

Abstract
Whole genome analysis, now including whole genome sequencing, is moving rapidly into the clinical setting, leading to detection of human variation on a broader scale than ever before. Interpreting this information will depend on the availability of thorough and accurate phenotype information, and the ability to curate, store, and access data on genotype-phenotype relationships. This idea has already been demonstrated within the context of chromosome microarray (CMA) testing. The International Standards for Cytogenomic Arrays (ISCA) Consortium promotes standardization of variant interpretation for this technology through its initiatives, including the formation of a publicly available database housing clinical CMA data. Recognizing that phenotypic data is essential for the interpretation of genomic variants, the ISCA Consortium has developed tools to facilitate the collection of this data and its deposition in a standardized, structured format within the ISCA Consortium database. This rich source of phenotypic data can also be used within broader applications, such as developing phenotypic profiles of emerging genomic disorders, the identification of candidate regions for particular phenotypes, or the creation of tools for use in clinical practice. We summarize the ISCA experience as a model for ongoing efforts incorporating phenotype data with genotype data to improve the quality of research and clinical care in human genetics.
Keywords: Genotype-Phenotype Correlation, copy number variation, CNV, Oligonucleotide array sequence analysis, Cytogenetics
Introduction
Techniques for genome-wide analysis are rapidly making their way into the clinical setting. Though these advances will make it possible to detect human genetic variation on a broader scale than ever before, the clinical interpretation of these variants may prove difficult, particularly for those genes that have not been well characterized. Thorough and accurate phenotype information, the outward manifestations of genomic variation, will be crucial not only in the clinical characterization of novel variants, but in the elucidation of genotype-phenotype relationships. Large-scale efforts are underway to aggregate the genotype and phenotype information generated through the course of clinical testing in order to provide the resources necessary to facilitate this type of discovery.
The Importance of Collecting Phenotype Information
The importance of obtaining quality phenotype information can be demonstrated through experiences with chromosomal microarray analysis (CMA). CMA is currently the most widely-used genome-wide assay in the clinical setting, and the lessons learned with this technology can easily be applied to emerging technologies, such as whole exome and whole genome sequencing. Originally developed in the research setting in the 1990s (Pinkel, et al., 1998; Solinas-Toldo, et al., 1997), CMA rapidly evolved into widespread clinical use as its potential to improve diagnostic yield was realized. Though some initial designs focused on identifying dosage imbalances within known syndromic regions by enriching probe coverage through these particular areas, designs eventually included evenly-spaced coverage throughout the genome. This genome-wide coverage allowed for the detection of novel imbalances as well as the more accurate delineation of breakpoints of copy number variants (CNVs); a combination of genome-wide and targeted coverage is now part of the recommended design guidelines for CMA in the clinical, postnatal setting (Kearney, et al., 2011). With genome-wide coverage, however, came genome-wide CNV identification; CNVs identified within areas of the genome that are not well described, particularly those that are rare, have historically been difficult to interpret, leaving open the potential for great inter-laboratory variability in terms of result interpretation (Tsuchiya, et al., 2009).
In response to this issue, the International Standards for Cytogenomic Arrays (ISCA) Consortium was established in 2008 with the ultimate goal of raising the standard of patient care by improving the quality of CMA testing (www.iscaconsortium.org). One of the major efforts of the ISCA Consortium has been to leverage data from the thousands of patients with intellectual disabilities and developmental delays being tested by clinical laboratories to accelerate our understanding of the clinical significance of novel CNVs. This aggregated, de-identified clinical data, collected from laboratories around the world, is stored in a publicly available database (http://www.ncbi.nlm.nih.gov/dbvar/studies/nstd37/; www.iscaconsortium.org). Further, laboratories have the option of submitting raw data files to a controlled-access database housed within the Database of Genotypes and Phenotypes (dbGaP) at the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000205.v1.p1); users that apply for and are granted access to this data have the ability to reanalyze submitted array data, a feature that differentiates the ISCA Consortium database from similar efforts such as DECIPHER(Firth, et al., 2009). From the ISCA Consortium website interface, users are able to search by genome coordinates, cytogenetic band, or gene name; corresponding cases that have been submitted to the database are displayed within a genome browser, complete with the submitting laboratory’s clinical interpretation. The intent of the ISCA database is to become a “CNV atlas” of human development; the collective observations obtained by combining data from multiple laboratories will provide the large sample size needed to aid in the more standardized interpretation of array data.
Though large case numbers will ultimately help us to determine which CNVs are observed statistically more often in cases vs. controls, these observations do not provide information regarding the phenotypic spectrum associated with particular copy number gains and losses, information that is an integral part of determining the clinical significance of certain CNVs (Riggs, et al., 2011). Recognizing this, the ISCA Consortium encourages the submission of phenotypic information in conjunction with genotypic information. There is a compelling need for ordering clinicians to submit thorough, accurate phenotype information in conjunction with laboratory samples, and for clinical laboratories to submit this type of information to publicly available databases. The ability to correlate genomic findings with a demonstrable phenotype is essential both at the individual level, in the interpretation of genomic imbalance events for individual patients, and at the global level, for the investigation of relationships between dosage sensitivity and gene function.
Need for Genotype-Phenotype Correlations at the Individual Level
At the individual level, phenotype information provided to the clinical laboratory at the time of testing can influence the interpretation of laboratory results. Having this information available may result in a copy gain or loss that might have been classified as being of uncertain significance on its own being classified as “likely pathogenic” or “pathogenic” based on the correlation between genomic content and clinical presentation. For example, in one clinical laboratory, a patient referred for CMA testing with developmental delay and spherocytosis was found to have a deletion encompassing the SPTB gene (MIM# 182870) associated with autosomal dominant spherocytosis type 2. Knowing that the patient was affected with this condition provided the laboratory with more confidence to classify this particular CNV as pathogenic (personal communication, C.L. Martin). Other examples include small, intragenic deletions/duplications that may be below a laboratory’s standard reporting threshold for size; knowing that a patient exhibits features of the phenotype associated with the gene in question may change how a variant like this is reported. This point is illustrated by a laboratory with an established reporting threshold for copy number gains/losses of 500 kilobases (kb) in size or greater, unless the variant is within a known microdeletion/microduplication region, or involves a “clinically relevant” gene. When a 173 kb deletion involving VPS13B, the gene associated with Cohen syndrome (MIM# 216550), was detected in a child referred for hypotonia and developmental delay, the laboratory opted to contact the referring clinician for additional information. Though hypotonia and developmental delay are features that overlap those of Cohen syndrome, they are relatively nonspecific; further, Cohen syndrome is an autosomal recessive condition and the deletion in and of itself would not be sufficient to cause disease. Additional information provided by the clinician, however, noting the presence of particular dysmorphic features and high myopia, more specific features of Cohen syndrome, prompted the laboratory to report this finding and encourage further diagnostic evaluation. Subsequent molecular analysis identified a second mutation on the other allele, confirming the diagnosis of Cohen syndrome (Sebold, et al., 2009).
Once a laboratory submits a case to the ISCA database, clinicians around the world querying that particular region will be able to view available phenotype information and apply it to their individual patients. This has the potential to improve quality of care and impact medical management. First, the availability of phenotypic data can help improve the accuracy of classification of variants (as noted above); accurate and consistent cross-laboratory classification of variants has historically been a concern in regards to CMA testing (Tsuchiya, et al., 2009). Also, if a clinician consults the ISCA database for information regarding a particular CNV found in a patient and notices that most of the cases involving that region demonstrate a particular phenotype for which their patient has not been evaluated, such as renal malformations in the17q21 deletion, he or she may be inclined to screen their patient for that particular issue (Koolen, et al., 2008).
The systematic collection of phenotypic information will not only aid in the interpretation of CMA results in the postnatal setting, but will also contribute to our understanding of copy number variation in the prenatal setting. Currently, prenatal diagnostics constitute the largest clinical service application of conventional karyotype analysis. As CMA becomes more widely used in prenatal diagnostics, the importance of phenotype data for interpretation of variants found by array, but not previously seen by karyotype, will be realized. Many of the more recently described copy number changes may be unfamiliar to clinicians involved in prenatal diagnosis, and the present understanding of the variation in clinical phenotype, frequency and age of onset of some of these conditions is insufficient to provide accurate genetic counseling to the prospective parents. Further, much of the literature regarding well-described CNVs focuses on individuals ascertained postnatally, or prenatally due to family history. As prenatal CMA becomes more widespread, these CNVs (and others) will be ascertained for different reasons (e.g. advanced maternal age, etc.), a situation which may ultimately change the natural history of some of these disorders. As such, counseling in these scenarios may prove difficult – will the postnatal course as it is currently known be applicable to individuals ascertained prenatally? Only by development of accessible phenotype information in ongoing databases will this important clinical activity be supported and resolved.
Need for Genotype-Phenotype Correlations at the Global Level
At the global level, phenotype data collected in conjunction with genotype data in databases such as that of the ISCA Consortium can be used to inspire future research. Genotype-phenotype correlations have been used to inform discovery related to gene function. By analyzing patterns between observed phenotypes and associated genes, researchers have been able to uncover new or previously unappreciated relationships between and within gene families, adding to our understanding of the pathophysiology of disease (Goh, et al., 2006; Groth, et al., 2008; Perez-Iratxeta, et al., 2002). In terms of copy number variation, a database associating detailed phenotypic information with particular CNVs can be used to hasten the description of new microdeletion and microduplication syndromes. This kind of database can also be mined to show regions of the genome potentially associated with phenotypes of interest, aiding in candidate gene identification.
Collecting Phenotype Information in the Context of the ISCA Consortium Project
Given the many potential applications of phenotypic information in association with particular CNVs, it became clear that the collection of this information for the ISCA database needed to be in a format that allowed for maximum utility. It was important that a standardized vocabulary was utilized. Employing a standardized vocabulary allows for the database to be easily indexed and searched, and removes the risk of potentially identifying free text descriptions of patients entering the database.
The Human Phenotype Ontology (HPO) was developed in order to describe the attributes of and relationships between terms frequently encountered in human genetic disease (Robinson, et al., 2008). Initially developed using terms appearing in the Online Mendelian Inheritance in Man (OMIM), the HPO currently has over 10,000 terms; over 50,000 annotations between these terms and specific diseases within OMIM have been made, allowing for searches relying on the relationships between terms and diseases to generate differential diagnoses for use in the clinical setting (Köhler, et al., 2009; Robinson and Mundlos, 2010). These relationships can also be used to explore associations between phenotypic traits and gene families, possibly revealing clues about gene function and interactions. Terms are associated with parent terms in an “is_a” type of relationship, and the “true-path” rule applies, meaning that phenotype terms are annotated to the most specific HPO term available, yet automatically also annotated to the more general terms in that family (Gkoutos, et al., 2009). For example, “horseshoe kidney” (HP: 0000085) is related to its parent term “abnormal localization of kidneys,” as well as to further removed ancestor terms such as “abnormality of the kidney,” “abnormality of the upper urinary tract,” and “abnormality of the genitourinary system.” This allows for the potential for searches using HPO terms to be informative, despite the specificity of the available phenotype information. The developers of the HPO have also been proactive in developing mapping files between HPO terms and other widely accepted dysmorphology lexicons, such as the London Dysmorphology Database (http://www.lmdatabases.com/) and the Elements of Morphology: Human Malformation Terminology (http://elementsofmorphology.nih.gov/) system. While the ideal scenario would be for all projects to utilize one set of standard phenotypic terms, these types of mapping files make it possible to convert phenotypic data from a project utilizing one system to another for combined analyses.
Once the desired format of the phenotypic data was determined, systems were developed to facilitate its actual collection. Recognizing that some test requisition forms are submitted to laboratories without phenotype information, this type of information is not a requirement to submit genotype data to the ISCA database. The success of efforts to include phenotypic data in the ISCA database is dependent upon the willingness of referring physicians and submitting laboratories to provide this information. In an effort to make the submission of this information as user-friendly as possible, the ISCA Consortium developed several different approaches to capturing phenotypic data, allowing users to choose the option that best fits with their current work flow (Figure 1).
Figure 1.
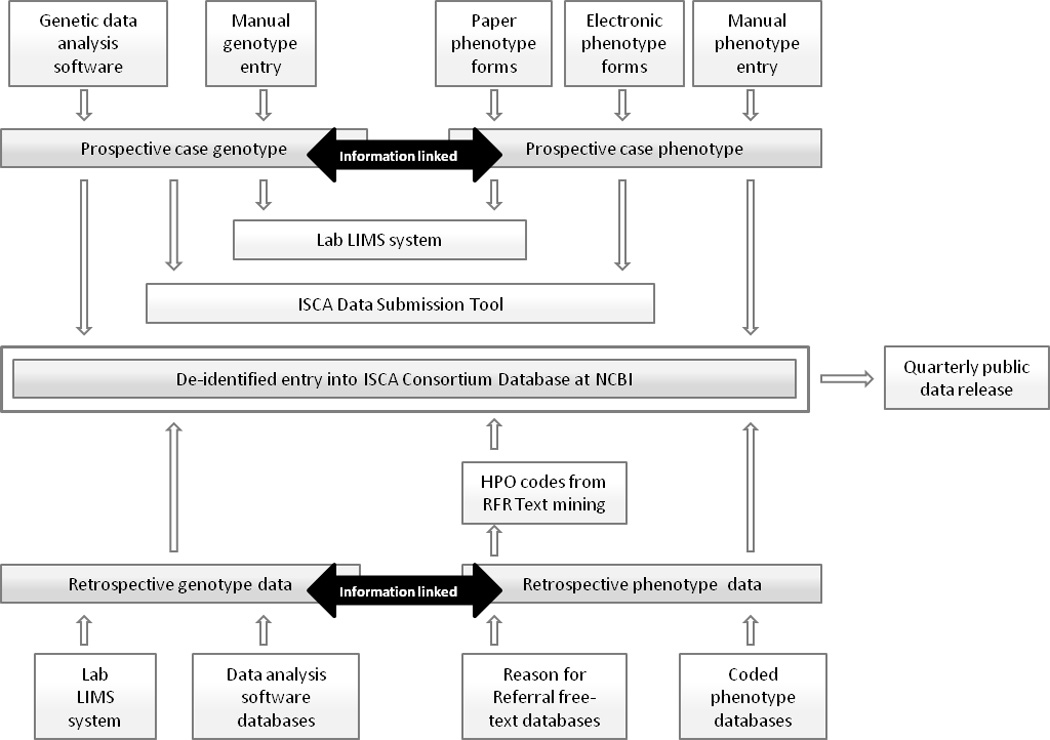
Different processes by which data is deposited into the ISCA Consortium database. (LIMS = Laboratory information management system; RFR = reason for referral)
ISCA Consortium Phenotype Collection Tools: One-Page Phenotype Forms
One-page phenotype collection forms (separate forms for use in the postnatal and prenatal settings) were developed with input from practicing clinicians, including clinical geneticists and maternal-fetal specialists. These forms include commonly encountered terms organized by body system (e.g. “cardiovascular,” “musculoskeletal,” etc.) in a simple, check-box format. The forms also include extra space for free text descriptions of phenotypic traits not represented on the form. Each term represented on the phenotype form has been mapped to a specific HPO code. The forms are available through the ISCA Consortium website (within the section entitled “The Importance of Submitting Phenotypic Data”) for public use, and have been integrated into the test requisition forms of several ISCA Consortium laboratories.
In order for ISCA labs to be able to easily transfer genotype and phenotype data into the database, the ISCA Consortium has partnered with a commercial provider to develop an ISCA Data Submission Tool (Cartagenia, N.V, providers of the BENCH software and database platform). This tool integrates with array vendor software, provides electronic versions of the postnatal and prenatal phenotype forms, and links patient genotype and phenotype information together in a de-identified manner. Once a phenotype form filled out by a client is received by a submitting laboratory, they may incorporate this information into their manual submission of genotype data, or input this data into the ISCA Data Submission Tool. Participating laboratories can also opt to make these forms available to their customers online for electronic completion and transmission. The ISCA Consortium is committed to support the continued availability of such submission tools to the community on the longer term, facilitating the aggregation of valuable information from a routine diagnostic context into public repositories.
To demonstrate that the ISCA phenotype forms can successfully be used within clinical practice, an analysis was performed on data provided by two main contributing laboratories that work with the one-page phenotype forms in daily use. Here, 3704 phenotype annotations were made for 1518 cases during the course of routine clinical work flow over approximately 8 months (1207 annotations on 440 clinical cases performed at Lab A, and 2497 annotations on 1078 clinical cases performed at Lab B). These 3704 phenotype annotations represent 637 unique HPO terms, mostly annotated through use of the electronic version of the postnatal ISCA phenotype form, sometimes also added manually through a detailed phenotype search interface available within the ISCA Data Submission Tool. The top 50 most frequently used terms within this sample (each occurring ten or more times) are listed in Table 1.
Table 1.
The 50 most frequently used phenotype terms amongst a sample of prospectively acquired cases submitted to the ISCA Consortium database
| Term | # | Definition | Term | # | Definition | Term | # | Definition |
|---|---|---|---|---|---|---|---|---|
| HP:0001263 | 499 | Developmental delay | HP:0007018 | 32 | Attention deficit hyperactivity disorder | HP:0000028 | 14 | Cryptorchidism |
| HP:0000717 | 243 | Autism | HP:0000204 | 29 | Cleft lip | HP:0000707 | 14 | Neurological abnormality |
| HP:0001999 | 179 | Facial dysmorphism | HP:0002650 | 27 | Scoliosis | HP:0001275 | 14 | Epilepsy |
| HP:0001250 | 137 | Seizures | HP:0001629 | 25 | Ventricular septal defect | HP:0000047 | 13 | Hypospadias |
| HP:0004322 | 126 | Short stature | HP:0001631 | 24 | Atrial septal defect | HP:0000238 | 13 | Hydrocephalus |
| HP:0001252 | 123 | Muscular hypotonia | HP:0000316 | 21 | Hypertelorism | HP:0000347 | 13 | Mandibular hypoplasia |
| HP:0000750 | 113 | Impaired language development | HP:0001290 | 21 | Generalized hypotonia | HP:0001762 | 12 | Talipes equinovarus |
| HP:0000252 | 105 | Microcephaly | HP:0000729 | 20 | Pervasive developmental disorder | HP:0002370 | 12 | Poor coordination |
| HP:0001249 | 92 | Intellectual disability | HP:0001298 | 19 | Encephalopathy | HP:0100021 | 12 | Cerebral paralysis |
| HP:0001508 | 81 | Failure to thrive | HP:0000369 | 18 | Low-set ears | HP:0000598 | 11 | Abnormality of the ears |
| HP:0001328 | 70 | Learning disability? | HP:0000708 | 18 | Behavioural/Psychiatric Abnormality | HP:0001251 | 11 | Ataxia |
| HP:0000256 | 60 | Macrocephaly | HP:0002117 | 18 | Speech delay | HP:0002011 | 11 | Abnormality of the central nervous system |
| HP:0002194 | 52 | Delayed gross motor development | HP:0000126 | 16 | Hydronephrosis | HP:0001274 | 10 | Agenesis of corpus callosum |
| HP:0010862 | 50 | Delayed fine motor development | HP:0001159 | 16 | Syndactyly | HP:0001363 | 10 | Craniosynostosis |
| HP:0000175 | 46 | Cleft palate | HP:0001622 | 16 | Premature birth | HP:0001636 | 10 | Tetralogy of Fallot |
| HP:0007228 | 41 | Global developmental delay, severe | HP:0001513 | 15 | Obesity | HP:0002575 | 10 | Tracheoesophageal fistula |
| HP:0001511 | 39 | Intrauterine growth restriction | HP:0007281 | 15 | Developmental arrest |
Number of occurrences within the sample
To assess whether the use of ISCA Consortium phenotype forms resulted in the collection of more detailed phenotypic information in routine clinical practice than the free-text information typically provided on test requisition forms, the origin of phenotype information was analyzed for a single ISCA Consortium laboratory. The number of HPO codes generated for patients for whom an ISCA phenotype form had been submitted by their clinician was compared to the number of HPO codes gleaned from free text on test requisition forms submitted to the same laboratory. Of note, because of a designated “free text” area within the phenotype form, free text was still present on some forms; this free text was annotated to appropriate HPO terms using the phenotype search interface within the ISCA Data Submission Tool. The same method was used to annotate the free text appearing on standard test requisition forms. All cases were collected over the same 8-month time period. A total of 85 cases were submitted with an ISCA phenotype form, while 372 cases were submitted without; 449 HPO terms were annotated to the cases with phenotype forms, while 868 HPO terms were annotated to the cases with only free-text phenotype information submitted on the requisition form. On average, cases submitted without ISCA phenotype forms had approximately 2.3 usable HPO terms per case, while those submitted with ISCA phenotype forms had approximately 5.3 useable HPO terms per case; in this example from this particular laboratory, the amount of quality phenotype information more than doubled when clinicians used the ISCA phenotype form to document their patients’ phenotypes. This could be due to the fact that clinicians may often fill out a test requisition form with what they feel is the “most important” phenotype, or the phenotype that they feel might be the most successful at securing insurance reimbursement for the test. A pre-populated list of commonly-used terms may serve as a visual reminder to busy clinicians, prompting them to check-off appropriate phenotypes that they otherwise may not have taken the time to hand-write on a test requisition form.
ISCA Consortium Phenotype Collection Tools: Text-Mining Algorithm
For those groups that are unable to use the phenotype forms, phenotype information can be collected through free-text information supplied on test requisition forms. Laboratories typically collect information on the reason for referral provided by the referring physician as part of the test requisition process. This information varies from very limited (such as a single, general phenotypic term or ICD-9 code) to extensive descriptions. This type of phenotype information may be stored in a number of different sources, such as within private laboratory databases or CMA analysis software packages, and are typically found in an unstructured free-text format.
Phenotype information in this type of free-text format is not amenable to public distribution: free-text annotations may contain patient identifying information; spelling errors; ambiguous lab-specific codes and/or abbreviations (e.g., “ASD” could stand for either atrial septum defect or autism spectrum disorder); and varying levels of detail. Further, free text information may refer to individuals other than the actual patient (e.g. “sibling with multiple congenital anomalies”) or may reflect hypotheses or uncertainties rather than factual patient descriptions (e.g., “rule-out DiGeorge syndrome;” “possible autism”). In order to address these issues, a text-mining algorithm was developed to automatically process Reason for Referral (RFR) text and align this text with HPO terms for submission to the ISCA Consortium database. HPO-vocabulary terms are detected in RFR text and form a useful alternate way to collecting phenotype information, particularly from retrospective data sets.
This automated text-mining algorithm is based on a list of “trigger phrases” that are mapped to the most specific representative HPO term. These “trigger phrases” are groups of words that, when detected within the RFR text by the algorithm, prompt the assignation of a specific HPO term. The initial list of trigger phrases was automatically generated from the HPO terms themselves, the terms’ synonyms, and their clinical descriptions. For example, the trigger phrases for the HPO term “developmental delay” (HP: 0001263) originally included the following words/phrases: developmental delay, global developmental delay, delayed milestones, etc. Following an initial run of the algorithm on the data described below, each trigger list was then manually curated by clinicians in order to identify any errors or missed terms, and to increase sensitivity and specificity. Returning to the “developmental delay” example above, terms such as “DD,” “dev delay,” and “unspecified delays in development” were added in manual curation to represent some abbreviations and ICD-9 code text commonly used to refer to generalized developmental delay. Suggested changes were incorporated into subsequent versions of the algorithm.
The text matching algorithm also corrects for alternate spellings (tumor vs. tumour), takes synonyms and alternate wording into account, works around inflections and conjugations, and can be configured to match annotations from other coding systems (such as ICD-9), increasing sensitivity. To decrease the false positive rate in annotation pickup, the algorithm picks up on negations (through detection of modifying phrases such as “without,” “no,” “ruled out,” “not present,” etc.) and on uncertain findings (such as “suspected,” “probable,” “concern for,” “likely,” etc.). Additionally, the algorithm detects whether the RFR text describes the patient or his / her parents, siblings, etc. For example, a free-text RFR stating something like “suspected autism with dysmorphic facial features, mother with cleft palate, no cardiac abnormalities” would be annotated to HP: 0001999 (facial dysmorphism), but not to cleft palate, cardiac abnormalities, or autism. At this time, entries containing “uncertain” language, such as “suspected,” “probable,” etc., are not included in the current release of the ISCA database; methods for clearly depicting the level of uncertainty associated with entries like these are under development.
For proof-of-concept purposes, the text-mining algorithm was run on a subset of 8584 retrospectively collected patient records contributed by 12 different participating ISCA laboratories, many of which contained free-text RFR descriptions. From this sample, 6770 cases contained a description from which the text-mining algorithm was able to extract at least 1 HPO phenotype term. Amongst these 6770 RFR records, a total of 13,493 HPO terms were detected, with approximately 2 phenotype traits per patient on average, and a maximum of 16 HPO terms describing a single patient. While the HPO vocabulary reflects thousands of terms, only 800 were detected at least once within this sample. The ten most frequently used terms are listed in Table 2. After careful manual review of the resulting HPO phenotype annotations, the identified terms proved to be an accurate representation of the information provided in free-text form, and all annotations were submitted to the ISCA database.
Table 2.
The 10 most frequently used phenotype terms amongst a sample of retrospectively acquired cases submitted to the ISCA Consortium database
| Term | Definition | # |
|---|---|---|
| HPO:0001263 | Developmental delay | 934 |
| HPO:0001999 | (Facial) dysmorphism | 328 |
| HPO:0001250 | Seizures | 212 |
| HPO:0004322 | Short stature | 197 |
| HPO:0000252 | Microcephaly | 192 |
| HPO:0001252 | Muscular hypotonia | 181 |
| HPO:0000717 | Autism | 169 |
| HPO:0001249 | Intellectual Disability | 163 |
| HPO:0001627 | Cardiac abnormality | 150 |
| HPO:0001508 | Failure to thrive | 147 |
Number of occurrences within the sample
Through the above heuristics and a well-devised list of phrases mapped to HPO terms, the algorithm is able to reliably match phenotype annotations to HPO terms in free-text RFR descriptions. However, this algorithm is limited to terms that are available in the HPO ontology, which only include terms that are granular, well defined, and clinically distinguishable, leaving descriptions that are vague but still very current in practical use undetected (e.g. ”multiple congenital anomalies”). Though discussions regarding how to incorporate commonly used terms such as these into the ISCA database are underway, the ultimate solution is to encourage clinicians to be as specific as possible in their descriptions of patient phenotypes, as this will result in more rich, usable data in the long term.
Though efforts have been made by the ISCA Consortium to incorporate the submission of genotype and phenotype information to our publicly available database, the nature of our current data flow includes the inherent limitation of only capturing phenotype data at the time of CMA testing. In the current iteration of the ISCA database, the phenotype and genotype data must come from the same source (i.e., the testing laboratory); allowing clinicians and laboratories to separately submit data on the same individual would require the use of an identifier, which may compromise our current consent process (discussed below) and our ability to obtain the number of samples necessary for robust statistical analyses. Important information would certainly be gained with the ability to record a particular individual’s phenotype over time, and possibilities for capturing this type of information will be a consideration for future genomic variation databases.
Applications of the ISCA Consortium phenotype data
Phenotype information being collected as part of the ISCA Consortium database is being used in a variety of different ways. The first is to inform future versions of our phenotype collection forms. After analyzing the initial set of over 6000 cases with the text-mining algorithm, a frequency plot was generated representing the most frequently used HPO terms in the sample. Nineteen terms were represented over one hundred times each. Of these, only two were not represented on the original phenotype form: developmental delay (HPO: 0001263) and hearing loss (HP: 0000365). Though developmental delay is by far the most common reason for referral received in our sample, it was represented on the original phenotype form by the more specific terms speech delay (HP: 0002117), gross motor delay (HP:0002194), and fine motor delay (HP:0010862). The results of our pilot project demonstrated that clinicians used the more general, all-encompassing “developmental delay” more often than specifying the specific areas of delay, and we have edited our phenotype form to reflect this pattern of usage. The usage frequency of the term “hearing loss” was surprising, as this was not believed to be a common reason for CMA testing; the term has nonetheless been added to our form, and it will be interesting to record how often this and other terms are used in the future. Additionally, 11 new terms that were represented more than twenty times in the pilot sample were added, and 13 terms appearing less than twenty times were removed. Spaces for information not mappable to HPO terms (actual head circumference measurement, actual IQ measurement) were also removed. The ISCA Consortium will revise the phenotype form as needed to represent the usage patterns of referring clinicians.
The HPO-encoded phenotype data generated from the text-mining algorithm as well as phenotype data submitted in HPO format through the ISCA Data Submission Tool is being made publicly available through dbVar and UCSC custom tracks available on the ISCA website. Within the ISCA database custom tracks in UCSC, clicking on an individual CNV will take the user to a “details page” where the associated HPO terms are displayed. Updated information is released quarterly.
On a broader scale, the ISCA Consortium database phenotype information can be used to describe the phenotypic profile of emerging microdeletion/microduplication syndromes. This can be approached from both the genotype-first perspective and from the phenotype-first perspective.
From the genotype-first perspective, a clustering analysis was performed on all CNVs interpreted as “pathogenic” or “likely pathogenic” within the ISCA Consortium database: for those regions in which CNVs occurred at high frequencies, the most narrow consensus regions were defined. For each chromosome, the most frequently occurring region was selected and used to retrieve all phenotype annotations associated with overlapping CNVs within the ISCA database. These annotations were collected and aggregated into phenotype profiles by counting the occurrences of the different HPO terms. Three representative examples are shown in Table 3.
Table 3.
Comparison of phenotypic profiles generated from ISCA Consortium database data to clinical descriptions of overlapping genomic disorders found in the literature
| Wolf-Hirschhorn Syndrome (chr4:72449-2327204) |
Cri-du-Chat Syndrome (chr5:37694-11347262) |
22q11.2 Deletion syndrome (chr22:18661726-21561514) |
|||
|---|---|---|---|---|---|
| MIM# 194190 | MIM# 123450 | MIM# 188400 | |||
| Region analyzed: Chr4:329980-1399150 | Region Analyzed: Chr5:547872-1429714 | Region analyzed: Chr22: 19358153- 20229017 |
|||
| Observed Phenotype (# of instances) |
Documented? | Observed Phenotype? (# of instances) |
Documented? | Observed Phenotype? (# of instances) |
Documented? |
| Developmental Delay (7) | Yes | Developmental Delay (7) | Yes | Developmental Delay (41) | Yes |
| Intrauterine Growth Restriction (4) | Yes | Microcephaly (2) | Yes | Facial dysmorphism (17) | Yes |
| Seizures (3) | Yes | Mandibular Hypoplasia (2) | Yes (as synonym micrognathia) | Cardiac abnormality (10) | Yes |
| Facial Dysmorphism (3) | Yes | Muscular hypotonia (2) | Yes | Intellectual disability (6) | Yes |
| Short stature (2) | Yes (generalized growth delay) | Cardiac abnormality (2) | Yes2 | Cleft palate (5) | Yes |
| Cleft Palate (1) | Yes | Hypertelorism (1) | Yes | Short stature (5) | Yes |
| Neurological Abnormality (1) | Yes (various) | Hearing loss (1) | Yes | Cleft lip (4) | Yes4 |
| Muscular Hypotonia (1) | Yes | Behavioral/ Psychiatric Abnormality (1) | Yes | Autism (4) | Yes4 |
| Failure to thrive (1) | Yes | Sacral dimple (1) | No | Muscular hypotonia (4) | Yes4 |
| Growth retardation (1) | Yes | Seizures (1) | Yes3 | Microcephaly (3) | Yes |
| Feeding difficulties (1) | Yes1 | Hypertonia (1) | Yes3 | Impaired language development (3) | Yes4 |
| Speech Delay (1) | Yes1 | Encephalopathy (1) | No | Seizures (3) | Yes |
| Unsteady Gait (1) | No | Failure to thrive (1) | Yes2 | Failure to thrive (3) | No |
| Febrile Seizures (1) | Yes | Facial dysmorphism (1) | Yes | Ventricular septal defect (3) | Yes |
| Feeding difficulties (1) | Yes2 | Feeding difficulties (3) | Yes4 | ||
| Recurrent respiratory infections (1) | Yes2 | Speech delay (3) | Yes4 | ||
| Duodenal atresia (1) | No | Tetralogy of Fallot (2) | Yes | ||
All references are from OMIM unless otherwise noted. The phenotypic profile of the 22q11.2 deletion syndrome was abbreviated due to space; phenotypes not documented here were observed 2 times or less in the sample (available upon request). All genome coordinates are in GRCh37 (hg19).
To demonstrate the validity of this concept, the phenotypic profiles generated from regions within selected known genomic disorders were evaluated. As described in Table 3, the HPO codes annotated to those particular regions were in alignment with predicted phenotypes based on the well-accepted clinical descriptions of these disorders. In general, CMA analysis is ordered when patients do not clearly demonstrate phenotypes associated with well-described genetic disorders; if an individual did exhibit classic features for a disorder such as one of those described above, their clinician would have likely ordered targeting FISH testing to confirm the diagnosis (though this is not always the case). It is of great interest that, despite the assumption that the individual cases within our sample likely did not have a constellation of absolutely classic features for these disorders, their combined phenotypic profiles do indeed mirror the classic features reported in the literature. This demonstration lends confidence to the idea that phenotypic profiles such as these generated from regions without well-described phenotypes may paint an accurate portrayal of the actual phenotypic consequences of that particular deletion/duplication.
From the phenotype-first perspective, the data can also be analyzed by observing genomic “hotspots” where particular phenotypes appear to cluster. For example, in order to identify areas throughout the genome where the phenotype of “autism” (HP: 0000717) appears to be overrepresented, all CNVs annotated with that term and interpreted as “pathogenic,” “likely pathogenic,” or “uncertain” were identified, and the most narrow regions of overlap were calculated. Deletions and duplications were considered separately. Amongst the cases currently in the ISCA Consortium database, the phenotype of “autism” was most frequently annotated to duplications within the 16p13.11 region, specifically chr16:15551302-16194578 (GRCh37/hg19) (the narrowest region of overlap). The term “autism” was annotated to a non-benign CNV within the ISCA Consortium database a total of 307 times, corresponding to 63 unique consensus regions seen at least twice (27 deleted, 36 duplicated), but was associated with the aforementioned 16p13.11 region nine of those times. Interestingly, though duplications within this region were initially proposed to be benign variants, and have not reached statistical significance for pathogenicity amongst two large-scale case-control studies (Cooper, et al., 2011; Kaminsky, et al., 2011), reports suggest that they may play a role in the development of neurocognitive phenotypes such as autism (Hannes, et al., 2009; Ramalingam, et al., 2011; Ullmann, et al., 2007). This type of analysis could conceivably be done on any phenotype of interest, identifying genomic regions that may warrant further investigation as possible “candidate” regions for the particular phenotype. Though preliminary analyses such as the one described above can be done on the data currently within the ISCA database, more robust and meaningful associations will come with the submission of more rich, detailed phenotypic data.
Future Prospects: Using Phenotype Information from Public Repositories to Aid Clinical Practice
Aside from its immediate utility to the particular project for which it was collected, phenotype information annotated using standardized vocabulary and stored in public repositories can be mined, aggregated, and used within broader clinical applications. In this way, the phenotype information clinicians have carefully collected on their individual patients and submitted to databases can be returned to them in the form of tools to benefit their clinical practice. Examples of such tools include case-based reasoning, variant prioritization, and differential diagnosis support.
Case based reasoning
Through the use of comparison algorithms that take genotype information as well as structured phenotype annotation into account, software platforms on which clinical diagnostic laboratories organize genetic assay analysis and interpretation workflow can be enabled to allow users to identify cases that are similar to one another. When evaluating a particular case, algorithms can be set up to alert users if other cases with an overlapping copy number variant in combination with a similar clinical phenotype have been encountered previously.
As the HPO vocabulary is an ontology, it represents both concepts (such as “ventricular septal defect”) and relationships between concepts (ventricular septal defect “is a” heart defect). Interpretation software implementing similarity measures for patient records can use the structure of the vocabulary to infer which patients are clinically similar. In this way, relevant previous patient records can be identified within a laboratory database, even if their phenotype annotations are not exactly the same. While computer systems cannot replace the finesse and nuance inherent in a clinician’s characterization of an individual’s phenotype, their ability to process vast amounts of information quickly can greatly facilitate the process of information triage, highlighting potentially relevant previous cases and supporting case-based reasoning. With standard phenotype nomenclatures and ontology-based phenotype comparison algorithms in place, automated searches for relevant cases do not have to be limited to local laboratory databases. With the increasing size of well-phenotyped cohorts of de-identified and published case records inside public databases such as the ISCA Consortium database, DECIPHER (Firth, et al., 2009), and ECARUCA (Feenstra, et al., 2006), genotype and phenotype information from these initiatives can be leveraged in the routine interpretation workflow of laboratories.
Variant prioritization
The use of standard phenotype nomenclatures to describe patient case records allows computer-assisted genotype-phenotype correlation tools to interpret variants seen in a patient assay in context of their phenotype. Such prioritization algorithms allow a computer system to highlight those variants or affected genes that, according to findings published in biomedical literature or extracted from public case registries or genotype-phenotype databases, are more likely to explain the patient’s phenotype. In this way, the variants of unknown significance identified in a patient sample can more easily be triaged and interpreted. Examples of prioritization algorithms include GECCO (designed to identify CNVs likely to cause intellectual disability) (Hehir-Kwa, et al., 2010), and the HPO-based candidate gene prioritization available within the Cartagenia BENCH platform for routine clinical interpretation of genomic variation.
Differential diagnosis support
When patient case records are adequately annotated with phenotype ontology terms, relations between phenotype concepts defined by the HPO can be used to support the differential diagnostic process, which attempts to identify candidate diseases that best explain a set of clinical features. The Phenomizer (Köhler, et al., 2009) is a web-based software tool that aims to prove this concept by taking a combination of phenotype traits queried by the user and applying semantic similarity metrics to measure phenotypic similarity with hereditary diseases annotated with the use of HPO. The tool assigns p-values to rank differential diagnoses according to their correspondence with queried phenotype traits. Such a tool can equally be used to refine the differential diagnosis process by suggesting clinical features that, if present, best differentiate among the candidate diagnoses. Ontology-based tools for differential diagnosis will become of increasing importance in supporting diagnostic interpretation as other genome-wide assays move into the clinical arena, and these tools will become more powerful when combined with structured and annotated case data from registries such as the ISCA Consortium database.
Conclusions
Our experiences in phenotypic data collection demonstrate that there are many challenges. Included among these are privacy concerns, complexity of data, lack of uniform methods for collection of phenotypic data, and inability to automate data collection to increase throughput.
Privacy
In generally, privacy concerns represent a serious challenge in collecting large-scale phenotypic data. An ideal database would include photographs and detailed phenotype information collected over time, associated with variants identified from all genetic tests performed. A collection of such information, however, would pose significant risks of individual identifiability, severely limiting the feasibility of making it publicly available. For example, ECARUCA, a valuable European-based database cataloging cytogenetic aberrations, collects detailed phenotypic information on individual cases, as well as photographs, results of other clinical examinations, and pedigree information; this rich resource, however, is only available to those that have been approved for membership, and at present only contains information on a few hundred microarray aberrations (www.ecaruca.net) (Feenstra, et al., 2006). A balance is necessary between collecting extremely detailed genotype and phenotype data on a small number of individuals and collecting less specific (yet still clinically relevant) data on a large number of individuals. While other groups have made significant efforts regarding the former, the ISCA Consortium has focused its efforts at this time on the latter; both are important in terms of our understanding of the phenotypic manifestations of human disease.
It is not difficult to maintain the level of confidentiality neccessary to make the type of deidentified information in databases such as that of the ISCA Consortium robust; for example, photographs are not collected and phenotype information, coded in standardized HPO terminology, is not easily identifiable. As a practical matter, however, meeting the requirements of local institutional review boards (IRBs) represents an obstacle, as the burden of consenting patients will fall on busy clinical providers. Even research projects dedicated to building genotype-phenotype databases are typically not able to allocate funding to support research staff to collect full consent for phenotypic data on all patients, limiting the number of patients that ultimately participate in the database. For example, the DECIPHER project, a trailblazing effort focused on, amongst other things, collecting quality phenotypic data on individuals with CNVs, utilizes a traditional, full consent process. This group reports on their website (http://decipher.sanger.ac.uk/) that information has been entered by clinicians on over 12,000 patients, but that only about 5600 had provided the full consent necessary to make the information available for public use. Appropriate alternatives to full consent must be considered in order to increase the amount of publicly available data.
Currently, for the submission of raw data files, the ISCA Consortium process uses an “opt-out” method of consent, a method that has been used in several prior studies (Clark, et al., 2004; Dziak, et al., 2005; Littenberg and MacLean, 2006). This is essentially a passive form of consent, whereby patients are informed of the research project, and informed that they are automatically enrolled unless they alert the study personnel otherwise. In terms of the ISCA Consortium, member laboratories utilizing this method of consent are required to alert patients to the study by notices on their test requisition forms, test results, and on their laboratory website. Patients are offered several different methods of “opt-out,” including calling a toll-free number, checking a box on a paper form and returning it to the testing laboratory via mail or fax, or through a web portal (for certain laboratories). Patients may opt out at any time, and their information is removed from the ISCA Consortium database. To date, no significant issues have arisen as the result of this method of consent for this project.
This method of consent has been deemed appropriate by the IRBs of several participating laboratories, as the ISCA Consortium project meets the criteria for waiver or alteration of consent as set forth by the Health Insurance Portability and Accountability Act (HIPAA). First, the research involves no more than minimal risk to the subjects. Data being submitted to this database are de-identified, and the likelihood that an individual could be re-identified based on their CNV profile and HPO-coded phenotype information is minimal. Second, an alteration of consent does not adversely affect the rights of the participants. The participants have the opportunity to remove themselves from the database at any time if they choose. Next, this research could not be reasonably carried out without this alteration of consent. Very large sample sizes are needed in order to effectively investigate the effects of certain CNVs; further, these samples are collected by multiple clinical laboratories around the world. Trying to obtain full informed consent on this vast number of individuals spread amongst a variety of locations would be exceedingly difficult and would limit the efficacy of this research. Further, clinical laboratories may not have access to contact information on every patient; only allowing those patients we could contact participate in the study would also bias our sample. Finally, efforts are made to ensure that participants are informed about the study. As above, patient-friendly information appears in several different places, and referring physicians are encouraged to discuss this research possibility with their patients in the clinical setting.
Though this method of consent has been successful thus far for the ISCA Consortium, it may not be appropriate for all projects. Respect for patient privacy and rights must be maintained, and ongoing dialogues will be needed to ensure that the goals of the research and patient communities remain aligned.
Data collection: quantity vs. quality
The utility of databases is only as good as the quality of data being submitted. Generating massive quantities of genetic variant data is becoming easier as technology for whole genome sequencing advances. Matching phenotypic information to all these genomic variants is an enormous challenge for several reasons. Collecting phenotypic information is much less amenable to automation. Approaches such as data mining of existing patient databases and/or electronic medical records may be considered, but one must not lose sight of the fact that the information in those resources required a trained clinical expert to collect on a case-by-case basis. The quality of sequencing data requires laboratory proficiency, and the quantity can be scaled massively based on new technology; however, the quality of phenotype data requires years of clinical training and dedicated time by individual experts, and the quantity can only scale linearly with the number of dedicated clinicians contributing to the effort.
Incentives for Collection of Phenotype Data
Recruiting more clinicians to contribute phenotype data will require appropriate incentives and recognition. Clinicians will benefit in various ways from these endeavors, and will hopefully recognize that they will “reap as they sow” on a community-wide basis. Access to rich genotype-phenotype data at the point of care will make it easier for clinicians to provide high quality clinical care for patients engaged in genetic testing. Beyond facilitating better interpretation of genetic test results, clinicians should be rewarded for their efforts in the collection of phenotype data in a way that allows them to demonstrate academic productivity. This does not necessarily mean that contributing data on a handful of cases should result in co-authorship on a journal article, but if clinicians can demonstrate a significant contribution in a particular area (or few areas) of clinical expertise, they will be better able to dovetail these activities with publication of disease-specific reviews of genotype-phenotype correlations that could be published in the medical literature. Additionally, smaller contributions can be recognized within the database itself, or through a system of “nanopublications” as proposed by Mons et al. (Mons, et al., 2011). Recognition of academically valuable contributions to genotype-phenotype databases will motivate clinicians to contribute their time and expertise to depositing and curating data. Distributing the work among many individuals is necessary due to the huge volume of data.
Though incentives are certainly necessary to incite participation in efforts such as the ISCA Consortium database, the ultimate solution to promote ongoing participation is to integrate the collection of phenotypic information into the routine workflow. As more clinic operational procedures move from paper-based systems to electronic ones (e.g., medical records, test ordering), the capture of phenotypic information should become part of these various electronic solutions. Clinicians must record at least some sort of phenotypic information as part of routine clinical care – the details of the visit are documented in some capacity. Sophisticated software systems and algorithms such as those described above could be configured to mine this information from wherever it is stored – be it in electronic medical records, or in laboratory information management systems, etc. – taking the onus off of the busy clinician to record the same information in a manner other than that prescribed by their standard clinical operating procedures. As discussed above, this information can then be used to assist the clinician, making their time researching and preparing for clinic more efficient. Integrating the phenotype information they have provided over time with genotype data will provide a powerful tool for variant interpretation; software solutions can be designed to present to them, at a glance, the phenotypes that have been reported with a particular variant, the literature relevant to that variant, and the number of times that variant has been observed in a particular dataset. All of this will allow clinicians and laboratorians alike to make more informed assertions regarding the clinical interpretation of variants identified from genome-wide analyses. Through the provision of high-quality, structured phenotypic data, resources can be built to assist with variant interpretation and raise the standard of patient care.
Acknowledgments
This work was supported, in part, by NIH Grant HD064525. The authors wish to thank David H. Ledbetter and Christa Lese Martin for review of the manuscript.
Footnotes
Conflict of Interest: SVV is an employee of Cartagenia, N.V. In the past 12 months, DTM has provided consulting services to Roche-Nimblegen regarding clinical issues related to chromosomal microarray testing. The other authors have no conflicts of interest to disclose.
Contributor Information
Erin Rooney Riggs, Department of Human Genetics, Emory University School of Medicine, Atlanta, GA.
Laird Jackson, Department of Obstetrics and Gynecology, Drexel University College of Medicine, and Division of Genetics, Children’s Hospital of Philadelphia, Philadelphia, PA.
David T. Miller, Division of Genetics, Department of Laboratory Medicine, Children’s Hospital, Boston, Boston, MA
Steven Van Vooren, Cartagenia, Leuven, Belgium.
REFERENCES
- Battaglia A, Carey JC, South ST, Wright TJ. Wolf-Hirschhorn Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 2010. [Google Scholar]
- Cerruti Mainardi P. Cri du Chat syndrome. Orphanet J Rare Dis. 2006;1:33. doi: 10.1186/1750-1172-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AM, Jamieson R, Findlay IN. Registries and informed consent. N Engl J Med. 2004;351:612–614. doi: 10.1056/NEJM200408053510621. author reply 612–4. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish K, Bramble D. Cri du chat syndrome: genotype-phenotype correlations and recommendations for clinical management. Dev Med Child Neurol. 2002;44:494–497. doi: 10.1017/s0012162201002419. [DOI] [PubMed] [Google Scholar]
- Dziak K, Anderson R, Sevick MA, Weisman CS, Levine DW, Scholle SH. Variations among Institutional Review Board reviews in a multisite health services research study. Health Serv Res. 2005;40:279–290. doi: 10.1111/j.1475-6773.2005.00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feenstra I, Fang J, Koolen DA, Siezen A, Evans C, Winter RM, Lees MM, Riegel M, de Vries BB, Van Ravenswaaij CM, et al. European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA); an online database for rare chromosome abnormalities. Eur J Med Genet. 2006;49:279–291. doi: 10.1016/j.ejmg.2005.10.131. [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkoutos GV, Mungall C, Dolken S, Ashburner M, Lewis S, Hancock J, Schofield P, Köhler S, Robinson PN. Entity/quality-based logical definitions for the human skeletal phenome using PATO. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:7069–7072. doi: 10.1109/IEMBS.2009.5333362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CS, Gianoulis TA, Liu Y, Li J, Paccanaro A, Lussier YA, Gerstein M. Integration of curated databases to identify genotype-phenotype associations. BMC Genomics. 2006;7:257. doi: 10.1186/1471-2164-7-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth P, Weiss B, Pohlenz HD, Leser U. Mining phenotypes for gene function prediction. BMC Bioinformatics. 2008;9:136. doi: 10.1186/1471-2105-9-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannes FD, Sharp AJ, Mefford HC, de Ravel T, Ruivenkamp CA, Breuning MH, Fryns JP, Devriendt K, Van Buggenhout G, Vogels A, et al. Recurrent reciprocal deletions and duplications of 16p13.11: the deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2009;46:223–232. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehir-Kwa JY, Wieskamp N, Webber C, Pfundt R, Brunner HG, Gilissen C, de Vries BB, Ponting CP, Veltman JA. Accurate distinction of pathogenic from benign CNVs in mental retardation. PLoS Comput Biol. 2010;6:e1000752. doi: 10.1371/journal.pcbi.1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777–784. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney HM, South ST, Wolff DJ, Lamb A, Hamosh A, Rao KW. American College of Medical Genetics recommendations for the design and performance expectations for clinical genomic copy number microarrays intended for use in the postnatal setting for detection of constitutional abnormalities. Genet Med. 2011;13:676–679. doi: 10.1097/GIM.0b013e31822272ac. [DOI] [PubMed] [Google Scholar]
- Köhler S, Schulz MH, Krawitz P, Bauer S, Dolken S, Ott CE, Mundlos C, Horn D, Mundlos S, Robinson PN. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destree A, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littenberg B, MacLean CD. Passive consent for clinical research in the age of HIPAA. J Gen Intern Med. 2006;21:207–211. doi: 10.1111/j.1525-1497.2006.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 2005. [PubMed] [Google Scholar]
- Mons B, van Haagen H, Chichester C, Hoen PB, den Dunnen JT, van Ommen G, van Mulligen E, Singh B, Hooft R, Roos M, et al. The value of data. Nat Genet. 2011;43:281–283. doi: 10.1038/ng0411-281. [DOI] [PubMed] [Google Scholar]
- Perez-Iratxeta C, Bork P, Andrade MA. Association of genes to genetically inherited diseases using data mining. Nat Genet. 2002;31:316–319. doi: 10.1038/ng895. [DOI] [PubMed] [Google Scholar]
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, Collins C, Kuo WL, Chen C, Zhai Y, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet. 1998;20:207–211. doi: 10.1038/2524. [DOI] [PubMed] [Google Scholar]
- Ramalingam A, Zhou XG, Fiedler SD, Brawner SJ, Joyce JM, Liu HY, Yu S. 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet. 2011;56:541–544. doi: 10.1038/jhg.2011.42. [DOI] [PubMed] [Google Scholar]
- Riggs ER, Church DM, Hanson K, Horner VL, Kaminsky EB, Kuhn RM, Wain KE, Williams ES, Aradhya S, Kearney HM, et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Mundlos S. The human phenotype ontology. Clin Genet. 2010;77:525–534. doi: 10.1111/j.1399-0004.2010.01436.x. [DOI] [PubMed] [Google Scholar]
- Sebold C, Graham L, McWalter K. Presented Abstracts from the Twenty-Eighth Annual Education Conference of the National Society of Genetic Counselors; November 2009; Atlanta, Georgia. 2009. pp. 622–691. [Google Scholar]
- Solinas-Toldo S, Lampel S, Stilgenbauer S, Nickolenko J, Benner A, Dohner H, Cremer T, Lichter P. Matrix-based comparative genomic hybridization: biochips to screen for genomic imbalances. Genes Chromosomes Cancer. 1997;20:399–407. [PubMed] [Google Scholar]
- Tsuchiya KD, Shaffer LG, Aradhya S, Gastier-Foster JM, Patel A, Rudd MK, Biggerstaff JS, Sanger WG, Schwartz S, Tepperberg JH, et al. Variability in interpreting and reporting copy number changes detected by array-based technology in clinical laboratories. Genet Med. 2009;11:866–873. doi: 10.1097/GIM.0b013e3181c0c3b0. [DOI] [PubMed] [Google Scholar]
- Ullmann R, Turner G, Kirchhoff M, Chen W, Tonge B, Rosenberg C, Field M, Vianna-Morgante AM, Christie L, Krepischi-Santos AC, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28:674–682. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]