

Summary
The intrinsic signaling networks of the coagulation pathways have recently emerged as crucial determinants for survival in sepsis and systemic inflammatory response syndromes. Protease activated receptor (PAR) 1 is central to both lethality promoting and vascular protective signaling. In the vascular anticoagulant pathway, EPCR/aPC-PAR1 signaling prevents vascular leakage and genetic or acute deficiencies in this pathway promote lethality. In addition, coagulation signaling acts directly on cells of the innate immune system. Dendritic cell (DC) thrombin-PAR1 signaling is coupled to the migration promoting sphingosine 1 phosphate receptor 3 (S1P3). Thrombin generated in the lymphatic compartment perturbs DCs to promote systemic inflammation and disseminated intravascular coagulation in severe sepsis. Signaling-selective aPC variants and selective modulators of the S1P receptor system attenuate sepsis lethality, suggesting novel therapeutic approaches that can be employed to rebalance alterations in the coagulation signaling pathways in severe inflammatory disorders.
Keywords: Tissue Factor, Thrombin, activated Protein C, DIC
Paradoxical roles for PAR1 in sepsis survival
Primate studies in lethal sepsis in the baboon demonstrated the importance of tissue factor (TF) initiated coagulation in the pathophysiology of sepsis syndrome and first established the sepsis protective effects of the anticoagulant protein C pathway [1]. Although genetic models of TF deficiency corroborated the crucial role of the TF pathway in murine sepsis, the relative contributions of TF-induced microthrombosis versus signaling of the coagulant and anti-coagulant pathways remained obscure. In a carefully titrated model of lipopolysaccharide (LPS) challenge, PAR1−/− mice showed a paradoxical survival response. Whereas wild-type mice died with increasing frequency as the LPS dose was escalated, ~ 50% PAR1−/− mice survived over the entire range of LPS concentrations (Fig. 1). Here, we discuss the underlying mechanisms for the sepsis protective and promoting roles of PAR1 signaling and the implication of the PAR1 signaling networks for sepsis therapy.
Fig. 1.
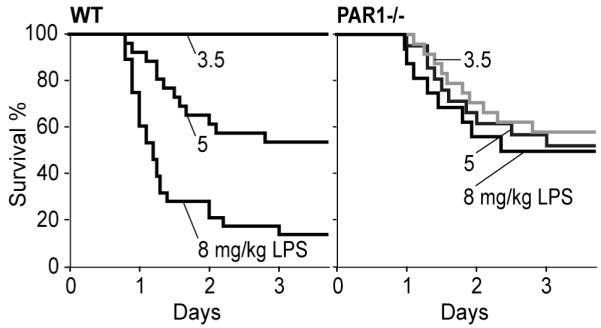
The PAR1 paradox in sepsis lethality.
Protease selectivity of PAR1 signaling in endothelial inflammatory responses, apoptosis, and barrier protection in vitro
PAR1 is cleaved by multiple coagulation serine proteases, including thrombin and aPC bound to the endothelial PC receptor (EPCR) [2]. Despite cleaving and activating the same receptor, aPC produces cellular responses that are distinct from thrombin signaling. The unique signaling specificity of aPC is likely dependent on combined effects of ligand occupancy of EPCR and restricted signaling in raft domains [3] and/or differences in PAR1 internalization [4]. Gene expression profiling has identified a signature of cellular responses consistent with anti-apoptotic effects of aPC signaling and suppression of inflammatory target genes downstream of NFκB [5,6]. Importantly, aPC counteracts the thrombin-mediated upregulation of pro-apoptotic genes specifically in inflamed endothelial cells [7].
Thrombin and aPC signaling produce particularly striking differences in their effects on endothelial barrier function. aPC potently inhibits thrombin-induced vascular hyper-permeability dependent on trans-activation of the sphingosine 1 phosphate (S1P) receptor 1 (S1P1) [8,9]. In contrast, thrombin induces vascular hyper-permeability dependent on another S1P receptor, S1P3 [10]. These in vitro studies suggest that G-protein receptor signaling crosstalk with lipid sensing S1P receptors is an important determinant for endothelial cell PAR1 signaling specificity (Fig. 2).
Fig. 2.

Coagulation signaling networks downstream of PAR1 in sepsis.
Prevention of inflammation-induced vascular leakage and lethality by endogenous EPCR/aPC-PAR1 signaling
The concept of differential S1P receptor coupling of thrombin-PAR1 versus EPCR/aPC-PAR1 signaling was supported in vivo by a combination of pharmacological interventions and genetic mouse models [11]. Genetic deficiencies in PC activation, as seen in thrombomodulin-mutated TMPro mice, or loss of EPCR/aPC signaling in EPCRlow and PAR1−/− mice resulted in increased vascular leakage and lethality following administration of a LPS dose that is typically non-lethal in wild-type mice. To exclude long term functional alterations due to these genetic deficiencies, we also acutely blocked the aPC pathway in LPS challenged mice with a potent monoclonal antibody that inhibits aPC’s catalytic activities. Blockade of the PC pathway with this antibody during established sepsis syndrome increased systemic markers of thrombin generation and resulted in severe vascular leakage and mortality in wild-type mice.
Remarkably, antibody-mediated disruption of the aPC pathway in mice lacking the S1P3 receptor also increased thrombin generation, but neither increased vascular leakage nor inflammation-associated lethality. PAR1−/− mice were also partially protected from the detrimental effects of administrating the blocking antibody to endogenous aPC. Together, these data supported the crucial connection between endothelial cell thrombin-PAR1 signaling and S1P3 in vivo. In addition, the complete survival of S1P3−/− mice after efficient aPC blockade indicated that anticoagulant effects of aPC contributed only minimally to survival of mice following LPS-induced systemic inflammation.
Experiments with variants of aPC corroborated the primary role of aPC signaling in the prevention of vascular leakage. Anticoagulant-selective aPC variants had no effect on inflammation-induced vascular hyper-permeability. In contrast, signaling-selective aPC prevented vascular leakage of TMPro mice, but no beneficial effects of aPC were seen in EPCRlow or PAR1−/− mice that lack the crucial endothelial receptors for aPC signaling, consistent with survival studies [12]. The identified role of S1P1 downstream of endothelial EPCR/aPC-PAR1 signaling predicted that a S1P1 agonist may show benefits in these models with defective aPC signaling. Indeed, TMPro, EPCRlow, PAR1−/−, as well as anti-PC treated wild-type mice were rescued from vascular leakage and lethality by therapy with a selective agonist of S1P1. This efficient reversal of vascular leakage due to genetic impairments or acute pharmacological blockade of the PC pathway provides indirect evidence that PAR1 signaling contributes to setting the tone of the vascular S1P1/S1P3 balance in inflammatory disorders.
PAR1-S1P3 proinflammatory signaling of dendritic cells
While the vascular protective effects of the EPCR/aPC-PAR1 signaling pathway explained increased lethality at low LPS doses (Fig. 1), the protection of PAR1−/− mice from lethality in severe sepsis was traced to a previously unrecognized role for PAR1 in regulating innate immunity [13]. Extensive chemokine and cytokine multiplex analysis showed that PAR1−/− mice responded to LD90 LPS challenge with an initial inflammatory response indistinguishable from wild-type animals. However, continuing elevation of dendritic cell (DC) and T cell-derived cytokines was drastically reduced at 18 hours in PAR1−/− relative to wild-type mice. DCs are key instructors of T cell activation, raising the possibility that PAR1 regulated DC function. Indeed, adoptive transfer of immature, bone marrow-derived wild-type DC populations into PAR1−/− mice was sufficient to restore wild-type levels of all inflammatory parameters, including T cell-derived cytokines. Thus, DCs orchestrate the inflammatory response in the late stages of these sepsis models.
Several experimental strategies supported the conclusion that DC PAR1 signaling was coupled to S1P3. Late stage inflammatory exacerbation was restored with a non-selective agonist for S1P receptors, but not with a selective agonist for S1P1, when these compounds were given to PAR1−/− mice 10 hours after LPS challenge. Similar to the PAR1 knock-out, late stage inflammation was reduced in mice lacking S1P3 or sphingosine kinase 1 (Sphk1) that phosphorylates sphingosine to S1P. In addition, S1P3−/− mice were markedly protected from LPS challenge and bacterial sepsis. This provided an opportunity for additional adoptive transfer experiments to directly probe the role of PAR1 signaling on DCs. Adoptive transfer of wild-type DCs into S1P3−/− mice produced severe inflammation, but adoptive transfer of PAR1−/− DCs had no adverse effect on late stage inflammation and lethality of S1P3−/− mice. Importantly, when S1P3−/− mice with PAR1−/− DCs received a S1P receptor agonist during the sepsis challenge, lethality was increased and indistinguishable from wild-type mice. Because this agonist had no effect in S1P3−/− mice and adoptively transferred PAR1 DCs were the only S1P3 expressing cells under these conditions, these data directly showed the essential role for DC PAR1-S1P3 signaling to cause sepsis lethality.
Further characterization of S1P3−/− revealed that circulating thrombin-antithrombin (TAT) levels were indistinguishable between S1P3−/− and wild-type mice 6 hours after LPS challenge, but TAT returned essentially to baseline levels at 18 hours in S1P3−/− mice. These data indicated that TF-initiated disseminated intravascular coagulation was driven by distinct cellular sources in early versus late stages of severe inflammatory disorders. Importantly, adoptive transfer of wild-type DCs into S1P3−/− mice restored circulating TAT to levels seen in wild-type mice, identifying DCs as the origin of both systemic coagulation activation and inflammation in late stage sepsis. Furthermore, treatment with high doses of the selective thrombin-inhibitor hirudin attenuated systemic inflammation and lethality of wild-type mice, identifying thrombin as the crucial protease that activates the DC PAR1-S1P3 pathway. Thus, DCs are the origin and the target of the vicious cycle that couples coagulation to amplified inflammation in sepsis.
Mechanistically, DCs exposed to LPS upregulate PAR1 and respond to thrombin by increased migration [14]. Pronounced fibrin deposition in lymphatic ducts of draining lymph nodes demonstrated that thrombin is generated in this unexpected anatomical location in severe sepsis [13]. Consistent with reduced mobility, more PAR1−/− than wild-type DCs were found in draining lymph nodes of challenged mice. All sepsis-protected knock-out strains as well as hirudin- or PAR1 antagonist-treated wild-type mice displayed draining lymph nodes that were markedly swollen with increased cellularity and elevated local levels of the DC-derived cytokine IL1β. Thus, disrupting the DC thrombin-PAR1-S1P3 signaling resulted in retention of DCs and local sequestration of inflammation to secondary lymphoid organs. In turn, inflammation was reduced in the lungs which are directly downstream of lymphatic draining of the thoracic duct into the central circulation. The uncovering of the sepsis promoting PAR1 signaling pathway thus indicated a link between DC-dependent inflammation and the almost invariable respiratory failure in severe sepsis syndrome.
Implications of PAR1 signaling networks for therapeutic interventions in sepsis
These studies uncovered a complex, but nevertheless crucial role of PAR1 in both sepsis protective as well as sepsis promoting host response pathways. Vascular EPCR/aPC-PAR1 signaling protects the host from inflammation-related vascular leakage that can cause mortality (Fig. 3). The role of PAR1 signaling is highly dependent on the severity and the stage of the inflammatory insult. PAR1’s lethality promoting effects are prominent in very severe sepsis and are triggered by thrombin signaling specifically in the lymphatic compartment. Here, DCs are perturbed by thrombin-PAR1-S1P3 signaling and disruption of this pathway achieves retention of DCs and containment of inflammation in secondary lymphoid organs, preventing systemic dissemination of inflammation and coagulation.
Fig. 3.
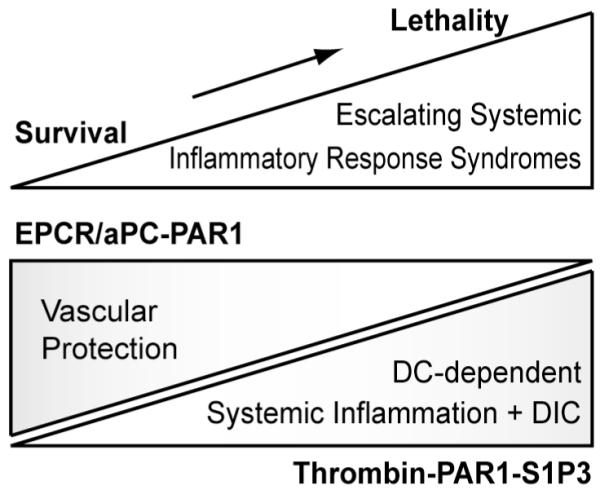
Vascular and DC PAR1 signaling in escalating inflammatory response syndromes.
Considering the importance of PAR1 in protection from vascular leakage, one may expect potential collateral damage of the therapy with a selective PAR1 antagonist. However, blocking the DC PAR1-S1P3 pathway with this approach was highly efficacious in attenuating DC-dependent systemic inflammation and lethality in sepsis [13]. The improved survival of PAR1 antagonist-treated versus PAR1−/− mice indicates compensatory effects, e.g. by PAR2 signaling. Indeed, PAR2 is essential for barrier protective effects of PAR1 activating pepducins [15]. Specific antagonists for PAR1 neither prevent proteolytic cleavage of PAR1 nor the cross-activation of PAR2 by the tethered ligand of cleaved PAR1, providing a safety margin for selective PAR1 antagonists that is not seen in PAR1−/− mice.
While extremely high doses of the thrombin inhibitor hirudin can interrupt the vicious cycle of lymphatic DC signaling in mice, concerns of bleeding risk prohibit this therapy for human sepsis. However, the signaling networks uncovered by these studies provide new potential avenues to rebalance vascular dysfunction and DC exacerbated severe inflammation. Signaling-selective aPC variants are as effective as wild-type aPC to prevent vascular leakage [11] and rescue mice from severe sepsis [12], promising potential improvements in aPC sepsis therapy by more optimal dosing while minimizing bleeding risk.
Vascular leakage of mice with impairments of the endogenous aPC pathway are also remarkably prevented by selective agonists of the S1P1 receptor, demonstrating that this presumed component of the vascular protective aPC signaling pathway can be effectively targeted to improve vascular dysfunction. Another potentially attractive therapeutic target is S1P3. Antagonists of this receptor are predicted to improve vascular dysfunction and simultaneously attenuate DC-dependent systemic inflammation and coagulopathy. Such alternative strategies with small molecule drugs are particularly attractive for chronic diseases in which coagulation signaling networks are implicated in disease progression [16,17].
Acknowledgements
We thank our collaborators for participation in our studies supported by NIH grants HL-16411 and HL-77753.
Footnotes
Conflicts of Interest: The authors have no financial conflicts of interest to declare.
References
- 1.Taylor FB., Jr. Staging of the pathophysiologic responses of the primate microvasculature to Escherichia coli and endotoxin: Examination of the elements of the compensated response and their links to the corresponding uncompensated lethal variants. Crit Care Med. 2001;29:S78–S89. doi: 10.1097/00003246-200107001-00026. [DOI] [PubMed] [Google Scholar]
- 2.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–2. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 3.Bae JS, Yang L, Manithody C, Rezaie AR. The ligand occupancy of endothelial protein C receptor switches the PAR-1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood. 2007;110:3909–16. doi: 10.1182/blood-2007-06-096651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuepbach RA, Feistritzer C, Brass LF, Riewald M. Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood. 2008;111:2667–73. doi: 10.1182/blood-2007-09-113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 6.Franscini N, Bachli EB, Blau N, Leikauf MS, Schaffner A, Schoedon G. Gene expression profiling of inflamed human endothelial cells and influence of activated protein C. Circulation. 2004;110:2903–9. doi: 10.1161/01.CIR.0000146344.49689.BB. [DOI] [PubMed] [Google Scholar]
- 7.Riewald M, Ruf W. Protease-activated receptor-1 signaling by activated protein C in cytokine-perturbed endothelial cells is distinct from thrombin signaling. J Biol Chem. 2005;280:19808–14. doi: 10.1074/jbc.M500747200. [DOI] [PubMed] [Google Scholar]
- 8.Finigan JH, Dudek SM, Singleton PA, Chiang ET, Jacobson JR, Camp SM, et al. Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem. 2005;280:17286–93. doi: 10.1074/jbc.M412427200. [DOI] [PubMed] [Google Scholar]
- 9.Feistritzer C, Riewald M. Endothelial barrier protection by activated protein C through PAR1-dependent sphingosine 1-phosphate receptor-1 crossactivation. Blood. 2005;105:3178–84. doi: 10.1182/blood-2004-10-3985. [DOI] [PubMed] [Google Scholar]
- 10.Singleton PA, Moreno-Vinasco L, Sammani S, Wanderling SL, Moss J, Garcia JG. Attenuation of Vascular Permeability by Methylnaltrexone: Role of mOP-R and S1P3 Transactivation. Am J Respir Cell Mol Biol. 2007;37:222–31. doi: 10.1165/rcmb.2006-0327OC. [DOI] [PubMed] [Google Scholar]
- 11.Niessen F, Furlan-Freguia C, Fernandez JA, Mosnier LO, Castellino FJ, Weiler H, et al. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009 doi: 10.1182/blood-2008-12-192385. in press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, et al. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–48. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, et al. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;109:654–8. doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Syrovets T, Paskas S, Laumonnier Y, Simmet T. Mature dendritic cells express functional thrombin receptors triggering chemotaxis and CCL18/pulmonary and activation-regulated chemokine induction. J Immunol. 2008;181:1215–23. doi: 10.4049/jimmunol.181.2.1215. [DOI] [PubMed] [Google Scholar]
- 15.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–12. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–81. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 17.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–58. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]