonal transport and phosphorylation of neurofilaments in cultured neurons. J Cell Science. 2004;117:933–941. doi: 10.1242/jcs.00785. [DOI] [PubMed] [Google Scholar]
- Stoltzfus RJ. Iron deficiency: global prevalence and consequences. Food Nutr Bull. 2003;24:S99–103. doi: 10.1177/15648265030244S206. [DOI] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]