

Abstract
Objective
Peroxisome Proliferator-Activated Receptorα (PPARα) is a ligand-activated transcription factor which controls lipid metabolism and inflammation. PPARα is activated by fibrates, hypolipidemic drugs used in the treatment of dyslipidemia. Previous studies assessing the influence of PPARα agonists on atherosclerosis in mice yielded conflicting results and the implication of PPARα therein has not been assessed. The human apoE2 knock-in (apoE2-KI) mouse is a model of mixed dyslipidemia, atherosclerosis and non-alcoholic steatohepatitis (NASH). The aim of this study was, using homo- and heterozygous PPARα-deficient mice, to analyze the consequences of quantitative variations of PPARα gene levels and its response to the synthetic PPARα agonist fenofibrate, on NASH and atherosclerosis in apoE2-KI mice.
Methods and results
Wildtype (+/+), heterozygous (+/−) and homozygous (−/−) PPARα-deficient mice in the apoE2-KI background were generated and submitted to a western diet supplemented or not with fenofibrate. Western diet-fed PPARα−/− apoE2-KI mice displayed an aggravation of liver steatosis and inflammation compared to PPARα+/+ and PPARα+/− apoE2-KI mice, indicating a role of PPARα in liver protection. Moreover, PPARα expression was required for the fenofibrate-induced protection against NASH. Interestingly, fenofibrate treatment induced a similar response on hepatic lipid metabolism in PPARα+/+ and PPARα+/− apoE2-KI mice, whereas, for a maximal anti-inflammatory response, both alleles of the PPARα gene were required.
Surprisingly, atherosclerosis development was not significantly different between PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice. However, PPARα gene level determined both the anti-atherosclerotic and vascular anti-inflammatory responses to fenofibrate in a dose-dependent manner.
Conclusions
These results demonstrate a necessary, but quantitatively different role of PPARα in the modulation of liver metabolism, inflammation and atherogenesis.
Keywords: Analysis of Variance; Animals; Anti-Inflammatory Agents; pharmacology; Aorta; drug effects; metabolism; pathology; Apolipoprotein E2; genetics; metabolism; Atherosclerosis; drug therapy; genetics; metabolism; pathology; Disease Models, Animal; Fatty Liver; drug therapy; genetics; metabolism; pathology; Female; Fenofibrate; pharmacology; Gene Expression Regulation; Gene Knock-In Techniques; Heterozygote; Homozygote; Humans; Hypolipidemic Agents; pharmacology; Inflammation; drug therapy; genetics; metabolism; pathology; Lipid Metabolism; drug effects; genetics; Lipids; blood; Liver; drug effects; metabolism; pathology; Mice; Mice, Inbred C57BL; Mice, Knockout; Mice, Transgenic; PPAR alpha; agonists; genetics; metabolism
Keywords: PPARalpha, fatty liver disease, atherosclerosis, inflammation, lipid metabolism, murine model
Introduction
Fibrates are lipid-lowering drugs widely used in clinical practice to treat dyslipidemia 1. Studies performed in Peroxisome Proliferator-Activated Receptorα (PPARα)-deficient mice have demonstrated that the hypolipidemic effects of fibrates are due to activation of PPARα, a ligand-activated transcription factor which modulates lipid metabolism 2. PPARα is expressed in many tissues, particularly in tissues with high fatty acid oxidation rates such as liver, kidney, heart and muscle. After activation by fibrates, PPARα binds as a heterodimer with the Retinoid X Receptor (RXR) to PPAR response elements (PPRE) in the promoters of genes implicated in lipid and lipoprotein metabolism. In addition to its effects on lipid metabolism, PPARα also inhibits pro-inflammatory pathways by negatively interfering with other signalling pathways such as NF-kB, STATs (Signal Transducer and Activator of Transcription) or AP-1 (Activator Protein-1). Consequently, through its effects on lipid metabolism and inflammation, PPARα may modulate pathophysiological pathways implicated in fatty liver disease and atherosclerosis. Data concerning the implication of PPARα in liver steatosis and inflammation in humans is scarce. However, it has been shown that PPARα agonist treatment decreases non-alcoholic steatohepatitis (NASH) development in wild-type mice fed a methionine choline-deficient (MCD) diet 3, 4 and apoE2 knock-in (apoE2-KI) mice or foz/foz mice fed a high-fat diet 5, 6. Moreover, PPARα is expressed in many cell types found in the atherosclerotic lesion such as macrophages, endothelial cells and smooth muscle cells (SMC) 7. In vitro and in vivo studies have suggested that fibrates could exert anti-atherogenic actions by improving lipid abnormalities and/or by modulating several steps of atherogenesis such as decreasing inflammation and thrombosis directly in the vascular wall. In humans, fibrates decrease cardiovascular disease especially in patients with high triglyceride (TG) and low high density lipoprotein-cholesterol (HDL-C) levels 1. Moreover, we have previously shown that fenofibrate treatment reduces macrophage-laden atherosclerotic lesions in apoE2-KI mice, a mouse model of NASH, atherosclerosis and mixed dyslipidemia 8. However, the role of PPARα in atherosclerosis is controversial, since PPARα-deficiency protects against atherosclerosis progression in apoE-deficient mice 9 and Tsukuba hypertensive mice 10, whereas macrophage-specific PPARα expression protects Low Density Lipoprotein-Receptor (LDL-R)-deficient mice from atherosclerosis 11, and PPARα agonist treatment increases 12 or decreases 8, 13, 14 atherosclerosis development in different murine models.
In the present study, we aimed to analyze the consequences of PPARα-deficiency on lipid metabolism and inflammation in the vascular wall and the liver using apoE2-KI mice, a model of mixed dyslipidemia, atherosclerosis and NASH, and to further explore the implication of PPARα in the response to fenofibrate treatment. Therefore, wildtype (+/+), heterozygous (+/−) and homozygous PPARα-deficient (−/−) apoE2-KI mice were fed a western diet with or without fenofibrate during 9 weeks. Surprisingly, homozygous PPARα-deficiency did not modify plasma lipid concentrations, however it aggravated liver steatosis and inflammation. Interestingly, PPARα gene levels differently affected the response to fenofibrate on hepatic lipid metabolism and inflammation. In addition, whereas PPARα-deficiency did not influence atherosclerosis development, the PPARα gene level dose-dependently controlled the response to fenofibrate on vascular inflammation and atherogenesis.
Materials and Methods
Animals
Homozygous PPARα-deficient mice on the C57BL/6 background 15 were crossed with homozygous human apoE2-KI mice 16, which express human apoE2 instead of mouse apoe under control of the endogenous promoter, to generate PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice. Three month-old female mice of the three genotypes (n=11 per group) were fed a Western diet containing 0.2% cholesterol and 21% fat (wt/wt) (UAR, France) without (control group, CON) or with fenofibrate (FF) for 9 weeks. Based on food consumption, the dose of fenofibrate corresponded to ~100 mg/kg of body weight. Mice were maintained under a 12 hour light/dark cycle and had free access to water. All animal experiments were conducted with the approval of the Pasteur Institute review board, Lille, France.
Plasma lipid and lipoprotein analyses
Mice were fasted for 4 hours before retro-orbital puncture under isoflurane-induced anesthesia. Plasma concentrations of total cholesterol (TC) and triglycerides (TG) were measured using commercially available kits (Biomerieux, France).
Hepatic lipid analysis
Frozen liver tissue (50 mg) was homogenized in SET buffer (1 mL; sucrose 250 mM, EDTA 2 mM and Tris 10 mM), followed by two freeze-thaw cycles and three times passing through a 27-gauge syringe needle and a final freeze-thaw cycle to ensure complete cell lysis. Protein content was determined with the BCA method and TG and cholesterol was measured as described above.
Liver immunohistochemistry
7 μm frozen-cut liver sections were fixed in acetone and stained with Mac1 (M1/70) antibodies, as described 5.
Isolation of primary hepatocytes
Primary hepatocytes were isolated from the livers of fed mice, as previously described 17.
RNA extraction and quantitative PCR analysis
RNA, isolated from livers using the acid guanidinium thiocyanate/phenol/chloroform method 18, was reverse transcribed using random hexamer primers and Moloney murine leukemia virus-reverse transcriptase (Invitrogen, France). RNA levels were determined by real-time quantitative PCR on a MX-4000 apparatus (Stratagene) using the Brilliant SYBR Green QPCR master mix (Stratagene) and specific primers. Results are expressed normalized to cyclophilin.
Atherosclerotic lesion analysis
At the end of the diet, mice were euthanized, the hearts were perfused with cold Krebs-Ringer buffer and fixed in a solution containing 4 % phosphate-buffered paraformaldehyde. Serial 10 μm-thick cryosections were cut between the valves and the aortic arch and atherosclerotic lesions were quantified by Oil-Red-O staining. Images were captured using a JVC 3-CCD video camera and analyzed using the computer-assisted Quips Image analysis system (Leica Mikroskopic und System GmbH, Germany). Cryosections from aortic lesions were stained with anti-mouse MOMA-2 (Santa Cruz Biotechnology) or anti-mouse MCP-1 (Santa Cruz Biotechnology). MCP-1 protein levels were semi-quantitatively scored for staining on lesions of 4 mice per group.
Statistical analysis
Results are expressed as the means ± SE. Data were compared by ANOVA. Significant differences were subjected to post-hoc analysis by using the Scheffe-test. A value of p<0.05 was considered as statistically significant.
Results
Homozygous PPARα-deficiency does not modify plasma lipid concentrations, but aggravates liver steatosis and inflammation in apoE2-KI mice fed a western diet
Female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice were generated and PPARα mRNA levels were found to be respectively intermediate and undetectable in the livers of PPARα+/− and PPARα−/− vs PPARα+/+ apoE2-KI mice (supplemental figure 1A). These mice were fed a western diet which was previously shown to induce liver steatosis and inflammation 5. After 9 weeks of western diet feeding, plasma TG and TC concentrations were similar in the three groups of mice (figure 1, A–B). Relative liver weight was not different between the three genotypes (supplemental figure 1B) and liver cholesterol content was slightly, but not significantly increased in PPARα−/− apoE2-KI compared to PPARα+/+ and PPARα+/− apoE2-KI mice (supplemental figure 1C). PPARα−/− apoE2-KI mice displayed more severe steatohepatitis, as illustrated by higher levels of liver TG (figure 1C) and increased numbers of Mac1-positive cells (figure 1D), compared to PPARα+/+ mice. Interestingly, heterozygous PPARα+/− apoE2-KI mice displayed a similar phenotype as PPARα+/+ mice (figure 1, C–D). These results demonstrate that PPARα-deficiency does not modify plasma lipid concentrations, and only homozygous PPARα-deficiency aggravates liver steatosis and macrophage content in western diet-fed apoE2-KI mice.
Figure 1. Homozygous PPARα-deficiency does not modify plasma lipid concentrations, but aggravates liver steatosis and inflammation in apoE2-KI mice.
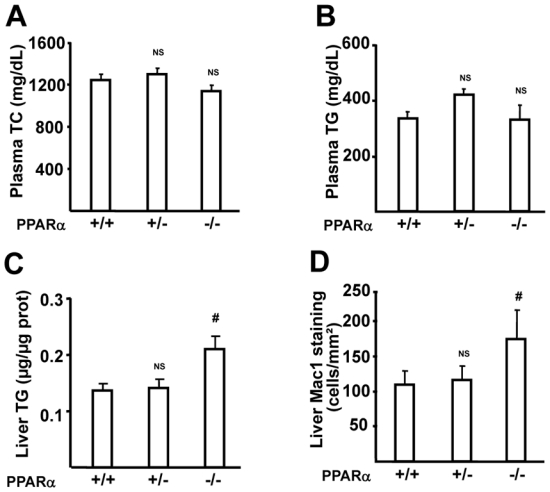
Blood samples were collected after a 4-hour fast for measurements of plasma TC (A) and TG (B) levels in female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet for 9 weeks. Liver TG content (C) and MAC-1 staining (D) were quantified in the liver. n=11 mice/group. Results are expressed as means α SE. NS=Non Significant. #p<0.05 versus PPARα+/+ apoE2-KI mice.
PPARα activation improves plasma and hepatic lipid homeostasis in PPARα+/+ and PPARα+/−, but not PPARα−/− apoE2-KI mice fed a western diet
To determine the role of the PPARα gene level on the hepatic and plasma response to its agonist fenofibrate, female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice were fed a western diet supplemented with fenofibrate for 9 weeks. The response to the PPARα agonist was compared to the respective placebo-treated PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice whose values were set at 100%. As expected 8, fenofibrate treatment decreased plasma TC and TG concentrations (figure 2, A–B) and liver TG content (figure 2C) in PPARα+/+ apoE2-KI mice. In marked contrast, fenofibrate-treated PPARα−/− apoE2-KI mice did not exhibit any significant decrease in plasma TC and TG concentrations and liver TG levels compared to placebo-treated mice, showing that fenofibrate improves dyslipidemia and hepatic steatosis in a PPARα–dependent manner in apoE2-KI mice (figure 2, A–C). Interestingly, fenofibrate reduced plasma TC and TG concentrations and liver TG levels to the same extent in PPARα+/− apoE2-KI as in PPARα+/+ apoE2-KI mice. Since PPARα is a transcription factor which regulates the expression of genes involved in fatty acid uptake and oxidation in parenchymal cells of the liver, mRNA levels for fatty acid translocase (FAT), very long chain acyl-CoA dehydrogenase (VLCAD), long chain acyl-CoA dehydrogenase (LCAD) and medium chain acyl-CoA dehydrogenase (MCAD) were measured (figure 3, A–D). Fenofibrate increased the hepatic expression levels of all these genes in PPARα+/+ but not in PPARα−/− apoE2-KI mice. However, in PPARα+/− apoE2-KI mice, the expression of these genes increased to a similar extent as in PPARα+/+ apoE2-KI mice upon fenofibrate treatment. A comparable induction of genes implicated in hepatic lipid metabolism was also observed in primary hepatocytes isolated from PPARα+/+ and PPARα+/− apoE2-KI mice and treated with the specific PPARα agonist, GW647, whereas this induction was not observed in PPARα−/− apoE2-KI hepatocytes (supplemental figure 2, A–B). Thus, only one allele of the PPARα gene is required for an optimal response to fenofibrate on dyslipidemia and liver steatosis.
Figure 2. Fenofibrate improves plasma and hepatic lipid homeostasis in PPARα+/+ and PPARα+/−, but not PPARα−/− apoE2-KI mice.
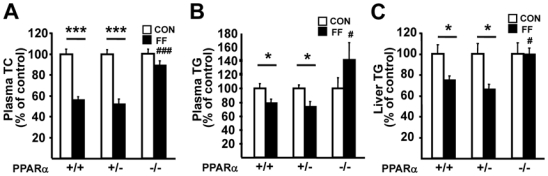
Plasma TC (A) and TG (B) levels as well as liver TG content (C) in female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet supplemented (FF, ■) or not (CON, □) with fenofibrate for 9 weeks. n=11 mice/group. Results are expressed as means α SE.*p<0.05, ***p<0.001 vs untreated mice; #p<0.05, ###p<0.001 versus fenofibrate-treated PPARα+/+ apoE2-KI mice.
Figure 3. Fenofibrate increases the expression of fatty acid uptake and oxidation genes in PPARα+/+ and PPARα+/−, but not PPARα−/− apoE2-KI mice.
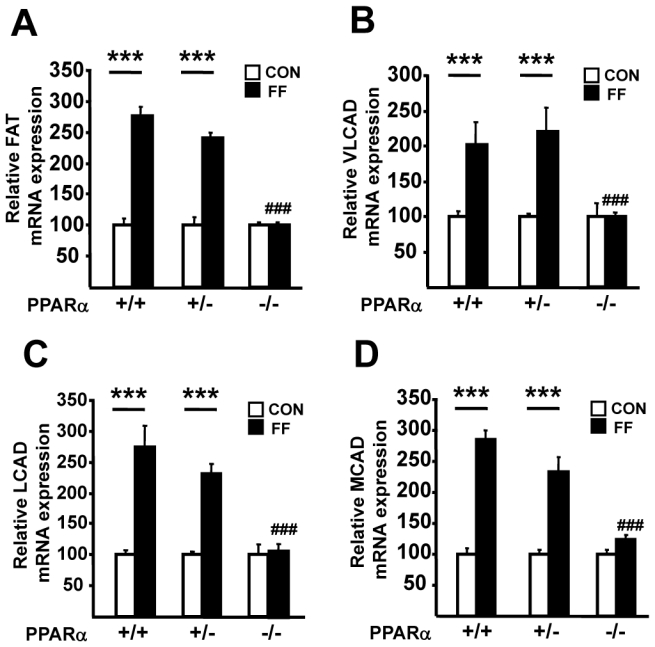
Hepatic mRNA levels of FAT (A), VLCAD (B), LCAD (C), MCAD (D) in female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet supplemented (FF, ■) or not (CON, □) with fenofibrate for 9 weeks. n=11 mice/group. Results are expressed as means ± SE. ***p<0.001 vs untreated mice; ###p<0.001 versus fenofibrate-treated PPARα+/+ apoE2-KI mice.
PPARα activation decreases hepatic inflammation and macrophage content in PPARα+/+, but not PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet
Hepatic inflammation was analysed in the fenofibrate-treated mice. Fenofibrate treatment decreased the number of Mac-1 positive cells, indicative of the number of macrophages, in livers of PPARα+/+ but not PPARα+/− or PPARα−/− apoE2-KI mice (figure 4, A–B). The expression of monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which are implicated in monocyte/macrophage recruitment in the liver (figure 4, C–E), were decreased in fenofibrate-treated PPARα+/+ apoE2-KI mice. By contrast, fenofibrate did not influence the expression of these inflammatory markers in livers of PPARα−/− apoE2-KI mice. Interestingly, fenofibrate-treated PPARα+/− apoE2-KI mice exhibited an intermediary gene expression level of MCP-1, VCAM-1 and ICAM-1. Of note, repression of LPS-induced MCP-1 and VCAM-1 expression by the specific PPARα agonist GW647 was most pronounced in isolated primary hepatocytes from PPARα+/+ apoE2-KI mice, whereas an intermediary response was seen in PPARα+/− apoE2-KI cells (supplemental figure 2, C–D). Thus, both PPARα alleles are necessary for an optimal inhibition of the hepatic inflammatory response by fenofibrate in apoE2-KI mice fed a western diet.
Figure 4. Fenofibrate decreases hepatic inflammation and macrophage content in PPARα+/+, but not PPARα+/− and PPARα−/− apoE2-KI mice.
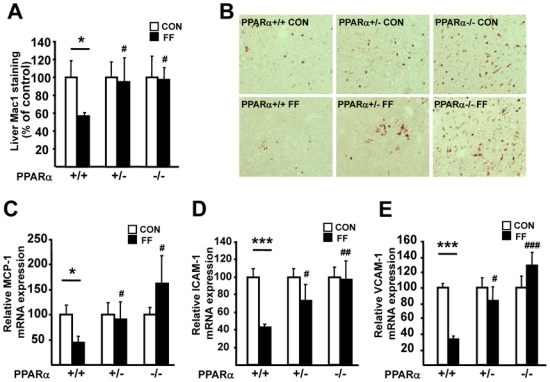
Liver MAC-1 staining (A) with representative microphotographs (B) and hepatic mRNA levels of MCP-1(C), ICAM-1(D) and VCAM-1 (E) in female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet supplemented (FF, ■) or not (CON, □) with fenofibrate for 9 weeks. n=11 mice/group. Results are expressed as means ± SE.*p<0.05, ***p<0.001 vs untreated mice; #p<0.05, ##p<0.01, ###p<0.001 versus fenofibrate-treated PPARα+/+ apoE2-KI mice.
PPARα gene levels do not influence atherogenesis, but determine the atheroprotective response to fenofibrate in apoE2-KI mice fed a western diet
Since non-alcoholic fatty liver disease is now considered a risk factor for cardiovascular disease 19, atherogenesis and vascular inflammation were assessed in PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet supplemented or not with fenofibrate. Interestingly, the mean aortic lesion area, as measured by lipid staining with oil-red-O, did not differ significantly between PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice (figure 5, A–B). As expected 8, mean lesion area was significantly reduced by about 80 % in fenofibrate-treated PPARα+/+ mice compared to controls (median: 0.030 mm2 in treated mice vs 0.166 mm2 in control mice, p<0.001) (figure 5, A–B). By contrast, no effect was observed in fenofibrate-treated PPARα−/− apoE2-KI mice (median: 0.137 mm2 in treated mice vs 0.123 mm2 in control mice, NS). Interestingly, treatment of PPARα+/− apoE2-KI mice with fenofibrate resulted in a significant, intermediary decrease in atherosclerotic lesion area (median: 0.138 mm2 in treated mice vs 0.185 in control mice, p<0.05), indicating a dose-response effect of PPARα gene expression on arterial wall lipid accumulation. To determine whether the modifications in atherosclerotic lipid accumulation were associated with altered inflammation in the arterial wall, immunostaining for MOMA-2 (specific of macrophages) and MCP-1 was performed in the lesions. Both MOMA-2 (figure 5C) and MCP-1(figure 5, D–E) co-localized with oil-red-O staining, were intense and did not differ between PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice, in accordance with the comparable lesion areas between the three genotypes (figure 5A). Treatment with fenofibrate strongly decreased MOMA-2 and MCP-1 staining in the lesions of PPARα+/+ but not PPARα−/− apoE2-KI mice, indicating PPARα-dependency. Interestingly, fenofibrate treatment of PPARα+/− apoE2-KI mice resulted in an intermediary phenotype with a slight decrease of MOMA-2 and MCP-1 staining in the lesions. Thus, while PPARα gene levels do not influence atherosclerosis development, they determine the response to fenofibrate on both lipid deposition and inflammation in the arterial wall of apoE2-KI mice.
Figure 5. PPARα gene levels do not influence atherogenesis, but determines the atheroprotective response to fenofibrate in apoE2-KI mice.
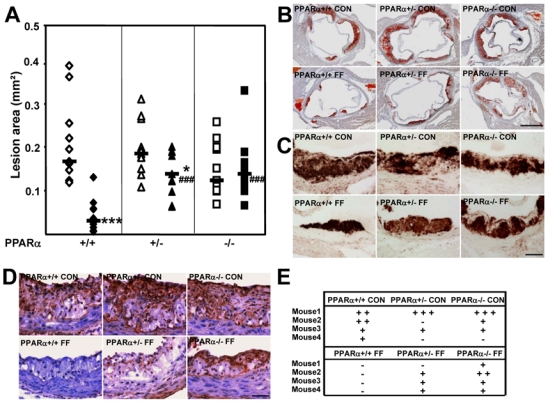
Atherosclerotic lesion area (A) in the aorta of female PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice fed a western diet supplemented (filled symbols) or not (open symbols) with fenofibrate for 9 weeks. The graph represents the mean area of lesions of the analysed sections and each symbol represents one mouse. The horizontal bar corresponds to the mediane of the values. n=11 mice/group. *p<0.05, ***p<0.001 versus untreated mice; ###p<0.001 versus fenofibrate-treated PPARα+/+ apoE2-KI mice. Representative microphotographs showing Oil Red-O staining (B) or MOMA-2 stained macrophages (C) in the atherosclerotic lesions. Representative photomicrographs showing MCP-1 staining of atherosclerotic lesions (D) and score of MCP-1 staining (E) graded as: (−) not detected, (+) slight, (++) marked, (+++) pronounced expression in lesions. n=4 mice/group. (B, scale bar = 500 μm; C–D, scale bar = 100 μm).
Discussion
Using apoE2-KI mice, a humanized mouse model of mixed dyslipidemia and NASH 16, we show that homozygous PPARα-deficiency aggravates western diet-induced steatosis and inflammation in the liver. This result is consistent with the reported increased hepatic steatosis observed in PPARα-deficient mice in response to the physiological stimulus fasting 20–22, further illustrating the role of PPARα in hepatic lipid metabolism. In apoE2-KI mice, aggravation of NASH induced by PPARα-deficiency was not accompanied by changes of plasma TC and TG concentrations nor atherosclerosis lesion development, suggesting a dissociation between both pathological states. We also analyzed the effects of PPARα gene level on the response to treatment with the PPARα agonist fenofibrate on NASH. Fenofibrate treatment protected against NASH in western diet-fed PPARα+/+ but not PPARα−/− apoE2-KI mice, showing that the previously reported effects of fenofibrate on NASH in apoE2-KI mice 5 occur via PPARα. Interestingly, fenofibrate treatment of PPARα+/− apoE2-KI mice resulted in decreased hepatic steatosis to an extent similar as PPARα+/+ apoE2-KI mice. In parallel, the hepatic expression of genes implicated in fatty acid uptake and oxidation increased to a similar extent in fenofibrate-treated PPARα+/− and PPARα+/+ apoE2-KI mice. However, in contrast with the regulation of lipid metabolism, the anti-inflammatory response to fenofibrate in the liver depends on both PPARα alleles. Indeed, fenofibrate treatment strongly reduced inflammation and macrophage content in livers of PPARα+/+ apoE2-KI mice (associated with decreased expression levels of MCP-1, ICAM-1, VCAM-1), whereas little or no response to fenofibrate was observed in livers of PPARα+/− apoE2-KI mice. Similar observations were made in isolated heterozygous primary hepatocytes treated with the specific PPARα agonist GW647. Thus, PPARα gene level differently influences fenofibrate’s effects on hepatic steatosis and inflammation. A single PPARα allele is sufficient for an optimal response of lipid metabolism to fenofibrate, whereas both alleles are required to obtain maximal anti-inflammatory effects in the liver.
Until now, no null mutations of PPARα have been identified in humans, but the hepatic PPARα expression levels vary largely among individuals 23 and several PPARα mutations have been reported 24, 25. Presently, the implication of PPARα in fatty liver disease and a possible modulation by PPARα agonists is still unclear. Magnetic Resonance Imaging (MRI) analysis of liver fat in fifteen type 2 diabetic patients has failed to show any changes in response to fenofibrate 26. A pilot study in NAFLD patients has shown that treatment with fenofibrate improves metabolic syndrome parameters, including the lipid profile, and has beneficial effects on certain liver function parameters, but its impact on liver histology was small 27. Another study has suggested a beneficial effect of fenofibrate treatment, particularly in combination with statins, in reducing fatty liver disease 28. Further studies are needed to evaluate the impact of PPARα agonists on NAFLD, and in particular on the inflammatory component of NASH.
We show that PPARα is required for the fenofibrate-induced improvement of dyslipidemia in apoE2-KI mice and a single PPARα allele is sufficient to mediate this effect, showing that PPARα gene level does not determine the plasma lipid-response of fenofibrate. In the Lower Extremity Arterial Disease Event Reduction (LEADER) trial, PPARα gene variation did not influence the magnitude of plasma TG lowering in response to bezafibrate, whereas genetic variation in the PPARα gene affected the changes in plasma fibrinogen, an inflammatory response marker, to fibrate treatment 29. Together with our data, variation in PPARα gene activity or expression level appears of lesser impact on the lipid response to fibrate, but of higher impact on the inflammatory response.
Since growing evidence links fatty liver disease to cardiovascular disease 19 and since apoE2-KI mice develop dyslipidemia and NASH as well as atherosclerosis, we also assessed the role of PPARα on vascular inflammation and atherosclerosis. Interestingly, despite the more severe NASH progression, PPARα+/+, PPARα+/− and PPARα−/− apoE2-KI mice developed quantitatively similar atherosclerotic lesion areas (as assessed by lipid, macrophage and MCP-1 content of the atherosclerotic plaques), indicating that PPARα does not modulate atherogenesis and vascular inflammation in apoE2-KI mice. This result is surprising since a study performed in LDLR-deficient mice demonstrated that macrophage PPARα confers anti-atherogenic effects via modulation of macrophage cholesterol trafficking and inflammatory activity 11. Similar as in apoE2-KI mice, plasma lipid concentrations were not modified by PPARα-deficiency in these LDLR-deficient mice, indicating that differences in plasma lipids do not explain the observed discrepancies of lesion formation between both models. The lesions in apoE2-KI mice mainly consist of foam cells, as occurs in the initial stages of atherogenesis in humans, whereas LDLR-deficient mice develop more advanced atherosclerotic plaques 30. Thus, the PPARα gene may not modulate the first stages of lesion formation, but could influence the progression of atherosclerosis to more complex stages. Surprisingly, PPARα-deficiency in apoE-deficient mice, another mouse model of atherosclerosis characterized by a wide spectrum of lesions going from fatty streaks to fibro-proliferative lesions 31, resulted in a protection against atherosclerotic lesion formation upon western diet feeding, notwithstanding a pro-atherogenic lipid profile characterized by higher levels of TG and TC 9. However, in contrast to apoE2-KI mice, apoE-deficient mice do not respond to PPARα agonists as humans, displaying no change 13 or an increase 12 in plasma lipids upon fibrate treatment, and may therefore not be a suitable model to study the impact of PPARα agonists and possibly other lipid-lowering drugs on atherosclerosis 30, 32.
PPARα activation with fenofibrate protected western diet-fed PPARα+/+ but not PPARα−/− apoE2-KI mice against atherosclerosis progression, showing PPARα-dependency of the response to fenofibrate. Treatment of PPARα+/− apoE2-KI mice with fenofibrate resulted in an intermediary level of atheroprotection and vascular anti-inflammatory response (assessed by MOMA-2 and MCP-1 staining). Thus, the PPARα gene level dose-dependently controls the response to its agonist on vascular inflammation and atherogenesis, despite a similar plasma lipid response in PPARα+/+ and PPARα+/− apoE2-KI mice. These observations suggest that atheroprotection upon PPARα activation, in addition to being determined by the plasma lipid concentrations, also depends on other parameters, such as inflammation in the artery wall and the liver.
The pathophysiological role of PPARα in atherosclerosis has been investigated using two different and complementary strategies: genetic deficiency and a pharmacological intervention. PPARα-deficiency did not result in the opposite phenotype of fenofibrate-induced activation of PPARα on atherosclerotic lesion areas. It is well established that nuclear-receptor deficiency does not always mirror ligand-activation, which may be due to possible compensation mechanisms and/or the absence of active repression of target genes by the unliganded nuclear receptor via co-repressor recruitment. However, the effects of fenofibrate did require PPARα expression, since it was absent in PPARα−/− apoE2-KI mice.
In the Lopid Coronary Angiography Trial (LOCAT) study, the PPARα V162 allele was associated with reduced progression of atherosclerosis, whereas the intron C allele was associated with greater progression of atherosclerosis 29. In vitro, it has been shown that the V162 variant displays higher PPRE-dependent transcriptional activity 24, 33, whereas the intron 7 C allele was hypothesized to be associated with lower expression levels of PPARα 29. Collectively, these results suggest that variations in PPARα gene level or activity are associated with differential progression of atherosclerosis in humans. Both PPARα variants did not influence plasma lipid concentrations in either study, suggesting that, in line with our results, PPARα gene variation may influence atherosclerosis via mechanisms complementary to the PPARα regulation of plasma lipid concentrations. Finally, since PPARα agonists may selectively modulate the different activities of PPARα (SPPARM effect) 34, the effects of PPARα variants on the response to its agonists could be different according to the PPARα agonist used. Thus, it will be of interest to study the impact of PPARα variants on the responses to different agonists on fatty liver disease and to assess the association of modifications of lipid metabolism and inflammatory parameters with PPARα polymorphisms.
Supplementary Material
Acknowledgments
Sources of funding
This work was supported by grants of ACI 02 20475 (French Research Ministery and Servier laboratory), Veni: 916.76.070 (2006/00496/MW); Maag Lever Darm Stichting (MLDS) (WO 08-16), the foundation Coeur et Artères, the EU grant Hepadip 018734, Région Nord-Pas de Calais/FEDER, and a Kootstra fellowship from Maastricht University.
Footnotes
Disclosure
None
Bibliography
- 1.Staels B, Maes M, Zambon A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2008;5:542–553. doi: 10.1038/ncpcardio1278. [DOI] [PubMed] [Google Scholar]
- 2.Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FJ, Auwerx J. Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J Biol Chem. 1997;272:27307–27312. doi: 10.1074/jbc.272.43.27307. [DOI] [PubMed] [Google Scholar]
- 3.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology. 2003;38:123–132. doi: 10.1053/jhep.2003.50307. [DOI] [PubMed] [Google Scholar]
- 4.Ip E, Farrell G, Hall P, Robertson G, Leclercq I. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 5.Shiri-Sverdlov R, Wouters K, van Gorp PJ, Gijbels MJ, Noel B, Buffat L, Staels B, Maeda N, van Bilsen M, Hofker MH. Early diet-induced non-alcoholic steatohepatitis in APOE2 knock-in mice and its prevention by fibrates. J Hepatol. 2006;44:732–741. doi: 10.1016/j.jhep.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 6.Teoh NC, Williams J, Hartley J, Yu J, McCuskey RS, Farrell GC. Short-term therapy with peroxisome proliferation-activator receptor-alpha agonist Wy-14,643 protects murine fatty liver against ischemia-reperfusion injury. Hepatology. 2010;51:996–1006. doi: 10.1002/hep.23420. [DOI] [PubMed] [Google Scholar]
- 7.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. 2006;116:571–580. doi: 10.1172/JCI27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hennuyer N, Tailleux A, Torpier G, Mezdour H, Fruchart JC, Staels B, Fievet C. PPARalpha, but not PPARgamma, activators decrease macrophage-laden atherosclerotic lesions in a nondiabetic mouse model of mixed dyslipidemia. Arterioscler Thromb Vasc Biol. 2005;25:1897–1902. doi: 10.1161/01.ATV.0000175756.56818.ee. [DOI] [PubMed] [Google Scholar]
- 9.Tordjman K, Bernal-Mizrachi C, Zemany L, Weng S, Feng C, Zhang F, Leone TC, Coleman T, Kelly DP, Semenkovich CF. PPARalpha deficiency reduces insulin resistance and atherosclerosis in apoE-null mice. J Clin Invest. 2001;107:1025–1034. doi: 10.1172/JCI11497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tordjman KM, Semenkovich CF, Coleman T, Yudovich R, Bak S, Osher E, Vechoropoulos M, Stern N. Absence of peroxisome proliferator-activated receptor-alpha abolishes hypertension and attenuates atherosclerosis in the Tsukuba hypertensive mouse. Hypertension. 2007;50:945–951. doi: 10.1161/HYPERTENSIONAHA.107.094268. [DOI] [PubMed] [Google Scholar]
- 11.Babaev VR, Ishiguro H, Ding L, Yancey PG, Dove DE, Kovacs WJ, Semenkovich CF, Fazio S, Linton MF. Macrophage expression of peroxisome proliferator-activated receptor-alpha reduces atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. 2007;116:1404–1412. doi: 10.1161/CIRCULATIONAHA.106.684704. [DOI] [PubMed] [Google Scholar]
- 12.Fu T, Kashireddy P, Borensztajn J. The peroxisome-proliferator-activated receptor alpha agonist ciprofibrate severely aggravates hypercholesterolaemia and accelerates the development of atherosclerosis in mice lacking apolipoprotein E. Biochem J. 2003;373:941–947. doi: 10.1042/BJ20030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duez H, Chao YS, Hernandez M, Torpier G, Poulain P, Mundt S, Mallat Z, Teissier E, Burton CA, Tedgui A, Fruchart JC, Fievet C, Wright SD, Staels B. Reduction of atherosclerosis by the peroxisome proliferator-activated receptor alpha agonist fenofibrate in mice. J Biol Chem. 2002;277:48051–48057. doi: 10.1074/jbc.M206966200. [DOI] [PubMed] [Google Scholar]
- 14.Srivastava RA, Jahagirdar R, Azhar S, Sharma S, Bisgaier CL. Peroxisome proliferator-activated receptor-alpha selective ligand reduces adiposity, improves insulin sensitivity and inhibits atherosclerosis in LDL receptor-deficient mice. Mol Cell Biochem. 2006;285:35–50. doi: 10.1007/s11010-005-9053-y. [DOI] [PubMed] [Google Scholar]
- 15.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan PM, Mezdour H, Quarfordt SH, Maeda N. Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J Clin Invest. 1998;102:130–135. doi: 10.1172/JCI2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mansouri RM, Bauge E, Staels B, Gervois P. Systemic and distal repercussions of liver-specific peroxisome proliferator-activated receptor-alpha control of the acute-phase response. Endocrinology. 2008;149:3215–3223. doi: 10.1210/en.2007-1339. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Targher G, Day CP, Bonora E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl J Med. 2010;363:1341–1350. doi: 10.1056/NEJMra0912063. [DOI] [PubMed] [Google Scholar]
- 20.Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci U S A. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hashimoto T, Cook WS, Qi C, Yeldandi AV, Reddy JK, Rao MS. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J Biol Chem. 2000;275:28918–28928. doi: 10.1074/jbc.M910350199. [DOI] [PubMed] [Google Scholar]
- 23.Gervois P, PinedaTorra I, Chinetti G, Grotzinger T, Dubois G, Fruchart JC, Fruchart-Najib J, Leitersdorf E, Staels B. A truncated human peroxisome proliferator-activated receptor alpha splice variant with dominant negative activity. Mol Endocrinol. 1999;13:1535–1549. doi: 10.1210/mend.13.9.0341. [DOI] [PubMed] [Google Scholar]
- 24.Flavell DM, Pineda Torra I, Jamshidi Y, Evans D, Diamond JR, Elkeles RS, Bujac SR, Miller G, Talmud PJ, Staels B, Humphries SE. Variation in the PPARalpha gene is associated with altered function in vitro and plasma lipid concentrations in Type II diabetic subjects. Diabetologia. 2000;43:673–680. doi: 10.1007/s001250051357. [DOI] [PubMed] [Google Scholar]
- 25.Lacquemant C, Lepretre F, Pineda Torra I, Manraj M, Charpentier G, Ruiz J, Staels B, Froguel P. Mutation screening of the PPARalpha gene in type 2 diabetes associated with coronary heart disease. Diabetes Metab. 2000;26:393–401. [PubMed] [Google Scholar]
- 26.Bajaj M, Suraamornkul S, Hardies LJ, Glass L, Musi N, DeFronzo RA. Effects of peroxisome proliferator-activated receptor (PPAR)-alpha and PPAR-gamma agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia. 2007;50:1723–1731. doi: 10.1007/s00125-007-0698-9. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Miranda C, Perez-Carreras M, Colina F, Lopez-Alonso G, Vargas C, Solis-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–205. doi: 10.1016/j.dld.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 28.Athyros VG, Mikhailidis DP, Didangelos TP, Giouleme OI, Liberopoulos EN, Karagiannis A, Kakafika AI, Tziomalos K, Burroughs AK, Elisaf MS. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin. 2006;22:873–883. doi: 10.1185/030079906X104696. [DOI] [PubMed] [Google Scholar]
- 29.Pineda Torra I, Jamshidi Y, Flavell DM, Fruchart JC, Staels B. Characterization of the human PPARalpha promoter: identification of a functional nuclear receptor response element. Mol Endocrinol. 2002;16:1013–1028. doi: 10.1210/mend.16.5.0833. [DOI] [PubMed] [Google Scholar]
- 30.Wouters K, Shiri-Sverdlov R, van Gorp PJ, van Bilsen M, Hofker MH. Understanding hyperlipidemia and atherosclerosis: lessons from genetically modified apoe and ldlr mice. Clin Chem Lab Med. 2005;43:470–479. doi: 10.1515/CCLM.2005.085. [DOI] [PubMed] [Google Scholar]
- 31.Zhang SH, Reddick RL, Burkey B, Maeda N. Diet-induced atherosclerosis in mice heterozygous and homozygous for apolipoprotein E gene disruption. J Clin Invest. 1994;94:937–945. doi: 10.1172/JCI117460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tailleux A, Torpier G, Mezdour H, Fruchart JC, Staels B, Fievet C. Murine models to investigate pharmacological compounds acting as ligands of PPARs in dyslipidemia and atherosclerosis. Trends Pharmacol Sci. 2003;24:530–534. doi: 10.1016/j.tips.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Sapone A, Peters JM, Sakai S, Tomita S, Papiha SS, Dai R, Friedman FK, Gonzalez FJ. The human peroxisome proliferator-activated receptor alpha gene: identification and functional characterization of two natural allelic variants. Pharmacogenetics. 2000;10:321–333. doi: 10.1097/00008571-200006000-00006. [DOI] [PubMed] [Google Scholar]
- 34.Duez H, Lefebvre B, Poulain P, Torra IP, Percevault F, Luc G, Peters JM, Gonzalez FJ, Gineste R, Helleboid S, Dzavik V, Fruchart JC, Fievet C, Lefebvre P, Staels B. Regulation of human apoA-I by gemfibrozil and fenofibrate through selective peroxisome proliferator-activated receptor alpha modulation. Arterioscler Thromb Vasc Biol. 2005;25:585–591. doi: 10.1161/01.ATV.0000154140.73570.00. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.