

Abstract
Mouse phenotype data represents a valuable resource for the identification of disease-associated genes, especially where the molecular basis is unknown and there is no clue to the candidate gene’s function, pathway involvement or expression pattern. However, until recently these data have not been systematically used due to difficulties in mapping between clinical features observed in humans and mouse phenotype annotations. Here, we describe a semantic approach to solve this problem and demonstrate highly significant recall of known disease-gene associations and orthology relationships. A web application (MouseFinder; www.mousemodels.org) has been developed to allow users to search the results of our whole-phenome comparison of human and mouse. We demonstrate its use in identifying ARTN as a strong candidate gene within the 1p34.1-p32 mapped locus for a hereditary form of ptosis.
Keywords: phenotype, candidate disease genes, model organism, mouse
Introduction
The primary reason for generating and studying animal models, is the ability to gather insights to our understanding of human disease and gene function. The mouse, with its fully sequenced genome, almost complete genetic orthology with the human, and numerous genetic manipulation tools has become the principal model organism for such purposes (Rosenthal and Brown, 2007).
A wealth of readily accessible mouse phenotype data already exists thanks to the researchers who have generated and characterised mouse mutants, and the manual curation of the literature and submitted data carried out by the Mouse Genome Informatics (MGI) group of the Jackson Laboratory and stored in their Mouse Genome Database (MGD; Blake et al., 2011). At the time of writing, MGD contains 26945 phenotyped mutant alleles representing 11356 markers, including heritable phenotypic markers, deletions, inversions and other complex genomic mutations, in addition to mutations in 8124 protein coding and RNA genes. Hence, phenotype data is already available for a large proportion of mouse genes although, for many, the published phenotypes focus on a specific area of research rather a broad phenotypic characterisation of the mutant.. Throughout this decade, phenotype data will be made available for all mouse protein-associated genes due to the efforts of the International Mouse Phenotyping Consortium (IMPC; http://www.mousephenotype.org; Abbott, 2010). The IMPC will implement a high-throughput phenotyping pipeline to characterise strains carrying the null mutations produced by the systematic efforts of the International Knockout Mouse Consortium (IKMC; Ringwald et al., 2011; Skarnes et al., 2011). The results of this will be available from the IMPC portal, as well as the Mammalian Phenotype Ontology (MPO; Smith and Eppig, 2009) annotated data being deposited at MGI as part of a merged dataset with the mouse phenotype data from publications and submissions. The approach taken by the IMPC or performing the same wide spectrum of phenotype assays on every line will hopefully provide more comprehensive coverage of the mouse phenome than the current approach of curating literature reported observations.
Until recently, systematic use of mouse phenotype data by the human clinical and research communities to identify candidate disease genes and gain knowledge of protein function has been a rarity, despite the potential power of such an approach. For example, Kitsios et al., 2010 identified mouse models for human genome-wide associations, which provide concordance of evidence and novel insights into the roles of the candidate genes. Another study identified candidates for autism spectrum disorders using mouse phenotype data, some of which overlapped with the results from a global copy number variation study (Meehan et al., 2011).
Where associated or candidate genes already exist for a human disorder, it is trivial to recover the mouse ortholog and any associated phenotype data. Similarly, if some knowledge of function or pathway involvement is known for the disease then searches for genes with these functions or pathway involvement can reveal candidates.
However, for many disorders only the observed phenotype is known and here the ability to identify equivalent phenotypes in model organisms with a known genotype becomes critical. The main impediment to this is the lack of direct mappings between the terms used for human disease and mouse phenotypes (Schofield et al., 2010). MGI address this issue by manually curating disease associations for published mouse models. However, this is a huge effort and is likely to be unscalable for the IMPC project. In addition, most publications involving mouse models are focussed on a particular disease and do not address whether a mouse could be a good or even better model for another disease. The systematic phenotype analysis performed by Meehan et al. identified several models in the MGD databases that had not previously been associated by the manual curation effort. Clearly, there is a requirement for computational methods for associating mouse models with human diseases, and systematic analysis of both human and mouse datasets.
The first step to automation is capturing the phenotype data in a computable form using ontologies and controlled vocabularies. The mouse community is already in a good position, as MGD and many other mouse databases use the well-established MPO. Although termed “Mammalian”, MPO has primarily been used to capture mouse phenotypic data at MGD and rat phenotypic data at the Rat Genome Database (RGD; Twigger et al., 2007). Other model organism databases, such as ZFIN (Sprague et al., 2008) for zebrafish and FlyBase (Tweedie et al., 2009) for Drosophila, do not use a “pre-composed” species-specific phenotype ontology but rather use a “post-composed” Entity-Quality (EQ) approach. In this, the Q variable comes from the Phenotype and Trait Ontology (PATO) and the E variable from one of the Open Biomedical Ontologies (OBO) such as Gene Ontology (GO), ZFA (zebrafish anatomy) or FBbt (Flybase anatomy ontology). For example, motor neuron degeneration is represented in the mouse by MP:0000938 (motor neuron degeneration) and in zebrafish by the combination of ZFA:0009052 (motor neuron) and PATO:0000639 (degenerate). The latter approach is termed a post-composed approach, as the terms are joined post curation to form a human readable text description (Washington et al., 2009; Gkoutos et al., 2009).
Use of ontologies to capture human phenotype data is a more recent activity, stemming from the development of the Human Phenotype Ontology (HPO; Robinson et al., 2008). Like MPO, HPO uses a pre-composed approach; e.g., HP:0007373 (atrophy/degeneration involving motor neurons) would be used for the motor neuron degeneration example above.
The mixed use of pre- and post-composed approaches and different ontologies would appear to hinder any cross-species phenotype querying. Lexical (text matching) based approaches can be used as demonstrated in PhenomicDB (Groth et al., 2006) and PhenoHM (Sardana et al., 2010) but will require non-trivial solutions where the same concept is described with different words (synonyms) or where the same word can refer to different concepts (homonyms) in the human and model organism communities. For instance, to a human reader MP:0000573 (enlarged hind paws) and HP:0001833 (large feet) clearly represent largely equivalent biological concepts, but to a computer using a purely lexical approach this association would be lost. In addition, the full semantic power of the ontologies is lost using a lexical approach. For example, the phenotypic consequence of the same genetic abnormality may be related but subtly different in diverse species; e.g., PAX6 mutations result in “small eyed” mice, “opaque cornea” in humans, a “malformed retina” in zebrafish, and “eyeless” Drosophila. A lexical, computational approach could not identify these related phenotypes but a semantic approach, using the structure and relationships of the phenotype ontologies and logical definitions, will identify that all involve “eye abnormalities”. Similarly, the human clinical community and the various model organism resources can annotate the same phenotype at different resolutions. This will present problems to a lexical approach but can be solved by the subsumptive power of an ontology approach.
A more logically rigorous approach to compare phenotypes in different species is to use a set of species-agnostic ontologies as the building blocks for logical definitions of terms in pre-composed species-specific ontologies. This approach is implemented by (i) generating EQ statements (known as logical definitions or equivalence axioms) for each of the terms used in the pre-composed phenotype ontologies such as MPO and HPO, and (ii) linking between the ontologies used in the EQ statements. Most of the ontologies used in the logical definitions are applicable to both species, but anatomy presents a special problem so the task is simplified to linking across the species-centric anatomical ontologies. Taking our example above of enlarged hind paws and large feet, the logical definition of the MPO term involves the PATO term “increased size” (PATO:0000586) and Mouse Anatomy term “foot” (MA:0000044) while the HPO logical definition involves the same PATO term and the human-centric Foundational Model of Anatomy term “foot” (FMA:9664). In this case, MA has already been made species-agnostic to some extent by referring to the foot of the hind limb rather than hind paw, so it is obvious that the MA and FMA terms refer to the same concept. However this is often not the case and we tackle this problem by using a bridging multi-species anatomy ontology to map between the individual species anatomy terms. Methods to generate these bridging ontologies, range from manually-assisted, automated matching, e.g., the UBERON unified metazoan anatomy ontology (Mungall et al., 2010), to relations based on the nearest common evolutionary ancestor of the structure in question, e.g, the Vertebrate Bridging Ontology (Ravensara et al., 2011).
This logical definition approach generated promising results in identifying gene candidates and animal models of human disease using 11 manually annotated diseases with known genes (Washington et al., 2009). Recently, a cross-species network built from the phenotype ontologies, logical definitions and UBERON has been shown to recall orthologues, genes involved in the same pathway and gene-disease associations (Hoehndorf et al., 2011).
We now have HPO annotations of almost all clinical OMIM entries representing Mendelian diseases and logical definitions available for a large proportion of the HPO and MPO terms. We can therefore extend our approach to nearly all known Mendelian diseases. Here, we present this extension using new semantic matching software (OWLSim) and report high recall of known disease genes. We describe a new web tool (MouseFinder), which allows anyone to mine the results of this analysis for the identification of new candidates for human disease and present some intriguing examples of this.
Implementation
HPO annotations of OMIM diseases, and the HPO ontology itself, were downloaded from http://www.human-phenotype-ontology.org/index.php/downloads.html. Known OMIM disease to gene associations are recorded in morbidmap and were downloaded from http://www.omim.org/downloads. MPO was obtained from http://obo.cvs.sourceforge.net/viewvc/obo/obo/ontology/phenotype/mammalian_phenotype.obo. MPO annotations of mouse models (MGI_PhenotypicAllele.rpt and MGI_GenePheno.rpt), MGI asserted disease models (ALL_OMIM.rpt) and OMIM human gene to MGI gene mappings (HMD_OMIM.rpt) were downloaded from the MGI ftp site (ftp://ftp.informatics.jax.org/pub/reports). Note, we used the MGI_GenePheno.rpt file, recently made available by MGI, rather than the larger MGI_PhenoGenoMP.rpt file used by most of the previously published studies. The latter file contains all phenotyped models including those with multiple genes mutated where it will be unclear which mutation is causative, conditional mutations which need further crossing to disrupt the gene and mutations of non-gene markers and complex/cluster/region markers (includes deletion regions, inversions).
All files were downloaded on August 7th, 2011, processed, and the contents stored in a simple database schema. The database stores the mappings from HPO annotated Mendelian diseases recorded in OMIM, through to mouse genes via orthology and thence to mutant allele and mouse model phenotype annotations. 5035 OMIM diseases (1858 with known gene association(s) and 3177 with no known gene) and 1791 OMIM genes with HPO annotation, along with the MPO annotations of 24904 mouse models and 8124 mouse genes, are stored in the database (Figure 1). In addition, 2624 associations between OMIM diseases and particular models from MGI curation of the literature are also captured (Figure 1).
Figure 1.
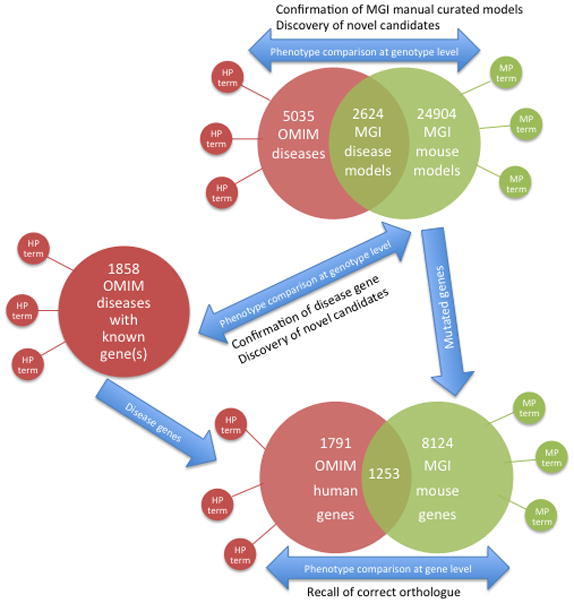
Schematic representation of the data used for the phenotypic comparisons described in this study. The comparisons were either at the gene or disease/mouse model (genotype) level. For the former, the original annotated data is projected to the gene level using the known mutated genes in the mouse models or the known disease-gene associations. Gene level comparisons are used as a positive control to assess how often we can recall the correct orthologue using the phenotype data alone. The genotype level comparisons are used to identify novel candidates for human disease utilising the recovery of the known disease-gene associations as a means to analyse the success of the approach.
OWLTools (freely available from http://owlsim.org) was used to prepare OWL representations of the human and mouse phenotype annotation at the genotype and the gene level. OWLTools provides convenience methods on top of the java OWL API (Horridge 2009), and includes the OWLSim package used for all the semantic comparisons described here. OWLSim uses the same metrics as described in Washington et al., 2009, and is, in fact, largely a re-implementation of the same system using a different underlying ontology model. Our previous approach was implemented on top of a relational database system called OBD, whereas, OWLSim is implemented on top of the OWL API and does not require an underlying database to run the semantic comparisons. This makes OWLSim easier to set up, and faster to run.
OWLSim was used to compare each of the HPO annotated OMIM gene or disease records against all the MPO annotated mouse genes or mutant lines. Pairwise comparisons were performed using a merged OWL file of PATO, UBERON, MPO plus logical definitions, HPO plus logical definitions and a mapping of HPO and MPO lexical matches. The logical definitions for HPO and MPO are available at http://phenotype-ontologies.googlecode.com/svn/trunk/src/ontology/mp/mp-equivalence-axioms.obo and http://phenotype-ontologies.googlecode.com/svn/trunk/src/ontology/hp/hp-equivalence-axioms.obo respectively. For a particular human and mouse comparison, each of the HPO annotations is compared to each of the MPO annotations and scored using either Information Content (IC) or Jaccard Similarity (SimJ) measures.
SimJ scores similarity as the ratio of shared attributes to total attributes. In the case of OWLSim, the attributes being compared are inferred attributes (for a full technical description see owlsim.org):
where ap is the inferred attributes of phenotype p.
The IC of a description is the negative log of the number of features annotated with that description over the total number of annotations in the dataset.:
In the case of OWLSim, IC is calculated for the Least Common Subsuming (LCS) phenotype of the HPO-MPO pair which is the most specific set of all shared attributes (the algorithm to identify the LCS is, again, more fully described at owlsim.org). The IC method provides a measure of how unusual or “surprising” the set of attributes in common is and the higher the score, the less frequent is the LCS. Thus, a match in which the combination of attributes in common is rare, or involves highly specific terms, will score more highly than those involving more frequent or less granular terms.
For each human-mouse comparison, we aggregate the measurements of individual HPO-MPO best matching pairs to give:
avgIC – average IC score across all the pairs
maxIC – maximum IC score across all the pairs
avgSimJ – average SimJ across all the phenotype pairs
maxSimJ – maximum SimJ across all the phenotype pairs
The mappings of human diseases/genes to mouse models/genes along with the various measures of semantic similarity were stored in the same database for further analysis of the results and are displayed in the MouseFinder tool.
Recall of known disease genes and models from mouse phenotype data
To test the potential of OWLSim human to mouse phenotype matches to recall genuine disease associations, we took advantage of OMIM morbid map which records known disease causative genes. In our database, 1858 of the 5035 OMIM disease records have a disease association with one or more of 1791 human genes (Figure 1). Where mouse models involving mutants of these genes have been phenotyped, we should be able to recall these disease associations with high specificity and sensitivity using just the phenotype comparison methodology. The subset of OMIM records with an associated gene with a mouse ortholog that has been phenotyped includes 1514 OMIM diseases and 1989 distinct disease to gene associations for 1253 unique human genes.
Figure 2 shows the results from the OWLSim phenotype comparison of HPO annotated human diseases and MPO annotated mouse mutant lines for recall of any of the associated gene(s) for the 1514 OMIM diseases. 58% of associations were recalled with most appearing in the top 50 hits. The maxSimJ metric performed best, followed by maxIC, avgIC and then avgSimJ. Figure 2 also shows the results of a 1000 random runs. For each random run, n mouse models were randomly selected for each OMIM disease and assessed for a mutation in the known disease associated gene to calculate the expected level of recall in the top n hits if there was no biological association between the HPO annotated diseases and MPO annotated mouse models. This was repeated 1000 times and the average result plotted on Figure 2. In all cases, OWLSim is recalling the disease gene associations at significantly higher levels than random. For example, the 53% recovery of the disease-gene associations in the top 10 hits by maxSimJ has a p value < 10−325 assuming a binomial distribution.
Figure 2.

Recall of known disease genes using phenotype comparisons between human OMIM diseases and mouse models for the 1514 diseases with gene associations described in OMIM morbid map. The graph shows the recall using OWLSim and maxIC, avgIC, maxSimJ and avgSimJ semantic measures as well as the results of 1000 runs where mouse models were randomly recalled.
Another test of the effectiveness of OWLSim is to utilise the manual, literature curation the MGI group performs to assert that particular mouse lines are models of a human diseases. For these associations we again assessed our success at recalling these models (Figure 3). 65% of these models could be retrieved, with most appearing in the top 50 hits. 20% were recalled as the top or joint top hit using maxSimJ. Again the recall is much higher than that shown by 1000 runs where mouse models were randomly chosen for each disease.
Figure 3.

Recall of mouse models of human disease as asserted by the MGI group using phenotype comparisons between human OMIM diseases and mouse models. The recall using OWLSim and maxIC and avgIC semantic measures is shown as well as the results of 1000 runs where mouse models were randomly recalled.
We can also test the recall using phenotype annotations projected onto the gene level rather than disease and mouse model (genotype) level as used above. 1253 HPO annotated human genes were compared to their MPO annotated mouse orthologs using OWLSim phenotypic comparisons to assess whether the mouse ortholog was recalled at significantly higher levels than expected by chance (Figure 4). Compared to the 1000 runs where the orthologs were randomly chosen, the recall was significantly higher. This time the avgIC metric performed best except for recall as the top hit or in the top 3. 78% of the orthologs could be recalled at highly significant levels, e.g., the 48% found in the top 50 by avgIC has a p value < 10−325. To give a measure of sensitivity versus specificity, Receiver Operating Characteristic (ROC) analysis was carried out on the avgIC ordered data from this human-mouse gene analysis. A highly significant area under the curve (AUC) value of 0.82 was obtained.
Figure 4.

Recall of correct orthologue using phenotype comparisons between human and mouse genes. The recall using OWLSim and maxIC, avgIC, maxSimJ and avgSimJ semantic measures as well as the results of 1000 runs where mouse genes were randomly recalled is shown.
Comparison with PhenomeNET
PhenomeNET (Hoehndorf et al., 2011) uses the same set of annotations, ontologies and logical definitions to compare human and mouse phenotype data. Although there are considerable similarities between the two approaches, the algorithms differ in a few key respects. PhenomeNET relies exclusively on subsumption between classes when calculating the least common ancestor: whereas, OWLSim makes use of other ontology relationships. In addition, OWLSim can generate class expressions on the fly whilst PhenomeNET relies on there being phenotype classes explicitly pre-coordinated in advance. PhenomeNET calculates the average of all pairs of phenotypes: whereas, the default algorithm in OWLSim is the average of best matches.
To compare the two approaches, we analysed the recall rates of known disease genes using OWLSim and the data available from the PhenomeNET site (files generated on 16th September 2011 at http://bioonto.gen.cam.ac.uk/phenomenet). As shown in Figure 5, the recall rates using OWLSim were considerably higher except outside the top 500, as OWLSim has a cutoff of 500 matches. The improved recall may be due to the algorithmic approach used and/or the fact that our OWLSim analysis makes use of simple lexical matching in addition to the ontological cross products; whereas, PhenomeNET uses a purely semantic approach. In addition, PhenomeNET also covers yeast, zebrafish, C. elegans and Drosophila phenotypes, and does not have the overhead of running the pairwise phenotype comparisons and storing the results in a database.
Figure 5.

A comparison of the OWLSim methodology used in this study with a similar approach (PhenomeNET) for the recall of any of the known disease genes using phenotype comparisons between OMIM diseases and mouse models. Both methods use the same phenotype annotations and ontology mapping files between human and mouse data and perform significantly better than a 1000 random runs.
MouseFinder web tool
Our web tool, MouseFinder (www.mousemodels.org), provides access to the phenotype comparisons described above. Users can identify a particular OMIM disease by browsing or searching by the disease name or OMIM ID, or by any of the associated genes or HPO terms. Once a disease is selected, a ranked list of the matching mouse models is displayed, ordered by avgIC as default (Figure 6A). Each row shows the allelic composition and genetic background of the mouse model, along with the mutated gene and the rank and score according to the chosen similarity measure. The disease, gene and mutant allele fields link out to more detailed data on the OMIM and MGI websites. Further tabs show the models ranked by maxIC, avgSimJ or maxSimJ, and the final tab reveals any known associated OMIM genes from morbid map or known, published mouse models of the disease as curated by MGI. Where a known OMIM gene exists, a red box in the gene symbol column indicates its mouse ortholog. Similarly, any MGI asserted mouse models are indicated by a green tick to the left of each row of results. The results can be restricted to hits involving matches to a limited set of the HPO annotated terms using the HP button at the top of the window.
Figure 6.

OWLSim phenotype comparisons between human and mouse presented in the mouseFinder web tool (www.mousemodels.org). OMIM diseases are chosen by name, OMIM ID, associated gene or HP annotation. (a) For a particular disease the matched mouse models ranked by phenotypic similarity are displayed. The similarity score used for ranking can be selected from avgIC, maxIC, avgSimJ or maxSimJ. The final tab in the interface shows any known genes for the disease from OMIM (indicated by a red box around the gene in the ranked list) and any associated mouse models from MGI (indicated by a green tick next to the match in the list). (b) Expanding any of the matched rows reveals the details on the matched HPO and MP paired terms that were used to associate the disease and mouse model.
In the example shown in Figure 6 for Craniosynostosis, Type 1 (MIM# 123100), a model involving the known causative gene (Twist1) is the top hit when ranked by avgIC. A MGI curated mouse model involving Axin2 is the 10th best hit, as indicated by the green tick in the screenshot. Expanding the detail for the top Twist1 match reveals further detail on the HPO and MPO annotation of the disease and mouse model, along with the phenotype terms and the IC and simJ measures for each paired match (Figure 6B). The MPO annotation of the mouse model (premature suture closure) matches the craniosynostosis (premature closure of the cranial sutures), turricephaly (high head resulting from premature closure of the cranial sutures) and dolichocephaly (long head resulting from premature closure of the cranial sutures) clinical features.
Novel candidates for human diseases
The rationale for developing our approach and MouseFinder is, of course, to identify novel candidates for human diseases. The recall analysis described above suggests that, for OMIM diseases with no known gene, the real causative gene should be present high up in our rankings. To explore this, we took the 468 OMIM diseases with a mapped locus but no known causative gene and looked for MouseFinder hits in the top 10 results by avgIC where the human orthologue maps to the correct genomic position. 9% of diseases had a candidate mapping to the correct locus in the top 10 hits (Table 1). This success rate was well above that seen in 1000 runs where the disease to locus mappings were randomised, strongly suggesting MouseFinder is discovering candidates worthy of further study for these uncharacterised diseases.
Table 1.
Candidate genes for OMIM diseases with a mapped locus but no known associated gene(s)
| MIM# | Disorder Name | Candidate Gene |
|---|---|---|
| 131400 | EOSINOPHILIA, FAMILIAL | IL5 |
| 300062 | MENTAL RETARDATION, X-LINKED 14 | TIMP1 |
| 221820 | GLIOSIS, FAMILIAL PROGRESSIVE SUBCORTICAL | GFAP |
| 159555 | MYELOID/LYMPHOID OR MIXED LINEAGE LEUKEMIA | MLL |
| 178300 | PTOSIS, HEREDITARY CONGENITAL 1 | ARTN |
| 156232 | MESOMELIC DYSPLASIA, KANTAPUTRA TYPE | HOXD11 |
| 156232 | MESOMELIC DYSPLASIA, KANTAPUTRA TYPE | HOXD13 |
| 600231 | PALMOPLANTAR KERATODERMA, BOTHNIAN TYPE | PTGES3 |
| 161950 | IGA NEPHROPATHY 1 | SGK1 |
| 105550 | AMYOTROPHIC LATERAL SCLEROSIS AND/OR FRONTOTEMPORAL DEMENTIA 1 | AGTPBP1 |
| 126900 | DUPUYTREN CONTRACTUREDUPUYTREN CONTRACTURE 1, INCLUDED | SALL1 |
| 102300 | RESTLESS LEGS SYNDROME, SUSCEPTIBILITY TO, 1 | KCNC2 |
| 153600 | MACROGLOBULINEMIA, WALDENSTROM, SUSCEPTIBILITY TO, 1 | NFKBIE |
| 310460 | MYOPIA 1 | OPN1LW |
| 601941 | DIABETES MELLITUS, INSULIN-DEPENDENT, 6 | MC4R |
| 607317 | SPINOCEREBELLAR ATAXIA, AUTOSOMAL RECESSIVE 4 | UBE4B |
| 603116 | CDAGS SYNDROME | MN1 |
| 609306 | SPINOCEREBELLAR ATAXIA 26 | CACNA1A |
| 313850 | THORACOABDOMINAL SYNDROME | GPC3 |
| 129900 | ECTRODACTYLY, ECTODERMAL DYSPLASIA, AND CLEFT LIP/PALATE SYNDROME1 | DLX5 |
| 145410 | OPITZ GBBB SYNDROME, AUTOSOMAL DOMINANT HYPERTELORISM WITH ESOPHAGEAL ABNORMALITY AND HYPOSPADIAS G SYNDROME HYPOSPADIAS-DYSPHAGIA SYNDROME OPITZ-FRIAS SYNDROME OPITZ-G SYNDROME, TYPE II TELECANTHUS W | TBX1 |
| 602483 | AURICULOCONDYLAR SYNDROME | LMNA |
| 149000 | KLIPPEL-TRENAUNAY-WEBER SYNDROME | GDF6 |
| 300652 | ANGIOMA SERPIGINOSUM, X-LINKED | EBP |
| 247200 | MILLER-DIEKER LISSENCEPHALY SYNDROME MILLER-DIEKER SYNDROME CHROMOSOME REGION, INCLUDED | HIC1 |
| 144120 | HYPERIMMUNOGLOBULIN G1(A1) SYNDROMEIMMUNOGLOBULIN HEAVY CHAIN REGULATOR, INCLUDED | KIAA1409 |
| 609625 | CHROMOSOME 10Q26 DELETION SYNDROME | FGFR2 |
| 109350 | GASTROESOPHAGEAL REFLUX | OLFM4 |
| 607498 | MIGRAINE WITH OR WITHOUT AURA, SUSCEPTIBILITY TO, 3 | POLH |
| 607516 | MIGRAINE WITH OR WITHOUT AURA, SUSCEPTIBILITY TO, 6 | TROVE2 |
| 607516 | MIGRAINE WITH OR WITHOUT AURA, SUSCEPTIBILITY TO, 6 | TROVE2 |
| 613096 | SPASTIC PARAPLEGIA 36, AUTOSOMAL DOMINANT; SPG36 | TRPV4 |
| 300125 | MIGRAINE, FAMILIAL TYPICAL, SUSCEPTIBILITY TO, 2 | OPN1LW |
| 148500 | TYLOSIS WITH ESOPHAGEAL CANCER | EVPL |
| 161550 | NASOPHARYNGEAL CARCINOMA | CDKN1A |
| 601846 | VACUOLAR NEUROMYOPATHY | CNN1 |
| 309605 | MILES-CARPENTER X-LINKED MENTAL RETARDATION SYNDROME | EFNB1 |
| 142470 | FETAL HEMOGLOBIN QUANTITATIVE TRAIT LOCUS 2 | L3MBTL3 |
| 166760 | OTITIS MEDIA, SUSCEPTIBILITY TO | CUZD1 |
The candidates shown appear in the top 10 hits by OWLSim using the avgIC metric.
An example of one of the candidates is shown in Figure 7. Here, a mouse model (Artntm1Jmi/Artntm1Jmi with a genetic background of 129X1/SvJ * FVB/N) is the top hit for Ptosis, hereditary congenital 1 (MIM# 178300). The causative locus for this disease has been mapped to 1p34.1-p32 and ARTN, the human orthologue of Artn, maps to 1p34.1. The clinical feature of congenital ptosis (drooping eyelids) matches the mouse phenotype of blepharoptosis (drooping eyelids) and clearly warrants further investigation of ARTN as a candidate for this disease. Ptosis is thought to result either from damage to the eyelid muscle (levator palpebrae superioris), the superior cervical sympathetic ganglion, or the oculomotor nerve (CNIII) which controls this muscle. ARTN is expressed in the nucleus of the oculomotor nerve in the pre- and perinatal period along wth neurturin, persephin; all three being members of the GDNF family (Quartu et al., 2007). Intriguingly, this mouse model was published as proof that ARTN is a neurotropic factor for developing sympathetic neurons (Honma et al., 2002) and the mice also show abnormalities in sympathetic ganglion morphology (in the small superior cervical ganglion) and sympathetic neuron morphology. This further strengthens the case for ARTN being the causative gene at this locus.
Figure 7.

MouseFinder results for Ptosis, hereditary congenital 1 (MIM# 178300) which has a mapped locus of 1p34.1-p32 but no known gene. Here a mouse line involving a mutation of Artn is the top hit by avgIC and the human orthologue ARTN is located at 1p34.1. The mouse model exhibits the same phenotype of blepharoptosis (drooping eyelids) and in addition reveals abnormalities in the small superior cervical ganglion. Damage to this ganglion is one of the known causes of blepharoptosis.
Conclusion and future directions
In this paper, we have described a novel approach and tool for the identification of candidate disease genes for human disease. The recall of known disease gene associations at highly significant rates demonstrates that we can start to fully utilise model organism phenotype data for this purpose. As shown above for a form of hereditary ptosis, MouseFinder can identify plausible candidates for the human disease using only the clinical phenotypic features. It should be borne in mind that for many diseases we have no information available regarding the protein function, biochemical pathway involvement or expression pattern of the affected gene. In these cases, our phenotype approach represents a viable alternative to the classical computational methods of candidate gene selection using Gene Ontology (GO) or pathway enrichment studies, or expression data analysis. However, an integrated approach using phenotype data alongside these other lines of evidence (when available), as well as the mapped locus, would of course improve the success rates in identifying disease genes. Future efforts will be focussed on developing this integrated analysis.
Despite the significant recall shown by all OWLSim analyses, there still remain some known disease gene and mouse model associations that were not recovered when using OWLSim to compare human and mouse phenotype annotations. At the genotype annotated level, some 40% of known gene associations were not recalled, at the gene level 22% of associations were missed, and for the MGI asserted models 35% were not recovered. This could be for a number of reasons, including:
need for improvement in the recently developed ontologies and logical definitions. Our analysis produces a tractable set of missed phenotype relationships that can be used for improvements by the groups developing HPO, MPO and the logical definitions. In addition, methods are being developed to automatically evaluate and improve the ontologies and logical definitions (Köhler et al., 2011).
limitations of the OWLSim approach which we can investigate and improve in the future.
informative phenotype assays not yet having been carried out on the mouse model.
under-representative annotation of the human disease and mouse models, e.g., for 4% of the disease genes we were trying to recall, the only MPO annotations for the mouse orthologs were “no abnormal phenotype” or “embryonic or postnatal lethal”. It will not be possible to recover these associations until more MPO annotation becomes available as a result of further curation or experimental work to generate further models and phenotype data.
for an unknown number of cases the mouse will prove not to be a good model for the particular human disease.
As highlighted by some of the papers in this special issue, this is an exciting time with rapid developments occurring in mouse phenotyping through the IMPC (see Schofield et al) and collection of human phenotype data through projects such as Orphanet (see Aymé et al). These new projects will generate a wealth of new phenotype data as well as physical mouse resources for the community to generate additional data. These initiatives can only improve the recall rates, and we envisage accurate, integrated phenotype querying across species becoming an essential tool for the clinical research community.
Acknowledgments
We thank the whole of the OMIM, HPO and MGI teams for the curation that made this work possible. This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 and NIH R01 grant HG004838-02.
Footnotes
The authors declare no conflict of interest in this work.
References
- Abbott A. Mouse megascience. Nature. 2010;465:526. doi: 10.1038/465526a. [DOI] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT Mouse Genome Database Group. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucl Acids Res. 2011;39:D842–D848. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkoutos GV, Mungall C, Dolken S, Ashburner M, Lewis S, Hancock J, Schofield P, Kohler S, Robinson PN. Entity/quality-based logical definitions for the human skeletal phenome using PATO. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:7069–72. doi: 10.1109/IEMBS.2009.5333362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth P, Pavlova N, Kalev I, Tonov S, Georgiev G, Pohlenz H-D, Weiss B. PhenomicDB: a new cross-species genotype/phenotype resource. Nucl Acids Res. 2006;35:D696–D699. doi: 10.1093/nar/gkl662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehndorf R, Schofield PN, Gkoutos GV. PhenomeNET: a whole-phenome approach to disease gene discovery. Nucl Acids Res. 2011:39. doi: 10.1093/nar/gkr538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma Y, Araki T, Gianino S, Bruce A, Heuckeroth R, Johnson E, Milbrandt J. Artemin is a vascular-derived neurotropic factor for developing sympathetic neurons. Neuron. 2002;35:267–82. doi: 10.1016/s0896-6273(02)00774-2. [DOI] [PubMed] [Google Scholar]
- Köhler S, Bauer S, Mungall CJ, Carletti G, Smith CL, Schofield P, Gkoutos GV, Robinson PN. Improving ontologies by automatic reasoning and evaluation of logical definitions. BMC Bioinformatics. 2011;12:418. doi: 10.1186/1471-2105-12-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsios GD, Tangri N, Castaldi PJ, Ioannidis JP. Laboratory mouse models for the human genome-wide associations. PLoS One. 2010;5:e13782. doi: 10.1371/journal.pone.0013782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan TF, Carr CJ, Jay JJ, Bult CJ, Chesler EJ, Blake JA. Autism candidate genes via mouse phenomics. J Biomed Inform. 2011 Mar 21; doi: 10.1016/j.jbi.2011.03.003. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall C, Gkoutos G, Smith C, Haendel M, Lewis S, Ashburner M. Integrating phenotype ontologies across multiple species. Genome Biol. 2010;11:R2. doi: 10.1186/gb-2010-11-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartu M, Serra MP, Boi M, Sestu N, Lai ML, Del Fiacco M. Tissue distribution of neurturin, persephin and artemin in the human brainstem at fetal, neonatal and adult age. Brain Research. 2007;1143:102–115. doi: 10.1016/j.brainres.2007.01.065. [DOI] [PubMed] [Google Scholar]
- Sardana D, Vasa S, Vepachedu N, Chen J, Gudivada RC, Aronow BJ, Jegga AG. PhenoHM: human-mouse comparative phenome-genome server. Nucleic Acids Res. 2010;38:W165–W174. doi: 10.1093/nar/gkq472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringwald M, Iyer V, Mason JC, Stone KR, Tadepally HD, Kadin JA, Bult CJ, Eppig JT, Oakley DJ, Briois S, Stupka E, Maselli V, Smedley D, Liu S, Hansen J, Baldock R, Hicks GG, Skarnes WC. Nucl Acids Res. 2011;39:D842–D848. doi: 10.1093/nar/gkq879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Koehler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travillian RS, Malone J, Pang C, Hancock J, Holland PWH, Schofield PN, Parkinson H. The Vertebrate Bridging Ontology (VBO) Journal of Biomedical Semantics. 2011 doi: 10.1186/2041-1480-2-S4-S3. (Accepted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9:993–999. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- Schofield PN, Gkoutos GV, Gruenberger M, Sundberg JP, Hancock JM. Phenotype ontologies for mouse and man: bridging the semantic gap. Dis Model Mech. 2010;3:281–289. doi: 10.1242/dmm.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Eppig JT. The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip Rev Syst Biol Med. 2009;1(3):390–9. doi: 10.1002/wsbm.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague J, Bayraktaroglu L, Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Haendel M, Howe DG, Knight J, Mani P, Moxon SA, Pich C, Ramachandran S, Schaper K, Segerdell E, Shao X, Singer A, Song P, Sprunger B, Van Slyke CE, Westerfield M. The zebrafish information network: the zebrafish model organism database provides expanded support for genotypes and phenotypes. Nucleic Acids Res. 2008;36:D768–D772. doi: 10.1093/nar/gkm956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, Zhang H FlyBase Consortium. FlyBase: enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigger SN, Shimoyama M, Bromberg S, Kwitek AE, Jacob HJ RGD Team. The Rat Genome Database, update 2007 – easing the path from disease to data and back again. Nucleic Acids Res. 2007;35:D658–62. doi: 10.1093/nar/gkl988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington NL, Haendel MA, Mungall CJ, Ashburner M, Westerfield M, Lewis SE. Linking human diseases to animal models using ontology-based phenotype annotation. PLoS Biol. 2009;7:e1000247. doi: 10.1371/journal.pbio.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]