

Abstract
Sympathetic stimulation enhances cardiac contractility by stimulating β-adrenergic signaling and protein kinase A (PKA). Recently, phospholemman (PLM) has emerged as an important PKA substrate capable of regulating cytosolic Ca2+ transients. However, it remains unclear how PLM contributes to β-adrenergic inotropy. Here we developed a computational model to clarify PLM's role in the β-adrenergic signaling response. Simulating Na+ and sarcoplasmic reticulum (SR) Ca2+ clamps, we identify an effect of PLM phosphorylation on SR unloading as the key mechanism by which PLM confers cytosolic Ca2+ adaptation to long-term β-adrenergic receptor (β-AR) stimulation. Moreover, we show phospholamban (PLB) opposes and overtakes these actions on SR load, forming a negative feed-forward loop in the β-adrenergic signaling cascade. This network motif dominates the negative feedback conferred by β-AR desensitization and accelerates β-AR-induced inotropy. Model analysis therefore unmasks key actions of PLM phosphorylation during β-adrenergic signaling, indicating that PLM is a critical component of the fight-or-flight response.
Keywords: phospholemman, Na+, β-adrenergic signaling, excitation-contraction coupling, negative feed-forward, computational model
Introduction
During the sympathetic fight-or-flight response, β-adrenergic receptor (β-AR) stimulation activates protein kinase A (PKA) to enhance cardiac inotropy and lusitropy [1]. The main PKA substrates responsible for these responses are the L-type Ca2+ channels (LCCs) and phospholamban (PLB), which regulate excitation-contraction (EC) coupling by increasing Ca2+ influx and increasing sarcoplasmic reticulum (SR) Ca2+ reloading, respectively [2]. PKA phosphorylates LCCs on both α and β subunits to both increase total current density and prolong LCC opening (Mode 2 gating). PLB phosphorylation releases inhibition of the SR Ca2+-ATPase (SERCA), increasing the rate of Ca2+ uptake into the SR during relaxation.
Recently, phospholemman (PLM) emerged as another PKA substrate capable of regulating cardiomyocyte Ca2+ during β-AR stimulation [3]. In the heart, PLM directly inhibits the Na+/K+-ATPase (NKA) [4]. PLM phosphorylation by PKA releases this inhibition, driving Na+ extrusion and indirectly augmenting Na+/Ca2+-ATPase (NCX) function via [Na+]i [5]. This is itself a clinically relevant element of β-adrenergic signaling as [Na+]i is elevated in heart failure [6] and PLM is phosphorylated during ischemia [7], identifying PLM as a candidate drug target [8]. Moreover, PLM is hypothesized to serve a protective role against arrhythmia by limiting the rise of intracellular Na+ and Ca2+ [5]. However, it remains unclear if PLM phosphorylation is central to β-adrenergic inotropy.
In this study, we construct a novel mechanistically detailed computational model of the mouse ventricular myocyte to quantitatively investigate the role of PLM in β-adrenergic regulation of Ca2+ handling and contractility. Because PLM is already identified as a key integrator of Na+ and Ca2+ in normal and failing cardiomyocytes [9], we hypothesized that PLM phosphorylation is critically important for the β-adrenergic signaling response. We asked the question, “how does PLM phosphorylation contribute to β-AR enhanced contractility?” Using the model, we show PLM forms a negative feed-forward loop with PLB and is necessary for producing rapid fight-or-flight responses.
Material and Methods
WT mouse ventricular myocyte model
During the cardiac action potential, membrane depolarization triggers the opening of L-type Ca2+ channels (LCCs), which in turn trigger the Ca2+ release from the SR through the ryanodine receptors (RyRs). Following this Ca2+-induced Ca2+-release (CICR), Ca2+ is resequestered by the SR Ca2+-ATPase (SERCA) and extruded from the myocyte by NCX [10]. CICR is a tightly regulated process, requiring local control and luminal Ca2+ sensitivity for maintaining graded Ca2+ release with high gain and rapid RyR refractoriness [11-12]. β-adrenergic signaling regulates CICR primarily through protein kinase A (PKA) phosphorylation of the LCCs (increasing Ca2+ influx) and PLB (releasing basal SERCA inhibition to further load the SR).
In order to quantitatively describe Ca2+ dynamics in the mouse ventricular myocyte accurately, we updated the Bondarenko, et al., model of mouse myocyte electrophysiology [13] with new descriptions for locally controlled CICR [14-16], luminal Ca2+ dependence of RyR gating [17], reversible SERCA activity [18-19] and cytosolic and SR Ca2+ buffering [18, 20]. In order to investigate β-adrenergic regulation of mouse EC coupling, we fully integrated a model of β1-adrenergic signaling [21-22]. This included PKA-mediated phosphorylation of LCC (increasing peak LCC current and prolonging LCC openings; Fig. S5C), PLB (increasing SERCA affinity for cytosolic Ca2+; Fig. S6A), PLM (increasing NKA affinity for cytosolic Na+; Fig. S6B) and troponin I (increasing troponin C affinity for cytosolic Ca2+). We also updated the NCX model [18] to better capture the interplay between Na+ dynamics and Ca2+ handling. Because EC coupling dynamics and β-adrenergic signaling dynamics vary over very different time scales (ms vs. min), we imposed ionic charge conservation on the stimulus current to overcome drift and help the model achieve stable steady-state behavior [23]. Detailed description of the equations, parameters and associated validations for this model are given in the Supplement. MATLAB (Mathworks, Natick, MA) code for this model is also included in the Supplement.
Transgenic myocyte models
In order to simulate Ca2+ dynamics in PLM knockout (PLM-KO) and PLB knockout (PLB-KO) myocytes, we modified the WT model by setting phosphorylated [PLM] or [PLB] to total [PLM] or [PLB[, negating their effects on NKA and SERCA, respectively. We further included relevant changes in gene expression to Na+ or Ca2+ handling proteins. For the PLM-KO case, this meant a 20% reduction in NKA expression [4]. For the PLB-KO case, this meant a 25% reduction in RyR expression [24]. In the case of the PLB/PLM double knockout, we applied both sets of modifications to the WT model. Validations for the PLM-KO and PLB-KO models are given in the Results (Figs. 2, 5).
Figure 2.
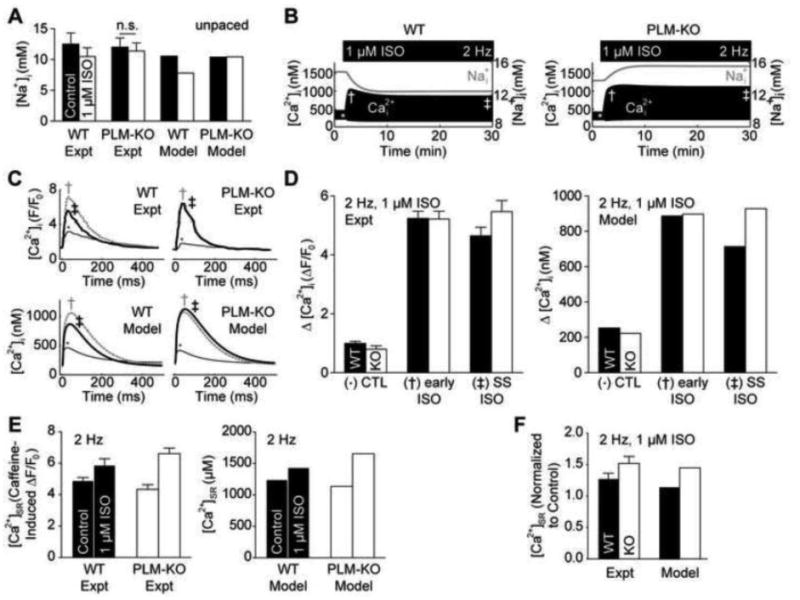
Model validation of PLM-KO cardiomyocytes using data from Despa et al. [4-5]. A, ISO stimulation reduces [Na+]i in resting WT, but not PLM-KO myocytes [4]. B, Cytosolic Ca2+ adaptation is uniquely present in the WT myocyte and coincides with the drop in intracellular Na+ [5]. C, Model Ca2+ transients from control (*), 2 min. early ISO stimulation (†) and 30 min. steady-state ISO stimulation (‡) are consistent with experimental findings [5]. D, Twitch Ca2+ amplitude decreases in the WT myocyte at steady-state, indicating Ca2+ adaptation [5]. E, ISO-stimulated SR load is larger in PLM-KO myocytes than WT myocytes [5]. F, ISO-stimulated SR loading is similar between model and experiment [5].
Figure 5.
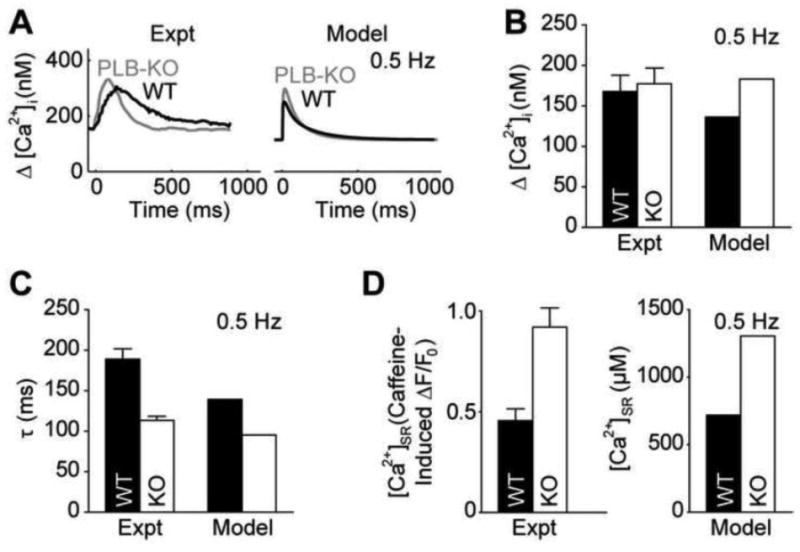
Model validation of PLB-KO cardiomyocytes. A, Model Ca2+ transients from WT and PLB-KO myocytes are qualitatively similar to experimental findings [27]. B, WT and PLB-KO Ca2+ transients have similar amplitudes [26]. C, PLB-KO myocytes exhibit accelerated relaxation [26]. D, PLB-KO myocytes exhibit significantly enhanced SR load [26].
Results
To quantitatively investigate the role of PLM in managing Ca2+ cycling under β-adrenergic signaling, we developed a new mechanistic computational model of Ca2+ dynamics in the mouse ventricular myocyte (Figs. 1, S2-S5). Because published models lack mechanistic descriptions for PLM phosphorylation and their downstream effects on intracellular Ca2+, we fully integrated a detailed model of β1-adrenergic signaling to simultaneously track PKA-mediated phosphorylation dynamics and functional effects on cardiomyocyte Ca2+ handling. This model serves as a suitable platform for studying the Na+/Ca2+ balance involved in PLM signaling.
Figure 1.
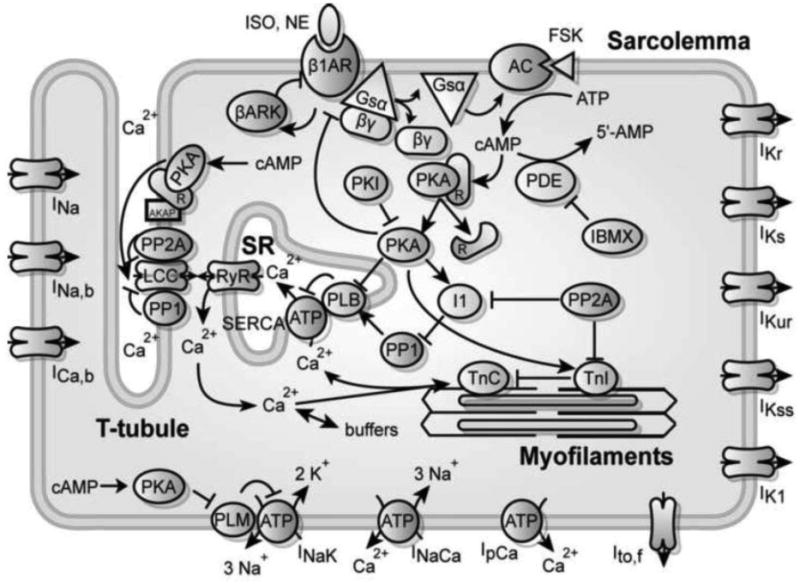
Schematic for the computational model of integrated β-adrenergic signaling and EC coupling in the mouse ventricular myocyte.
PLM confers adaptation to β-AR-stimulated Ca2+ transients
During the sympathetic fight-or-flight response, intracellular [Na+] is elevated by both increased Na+ channel firing frequency and enhanced Ca2+-driven Na+ influx via NCX [25]. Simultaneously, β-AR stimulation enhances Na+ efflux via PKA-mediated PLM phosphorylation. PLM phosphorylation stimulates NKA Na+ extrusion in a manner analogous to PLB enhancement of SERCA function – by releasing basal inhibition and decreasing the Km for intracellular Na+ [4]. In order to examine how β-AR stimulation coincidentally regulates Na+ and Ca2+, we derived new expressions for PKA phosphorylation of PLM and incorporated the effects of PLM on NKA into the integrated model (see Supplement). We then developed a PLM-KO version of this model (see Methods).
To validate the model, we simulated Na+ responses to the β-adrenergic agonist isoproterenol (ISO) in both resting (Fig. 2A) and 2 Hz paced (Fig. S7) wild-type (WT) and PLM-KO cells. Treatment with 1 μM ISO reduced resting [Na+]i in WT cells, but not in PLM-KO cells (Fig. 2A; WT: 10.5 mM control to 7.8 mM ISO-stimulated [Na+]I, PLM-KO: 10.4 mM control to 10.4 mM ISO-stimulated [Na+]i) [4]. Under 2 Hz pacing, [Na+]i increased for both WT and PLM-KO myocytes, but 1 μM ISO reversed this Na+ accumulation in WT cells only (Fig. S7; WT: 10.5 mM resting to 14.8 mM paced to 12.4 mM ISO-stimulated [Na+]i, PLM-KO: 10.4 mM resting to 13.8 mM paced to 15.5 mM ISO-stimulated [Na+]i) [5]. These results are consistent with the cited experimental data, demonstrating that the model accurately described Na+ dynamics.
Dynamically, we observed a [ Ca2+]i adaptation (the property of returning to a sub-maximal response following persistent biochemical stimulation) concurrent with the decline in [ Na+]i in simulated WT cells, but not in PLM-KO cells (Fig. 2B). This indicates a necessary role for PLM in conferring intracellular Ca2+ adaptation consistent with prior experiments [5]. Individual Ca2+ transients from unstimulated (*), 2 min early ISO-stimulated (†) and 30 min steady-state ISO-stimulated (‡) agreed with experimental measurements in both shape (Fig. 2C) and relative change in magnitude (Fig. 2D; WT: 253 nM control to 887 nM early ISO to 714 nM steady-state ISO twitch Ca2+ amplitude, PLM-KO: 223 nM control to 897 nM early ISO to 928 nM steady-state ISO twitch Ca2+ amplitude) for WT and PLM-KO myocytes [5]. Moreover, we also found similar increases in SR load to experimental observations (Figs. 2E and 2F; WT: 1242 μM control to 1435 μM steady-state ISO [ Ca2+]SR, PLM-KO: 1136 μM control to 1655 μM steady-state ISO [ Ca2+]SR) [5]. Thus, the model accurately predicts numerous aspects of Na+/Ca2+ handling and β-adrenergic regulation in both WT and PLM-KO myocytes.
Na+ manages β-AR-stimulated Ca2+ adaptation
Confident that the integrated model faithfully captured Na+ and Ca2+ dynamics, we interrogated the role of Na+ in managing Ca2+ adaptation by performing simulated intracellular Na+ clamp experiments (Fig. 3). First, we recorded the simulated normal Na+ transients from both WT and PLM-KO myocytes (Fig. 3A). We then repeated the 2 Hz pacing with 1 μM ISO simulations with WT and PLM-KO [ Na+]i clamped to the recordings from the opposite cell type. Switching the Na+ transients between WT and PLM-KO myocytes switched the ability of intracellular Ca2+ transients to adapt to ISO stimulation (Fig. 3B), giving direct evidence that PLM-mediated Ca2+ adaptation is managed via Na+. Clamping [ Na+]i to control concentrations (Fig. 3A; an intermediate between both ISO-stimulated conditions) prescribed an equivalent insignificant adaptation response (Fig. 3B). Together, these indicate that the PLM-mediated drop in intracellular Na+ is both sufficient and necessary to produce the Ca2+ adaptation observed in WT cells.
Figure 3.
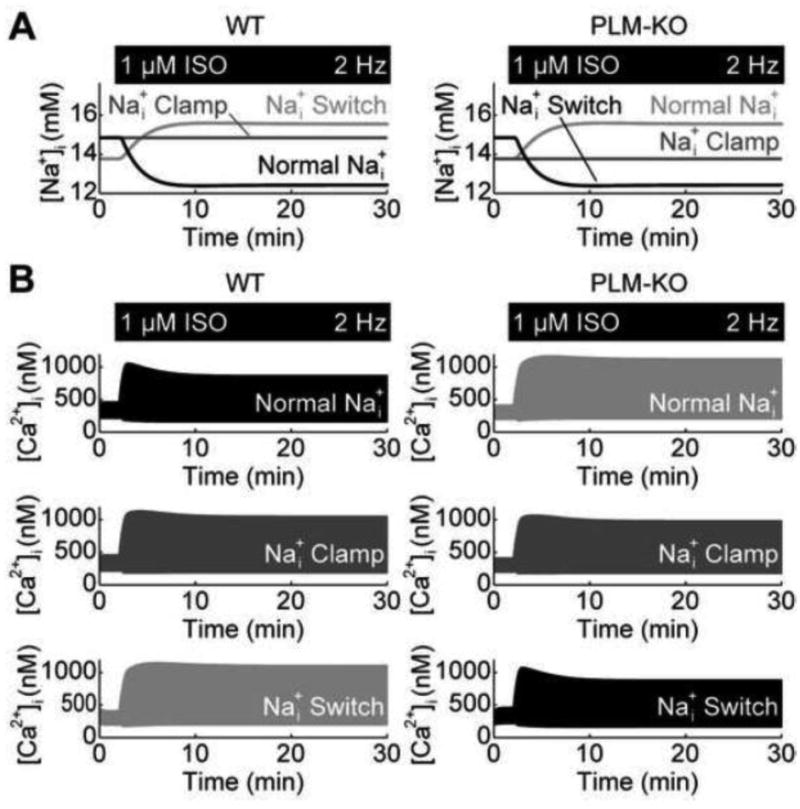
Role of Na+ in managing PLM-mediated Ca2+ adaptation. A, Na+ responses from simulated Na+ clamp experiments – ISO causes Na+ to decrease in WT myocytes but increase in PLM-KO myocytes B, Clamping Na+ in WT and PLM-KO myocytes to different Na+ transients indicate a necessary and sufficient role for Na+ in conferring Ca2+ adaptation, evidenced by switching of the Ca2+ adaptation phenomena between WT and PLM-KO myocytes.
Na+ regulates cytosolic Ca2+ by unloading SR Ca2+
To further understand how Na+ may regulate cytosolic Ca2+ dynamics, we investigated the effects of Na+ dynamics on SR load (Fig. 4). Concurrent with the adaptation in cytosolic Ca2+ transients, the model predicted an adaptation in SR Ca2+ in WT, but not PLM-KO cells (Fig. 4A). However, the correlation between adaptive cytosolic Ca2+ transients and SR Ca2+ alone does not indicate the direction of causality. Recording these SR Ca2+ dynamics and performing SR load switch-clamp simulations, we again observed switching of the cytosolic Ca2+ adaptation responses (Fig. 4B). These results give evidence that PLM-mediated Ca2+ adaptation to β-AR stimulation may be explained by Na+ indirectly unloading the SR.
Figure 4.

Role of SR Ca2+ load in managing PLM-mediated Ca2+ adaptation. A, SR Ca2+ loads for WT and PLM-KO myocytes. B, Switching SR load between WT and PLM-KO myocytes, switches the Ca2+ adaptation phenomena between WT and PLM-KO myocytes.
PLM opposes PLB-mediated SR Ca2+ loading
To further explore the role of SR unloading, we sought a system by which one could directly manipulate SR load. PLB-KO mice are one experimentally tractable way of achieving this. We therefore developed a PLB-KO version of our model (Fig. 5; see Methods). We validated this model by quantifying properties of SR load and cytosolic Ca2+ transients at rest and under 0.5 Hz pacing. At rest, PLB-KO myocytes had an SR load of 142 μM cytosol (vs. experimental 140 μM cytosol [26]). At 0.5 Hz pacing, cytosolic Ca2+ transients had similar amplitudes between WT and PLB-KO myocytes (Figs. 5A and 5B; WT: 136 nM twitch Ca2+ amplitude, PLB-KO: 183 nM twitch Ca2+ amplitude) [26-27]. We also observed faster relaxation in PLB-KO myocytes than WT control (Fig. 5C; WT: τWT 139.3 ms, PLB-KO: τKO =95.3 ms, 0.68 simulated τKO/ τWT vs. 0.60 experimental τKO/ τWT) [26]. Correspondingly, we also observed increased SR loads in PLB-KO myocytes (Fig. 5D; WT: 719 nM [ Ca2+]SR, PLB-KO: 1306 nM [ Ca2+]SR) [26]. Moreover, the simulated fraction of Ca2+ relaxation extruded by SERCA, NCX and IpCa was 94.0%, 5.7% and 0.3%, respectively, similarly biased to SERCA as in experimental measurements (96.4% SERCA, 3.4% NCX, 0.1% other [26]). Thus, the model captured fundamental features of Ca2+ handling in PLB-KO myocytes.
At 2 Hz pacing and under 1 μM ISO, the PLB-KO myocyte exhibited an adaptive response in the cytosolic Ca2+ transients in spite of the significantly elevated SR load (Fig. 6A, top). Upon further examination, we detected a decrease in SR load in PLB-KO myocytes when stimulated with ISO (Fig. 6A, bottom). This surprising result identified a hidden component of the β-adrenergic signaling response normally masked by PLB phosphorylation: SR unloading. To test if PLM phosphorylation was responsible for this SR unloading, we crossed the PLB-KO model with the PLM-KO model to derive a PLB-KO/PLM-KO mouse model (Fig. S8). In this double knockout simulation, cytosolic Ca2+ transients did not exhibit adaptation and SR Ca2+ content stayed elevated, indicating PLM phosphorylation underlied SR unloading (Fig. 6B).
Figure 6.
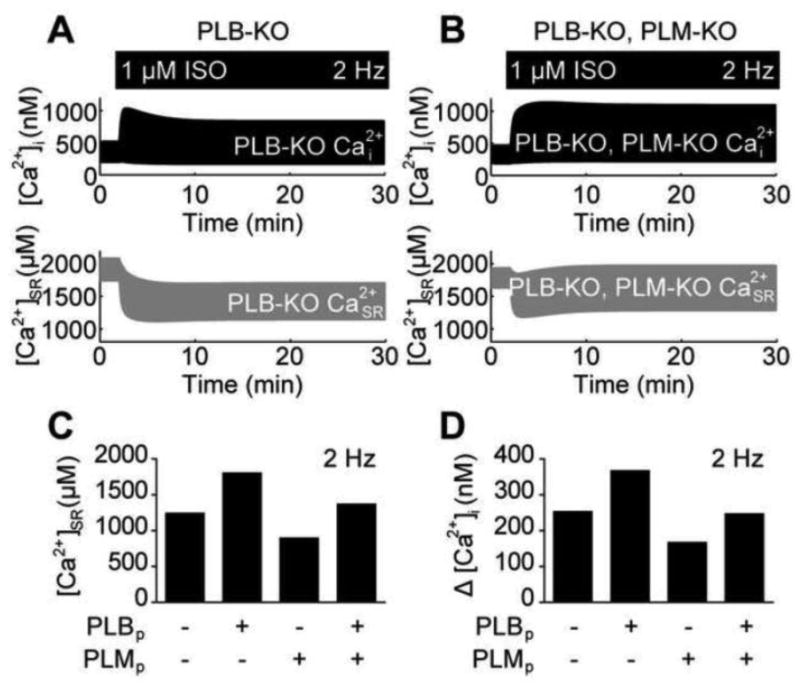
Opposing actions of PLB and PLM on EC coupling. A, PLB-KO myocytes also exhibit Ca2+ adaptation in spite of elevated SR load. SR load decreases with sustained β-AR stimulation. B, PLB-KO, PLM-KO myocytes do not exhibit Ca2+ exhibition. SR load does not decrease with sustained β-AR stimulation. C, PLB phosphorylation and PLM phosphorylation exert opposite effects on SR load. D, PLB phosphorylation and PLM phosphorylation exert opposite effects on cytosolic Ca2+ transients.
To further investigate the individual contributions of PLB and PLM to SR load under ISO stimulation, we simulated 2 Hz pacing, 1 μM ISO-stimulated responses in WT myocytes where PKA could only phosphorylate either PLB alone, PLM alone or both PLB and PLM only (Fig. 6C). When PLB alone was phosphorylated, the τ for [ Ca2+]i decline decreased to 113 ms (from 124 ms in unstimulated WT myocytes) and SR load increased to 1802 μM [ Ca2+]SR (from 1242 μM [ Ca2+]SR in unstimulated WT myocytes). This exceeded the steady-state SR load in fully stimulated WT myocytes (1435 μM [ Ca2+]SR). PLM phosphorylation alone also decreased the τ for [ Ca2+]i decline (to 118 ms), but in contrast to PLB phosphorylation alone, SR load decreased to 894 μM [ Ca2+]SR. When both PLB and PLM are phosphorylated (in the absence of LCC phosphorylation), the τ for [ Ca2+]i decline decreased to 100 ms and SR load reached 1369 μM [ Ca2+]SR. Looking at Ca2+ transient amplitudes, we observe similar relationships between these different PLB and PLM phosphorylation conditions (Fig. 5D), implying that cytosolic Ca2+ responses track with SR load. Together, these results demonstrate that while PLB and PLM both contribute to enhanced Ca2+ relaxation, PLB and PLM elicit opposite effects on both SR load and global Ca2+ dynamics.
Ca2+ adaptation is a negative feed-forward property of β-adrenergic signaling
In principle, only two network motifs are capable of giving rise to adaptation responses in a cell signaling network: negative feedback loops and negative feed-forward loops [28-29]. In the β-AR signaling pathway, both motifs are present (Fig. 1). First, β-ARs can be directly desensitized by both GRKs and PKA in a negative feedback loop [30] with significant implications for cardiac physiology [31]. Second, PKA phosphorylates both PLB and PLM, simultaneously increasing and decreasing total Ca2+ content. Both network motifs offer reasonable explanations for β-AR-stimulated Ca2+ adaptation.
To determine if the ISO-stimulated Ca2+ adaptation is a consequence of β-AR desensitization or PLM negative feed-forward control, we simulated 2 Hz pacing, 1 μM ISO-stimulated responses in WT myocytes with either PLM phosphorylation or β1-AR desensitization blocked, or both. Removing PLM phosphorylation by PKA significantly inhibited Ca2+ adaptation in both SR load (Fig. 7A) and cytosolic Ca2+ transients (Fig. 7B). Steady-state twitch Ca2+ amplitudes reached 988 nM [ Ca2+]i from 714 nM [ Ca2+]I in WT cells. Blocking β1-AR desensitization inhibited Ca2+ adaptation more weakly, with a steady-state twitch Ca2+ amplitude of 833 nM [ Ca2+]i. Blocking both PLM phosphorylation and β1-AR desensitization fully inhibited Ca2+ adaptation with a steady-state twitch Ca2+ amplitude of 1144 nM [ Ca2+]i. These results indicate that PLM phosphorylation accounts for most of the cytosolic Ca2+ adaptation.
Figure 7.
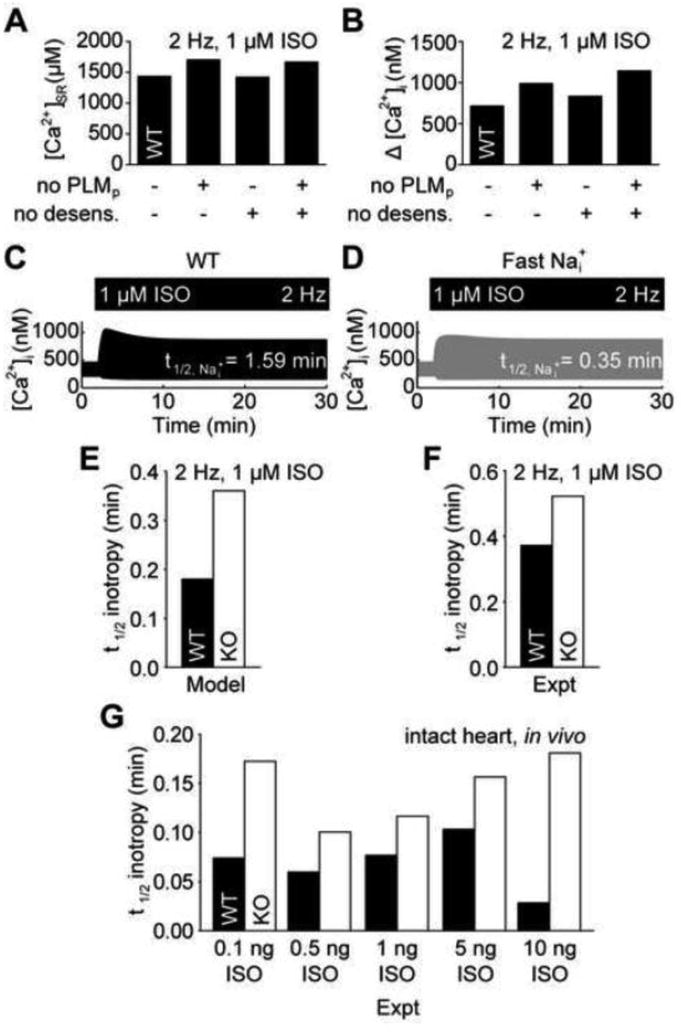
Ca2+ adaptation is explained by a negative feed-forward loop. A, Steady-state SR load is better explained by PLM phosphorylation than β-AR desensitization. B, PLM phosphorylation contributes more strongly limits cytosolic Ca2+ transients than β-AR desensitization. C, WT Ca2+ adaptation is associated with slow intracellular Na+ dynamics. D, Accelerating intracellular Na+ dynamics block Ca2+ adaptation in WT myocytes, indicating a negative feed-forward regulatory motif. E, The model predicts accelerated inotropic responses to ISO in WT myocytes over PLM-KO myocytes. F, Reanalyzed experimental data validates this model prediction in cardiac myocytes [5] and G, also indicates this acceleration occurs in the intact heart [33].
Adaptation in negative feed-forward loops requires a time delay between the fast positive signal transduction cascade and slow negative signal transduction cascade [29]. We therefore hypothesized that if the observed cytosolic Ca2+ adaptation is indeed a consequence of PLM-mediated negative feed-forward inhibition, then SR Ca2+ loading by SERCA (via PLB phosphorylation) must be fast with respect to Na+ extrusion by NKA (via PLM phosphorylation). Indeed, the t1/2 for SR Ca2+ loading was 0.36 min while t1/2 for Na+ extrusion was 1.59 min in ISO-stimulated WT cells (Fig. 7C). We then tested the hypothesis that accelerating Na+ extrusion (and therefore breaking the time delay between PLB- and PLM-mediated effects) would block the cytosolic Ca2+ adaptation. Indeed, increasing NKA activity to accelerate Na+ extrusion to the same rate as SR Ca2+ loading (t1/2 = 0.35 min) blocked Ca2+ adaptation to ISO stimulation (Fig. 7D). Together, these results offer strong evidence that Ca2+ adaptation is a negative feed-forward property of β-AR signaling, managed by PLM.
PLM phosphorylation accelerates β-adrenergic inotropy
In addition to providing mechanisms for adaptation, negative feed-forward loops are capable of accelerating cell signaling responses [32]. We hypothesized that PLM phosphorylation may be important for accelerating β-AR-stimulated inotropy. We measured the t1/2 for steady-state Ca2+ transient enhancement in the simulated WT and PLM-KO myocytes (Fig. 7E). Indeed, β-AR-stimulated inotropy was accelerated by 100% in WT myocytes over PLM-KO myocytes (WT: t1/2 = 0.18 min, PLM-KO: t1/2 = 0.36 min). We have not encountered any published reports of PLM-mediated acceleration of β-adrenergic inotropy, so we decided to experimentally validate this model prediction by reanalyzing published data. Digitizing and reanalyzing the work by Despa, et al. [5], we find 41% acceleration in their representative experiments of Ca2+ transients in isolated myocytes exposed to ISO (Fig. 7F; WT: t1/2 = 0.37 min, PLM-KO: t1/2 = 0.52 min). We further reanalyzed in vivo data from Wang, et al.[33], which measured the timecourse of left ventricular pressure in live WT and PLM-KO mice following serial injections of ISO. This analysis revealed an average 168% acceleration of left ventricular pressure inotropy in WT mice over PLM-KO mice across a 2-order of magnitude range of ISO concentrations (Fig. 7G). These results strongly support our hypothesis that PLM phosphorylation plays a central role in β-adrenergic signaling by accelerating steady-state inotropy in vitro and in vivo.
Discussion
The present study models the role of PLM phosphorylation in regulating cytosolic Ca2+ transients during β-adrenergic signaling. Extensive experimental work has demonstrated that PLM is important for managing intracellular Na+ and modulating EC coupling in normal and failing myocytes [4-6]. Here, we use a model to test our understanding of how PLM may regulate EC coupling. Model simulations indicate a necessary and sufficient role for Na+ to parlay PLM phosphorylation signals to the SR to confer long-term cytosolic Ca2+ adaptation to β-AR stimulation. The magnitude of this adaptation response cannot be explained by receptor-level negative feedback via β-AR desensitization, identifying PLM as an important negative feed-forward regulator of cytosolic Ca2+ (Fig. 8).
Figure 8.
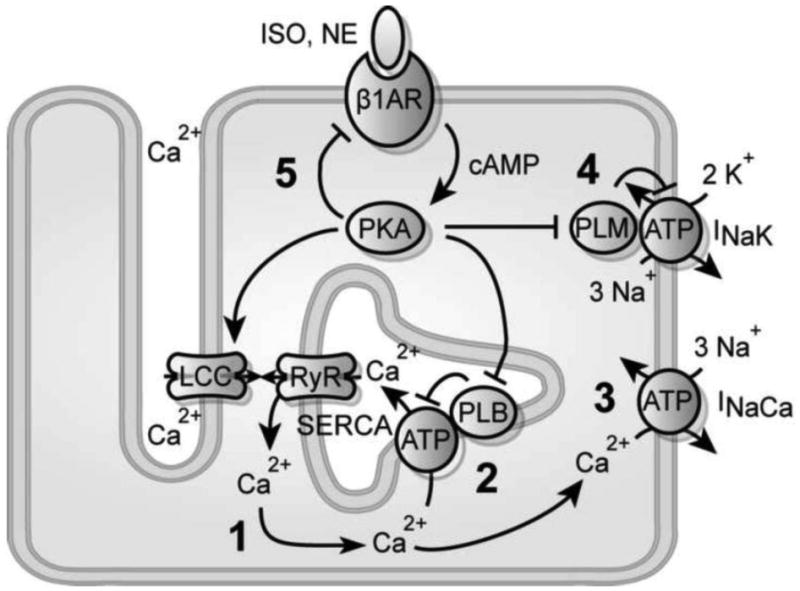
Overall schematic for PLM's role in β-adrenergic signaling. 1, SR Ca2+ load specifies the size of Ca2+ transients during CICR. 2, PLB increases SR load by biasing diastolic Ca2+ extrusion toward SERCA. 3, PLM decreases SR load by driving Na+ extrusion and biasing diastolic Ca2+ extrusion toward NCX. 4, Ca2+ adaptation occurs because Na+/K+ dynamics lag behind SERCA dynamics. 5, Receptor desensitization weakly contributes to Ca2+ adaptation, highlighting a negative feed-forward loop formed by PLB and PLM. This negative feed-forward network motif accelerates β-adrenergic inotropy.
PLM-mediated protection against spontaneous Ca2+ release
In the intact heart, β-adrenergic signaling simultaneously coordinates a number of events during the sympathetic fight-or-flight response [1]. While the net effect of these events is to enhance contractile function (via increased chronotropy, inotropy, lusitropy), persistent β-AR stimulation can itself drive cardiac pathologies. In addition to activating cardiac remodeling transcriptional programs, β-AR stimulation-induced chronotropy drives intracellular Na+ loading by accelerating Na+ entry with the increased frequency of myocyte depolarizations. Indeed, intracellular Na+ is elevated in the failing heart and may have important consequences on NCX function [6]. Despa, et al., hypothesized that PLM phosphorylation may play a protective role in the sympathetic fight-or-flight response by limiting the rise of intracellular Na+, thereby preventing Ca2+ overload and arrhythmic Ca2+ release [5]. Our results support this hypothesis, evidenced by the elevated [Na+]i and reduced [Ca2+]SR in our PLM-KO myocyte simulations (Fig. 2).
Interestingly, Despa, et al., observed an increased propensity for spontaneous ISO-stimulated Ca2+ transients in their PLM-KO experiments [5]. However, our model did not predict after-depolarizations in spite of the inclusion of a luminal RyR gating mechanism. There are a few possible explanations for this discrepancy. First, some evidence suggests that early after-depolarizations may be driven by stochastic LCC openings during β-AR stimulation [34], while our model is deterministic. Other evidence suggests RyR phosphorylation may lower the threshold for store overload-induced Ca2+-release by sensitizing luminal Ca2+ dependence for RyR gating [35]. But this mechanism is unclear as other evidence suggests luminal RyR gating may be protective against spontaneous Ca2+ release by accelerating Ca2+ regulation [17]. Our recent models have predicted that CaMKII-mediated RyR phosphorylation can play a key role in β-AR-induced delayed after-depolarizations [36]. However, these effects are not included here as we limited our study to focus on PKA substrate phosphorylation.
Systems understanding of PLM in β-adrenergic regulation of contractility
In the conventional understanding of β-adrenergic regulation of EC coupling, PKA phosphorylates many targets to collectively enhance cardiac inotropy and lusitropy. It is proposed that PLM may act as a cardiac stress protein that minimizes the risk of arrhythmogenesis at the expense of reduced inotropy [3]. Consistent with this proposition is our model prediction that PLB and PLM phosphorylation elicit opposite effects on SR load (Fig. 6C). Though these effects are similar in magnitude, release of PLB inhibition of SERCA overtakes release of PLM inhibition of NKA in regulating SR load, masking the PLM response. Simulations with PLB and PLM phosphorylation alone also unmask the relative effect of LCC phosphorylation. PLB and PLM are able to recapitulate most of the SR loading in ISO-stimulated WT cells (WT: 1435 μM [Ca2+]SR, PLB/PLM phosphorylation: 1369 μM [Ca2+]SR), but are unable to recapitulate the enhanced Ca2+ transients (WT: 725 nM Δ[Ca2+]i, PLB/PLM phosphorylation: 247 nM Δ[Ca2+]i).
From this perspective, the roles of LCC, PLB and PLM are more clearly defined. While Ca2+ influx through LCCs is thought to be the primary mechanism for regulating total cellular Ca2+ [37], our model suggests β-adrenergic increases in the total Ca2+ content in mouse are primarily explained by enhanced Ca2+ retention (via PLB phosphorylation and increased SERCA Ca2+ uptake), rather than enhanced Ca2+ influx. Moreover, enhancements to β-adrenergic Ca2+ transients are better explained by increased CICR (via LCC phosphorylation, increasing trigger Ca2+ flux to enhance RyR release), than by mere enhancements to sarcolemmal Ca2+ influx or global [Ca2+]. Indeed at 2 Hz pacing, 1 μM ISO increased steady-state RyR fractional release from 29.1% to 59.0%. However, when LCC phosphorylation was ablated, steady-state RyR fractional release only increased to 31.8%, highlighting the significance of high EC coupling gain under normal CICR (Fig. S2) and supporting the observation that β-adrenergic signaling enhances CICR by a local saturation of LCC trigger Ca2+ rather than an enhancement to SR load [38]. PLM moderates Ca2+ transients indirectly by enhancing NCX Ca2+ efflux, which reduces SR Ca2+ load and thus the extent of CICR.
If PLM acts to directly oppose PLB, what evolutionary advantage is gained by conserving this inefficient process? We show PLB and PLM form a negative (incoherent) feed-forward network motif [39], accelerating steady-state β-adrenergic inotropy (Fig. 7). This PLM-mediated acceleration was substantial in our model (100%) and reanalysis of published in vitro data by Despa, et al. [5] and in vivo data by Wang, et al. [33] validate this model prediction (Fig. 7). cAMP accumulation was previously shown to be a rate-limiting step in the β-adrenergic signaling response [40-41]. The current findings further elaborate this concept of pathway kinetics, showing that the downstream PLM feed-forward loop causes Ca2+ inotropy to reach steady state (t1/2 = 0.18 min) faster than the upstream cAMP (t1/2 = 0.41 min) and PKA (t1/2 = 0.33 min) signals. Thus paradoxically the β-adrenergic signaling pathway accelerates as the signal propagates downstream.
Moreover, the t1/2 for the WT myocyte in our model was 0.18 min shorter than the PLM-KO myocyte, implying these myocytes reach steady-state inotropy ∼40 beats faster at the cost of minor reductions in inotropy. This model prediction is only a lower-bound estimate since PKA also phosphorylates troponin I and myosin binding protein C to sensitize myofilaments to Ca2+ and accelerate stretch activation [42-43]. Together these indicate that in addition to its role in protecting against arrhythmia, PLM critically accelerates β-adrenergic signaling responses, overcoming slow cAMP and PKA dynamics to ensure a rapid fight-or-flight response. PLM simultaneously mediates both this acceleration and its anti-arrhythmic effects by adapting SR Ca2+ load.
Relevance to other species
Mouse EC coupling differs from EC coupling in other species with an increased heart rate and a more significant dependence on SERCA for Ca2+ relaxation. Mouse resting [Na+]i is also typically higher (10-15 mM) than that of other mammalian species (4-8 mM) [25], due to enhanced Na+ influx. In contrast, NKA function is similar across species and PLM phosphorylation induces similar enhancements to NKA activity [44]. While there are few published studies of PLM phosphorylation-mediated effects on Ca2+ handling in other species, it stands to reason that the results presented here would generalize to other species since NKA is the primary mechanism for Na+ extrusion in the cardiac myocyte. Because NCX more prominently regulates Ca2+ in human and other mammalian myocytes [2], PLM phosphorylation is expected to drive greater cytosolic Ca2+ unloading and greater Ca2+ adaptation in human than mouse, enhancing PLM-driven acceleration of β-adrenergic inotropy. Moreover, PLM phosphorylation may also have a stronger anti-arrhythmic role in human than mouse. New experimental work is needed to clarify the role of PLM in human cardiac Ca2+ handling.
Computational modeling of mouse EC coupling
Computational models have emerged as useful tools for interrogating cardiac signaling [45] and EC coupling [46]. There are several published computational models of the mouse ventricular myocyte [13, 47-51]. The present model improves upon existing mouse models by including mechanisms central to EC coupling and fully integrated descriptions of β-adrenergic signaling. Moreover, as Na+ dynamics are relevant to cardiac disease, our model is the first to explicitly represent Na+ regulation of Ca2+ transients. While this model is limited by variability in mouse strains and experimental data sources, this model captures many core components of cardiac Ca2+ handling, as evidenced by the ability to faithfully reproduce quantitative data from WT and two nontrivial transgenic knockout conditions.
One important component missing from this model is CaMKII regulation of EC coupling and frequency-dependent acceleration of relaxation (though the mechanism remains unknown [52]). However, for the purposes of this study we bounded the model at the level of PKA activation, as any model can always be improved without end. Indeed, the current model is already consistent with a wide range of experimental data. Modeling transgenic knockouts is also subject to gaps in knowledge of all the expression differences between knockout and WT – here we could only incorporate the primary known adaptive gene expression changes in PLM- and PLB-KO mice.
A second important consideration is that RyRs in the integrated CICR module can terminate Ca2+ release by Ca2+-dependent inactivation. While early work identified Ca2+-dependent inactivation as an important regulator of skeletal RyRs, there is little direct evidence to support this mechanism as a critical regulator of cardiac RyRs [53]. In the present model, steady-state Ca2+-dependent RyR inactivation is relatively minor (transitioning from 8.1% control to 19.6% under 1 μM ISO at 2 Hz pacing; Fig. S9). Adjusting RyR rate constants by one order of magnitude significantly reduced this fraction (transitioning from 2.3% control to 6.3% ISO) without significantly perturbing overall Ca2+ dynamics (Fig. S9), indicating Ca2+-dependent inactivation does not feature prominently to terminate Ca2+ release.
Finally, one global challenge in developing highly integrated models is finding the balance between capturing many behaviors under many conditions versus over-fitting available experimental data. For example, while model-predicted Fluo-3 F/F0s under-estimated the experimentally-measured F/F0s (Despa, et al. [5]) from ISO-stimulated myocyte (Fig. S10), simulated [Ca2+] varied qualitatively similar to experimental F/F0 (Fig. 2), indicating an over-estimate of diastolic Ca2+ (F0) in the model, which was constrained to fit experimental data from other studies. Computational models are therefore only useful when model predictions are within the boundaries set by model validation. In the present study we used separate data for model construction and model validation to give confidence for the model predictions. Indeed, while all of the data used for constructing this model was in vitro, the predicted β-adrenergic acceleration was independently validated by Wang, et al.,'s in vivo measurements [33]. Such validations help assign boundaries to a model's usefulness. Here, our model validations give supporting evidence that the present model is useful for simulating Ca2+ and Na+ dynamics in the mouse ventricular myocyte.
Conclusions
In summary, we have developed a new computational model of the mouse ventricular myocyte to investigate the role of PLM in regulating EC coupling responses to β-adrenergic signaling. Using this model, we have shown that PLM comprises a negative feed-forward loop with PLB, conferring both adaptation to cytosolic Ca2+ transients via Na+ effects on SR load and acceleration to β-AR-stimulated inotropy. In this way, PLM critically regulates the sympathetic fight-or-flight response.
Supplementary Material
Acknowledgments
Declaration of Funding Sources: This work was supported by the National Institutes of Health (HL094476 to J.S., Biotechnology Training Grant GM08715 to J.Y.) and the American Heart Association (0830470N to J.S., 0715283U to J.Y.).
Abbreviations
- β-AR
β-adrenergic receptor
- CaMKII
Ca2+/Calmodulin Protein Kinase II
- CICR
Ca2+-induced Ca2+-release
- ECC
excitation-contraction coupling
- ISO
isoproterenol
- LCC
L-type Ca2+ channel
- NCX
Na+/Ca2+-ATPase
- NKA
Na+/K+-ATPase
- PKA
protein kinase A
- PLB
phospholamban
- PLM
phospholemman
- RyR
ryanodine receptor
- SERCA
sarco-/endoplasmic reticulum Ca2+-ATPase
- SR
sarcoplasmic reticulum
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jason H. Yang, Email: jhyang@virginia.edu.
Jeffrey J. Saucerman, Email: jsaucerman@virginia.edu.
References
- 1.Saucerman JJ, McCulloch AD. Cardiac beta-adrenergic signaling: from subcellular microdomains to heart failure. Ann N Y Acad Sci. 2006 Oct;1080:348–61. doi: 10.1196/annals.1380.026. [DOI] [PubMed] [Google Scholar]
- 2.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2nd. Dordrecht; Boston: Kluwer Academic Publishers; 2001. [Google Scholar]
- 3.Cheung JY, Zhang XQ, Song J, Gao E, Rabinowitz JE, Chan TO, et al. Phospholemman: a novel cardiac stress protein. Clin Transl Sci. 2010 Aug;3(4):189–96. doi: 10.1111/j.1752-8062.2010.00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Despa S, Bossuyt J, Han F, Ginsburg KS, Jia LG, Kutchai H, et al. Phospholemman-phosphorylation mediates the beta-adrenergic effects on Na/K pump function in cardiac myocytes. Circ Res. 2005 Aug 5;97(3):252–9. doi: 10.1161/01.RES.0000176532.97731.e5. [DOI] [PubMed] [Google Scholar]
- 5.Despa S, Tucker AL, Bers DM. Phospholemman-mediated activation of Na/K-ATPase limits [Na]i and inotropic state during beta-adrenergic stimulation in mouse ventricular myocytes. Circulation. 2008 Apr 8;117(14):1849–55. doi: 10.1161/CIRCULATIONAHA.107.754051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002 May 28;105(21):2543–8. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 7.Fuller W, Eaton P, Bell JR, Shattock MJ. Ischemia-induced phosphorylation of phospholemman directly activates rat cardiac Na/K-ATPase. FASEB J. 2004 Jan;18(1):197–9. doi: 10.1096/fj.03-0213fje. [DOI] [PubMed] [Google Scholar]
- 8.Shattock MJ. Phospholemman: its role in normal cardiac physiology and potential as a druggable target in disease. Curr Opin Pharmacol. 2009 Apr;9(2):160–6. doi: 10.1016/j.coph.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 9.Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006 Oct;1080:165–77. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 10.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002 Jan 10;415(6868):198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 11.Cannell MB, Kong CH. Local control in cardiac E-C coupling. J Mol Cell Cardiol. 2011 May 14; doi: 10.1016/j.yjmcc.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 12.Gyorke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008 Jan 15;77(2):245–55. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 13.Bondarenko VE, Szigeti GP, Bett GC, Kim SJ, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004 Sep;287(3):H1378–403. doi: 10.1152/ajpheart.00185.2003. [DOI] [PubMed] [Google Scholar]
- 14.Greenstein JL, Hinch R, Winslow RL. Mechanisms of excitation-contraction coupling in an integrative model of the cardiac ventricular myocyte. Biophys J. 2006 Jan 1;90(1):77–91. doi: 10.1529/biophysj.105.065169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinch R, Greenstein JL, Tanskanen AJ, Xu L, Winslow RL. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys J. 2004 Dec;87(6):3723–36. doi: 10.1529/biophysj.104.049973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinch R, Greenstein JL, Winslow RL. Multi-scale models of local control of calcium induced calcium release. Prog Biophys Mol Biol. 2006 Jan-Apr;90(1-3):136–50. doi: 10.1016/j.pbiomolbio.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 17.Shannon TR, Wang F, Bers DM. Regulation of cardiac sarcoplasmic reticulum Ca release by luminal [Ca] and altered gating assessed with a mathematical model. Biophys J. 2005 Dec;89(6):4096–110. doi: 10.1529/biophysj.105.068734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004 Nov;87(5):3351–71. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000 Jan;78(1):322–33. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shannon TR, Bers DM. Integrated Ca2+ management in cardiac myocytes. Ann N Y Acad Sci. 2004 May;1015:28–38. doi: 10.1196/annals.1302.003. [DOI] [PubMed] [Google Scholar]
- 21.Saucerman JJ, Brunton LL, Michailova AP, McCulloch AD. Modeling beta-adrenergic control of cardiac myocyte contractility in silico. J Biol Chem. 2003 Nov 28;278(48):47997–8003. doi: 10.1074/jbc.M308362200. [DOI] [PubMed] [Google Scholar]
- 22.Saucerman JJ, McCulloch AD. Mechanistic systems models of cell signaling networks: a case study of myocyte adrenergic regulation. Prog Biophys Mol Biol. 2004 Jun-Jul;85(2-3):261–78. doi: 10.1016/j.pbiomolbio.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Hund TJ, Kucera JP, Otani NF, Rudy Y. Ionic charge conservation and long-term steady state in the Luo-Rudy dynamic cell model. Biophys J. 2001 Dec;81(6):3324–31. doi: 10.1016/S0006-3495(01)75965-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu G, Luo W, Slack JP, Tilgmann C, Sweet WE, Spindler M, et al. Compensatory mechanisms associated with the hyperdynamic function of phospholamban-deficient mouse hearts. Circ Res. 1996 Dec;79(6):1064–76. doi: 10.1161/01.res.79.6.1064. [DOI] [PubMed] [Google Scholar]
- 25.Bers DM, Barry WH, Despa S. Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res. 2003 Mar 15;57(4):897–912. doi: 10.1016/s0008-6363(02)00656-9. [DOI] [PubMed] [Google Scholar]
- 26.Li L, Chu G, Kranias EG, Bers DM. Cardiac myocyte calcium transport in phospholamban knockout mouse: relaxation and endogenous CaMKII effects. Am J Physiol. 1998 Apr;274(4 Pt 2):H1335–47. doi: 10.1152/ajpheart.1998.274.4.H1335. [DOI] [PubMed] [Google Scholar]
- 27.Santana LF, Kranias EG, Lederer WJ. Calcium sparks and excitation-contraction coupling in phospholamban-deficient mouse ventricular myocytes. J Physiol. 1997 Aug 15;503(Pt 1):21–9. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma W, Trusina A, El-Samad H, Lim WA, Tang C. Defining network topologies that can achieve biochemical adaptation. Cell. 2009 Aug 21;138(4):760–73. doi: 10.1016/j.cell.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Behar M, Hao N, Dohlman HG, Elston TC. Mathematical and computational analysis of adaptation via feedback inhibition in signal transduction pathways. Biophys J. 2007 Aug 1;93(3):806–21. doi: 10.1529/biophysj.107.107516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002 Sep;3(9):639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 31.Lefkowitz RJ, Rockman HA, Koch WJ. Catecholamines, cardiac beta-adrenergic receptors, and heart failure. Circulation. 2000 Apr 11;101(14):1634–7. doi: 10.1161/01.cir.101.14.1634. [DOI] [PubMed] [Google Scholar]
- 32.Mangan S, Itzkovitz S, Zaslaver A, Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J Mol Biol. 2006 Mar 10;356(5):1073–81. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Gao E, Song J, Zhang XQ, Li J, Koch WJ, et al. Phospholemman and beta-adrenergic stimulation in the heart. Am J Physiol Heart Circ Physiol. 2010 Mar;298(3):H807–15. doi: 10.1152/ajpheart.00877.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanskanen AJ, Greenstein JL, O'Rourke B, Winslow RL. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys J. 2005 Jan;88(1):85–95. doi: 10.1529/biophysj.104.051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao B, Tian X, Xie W, Jones PP, Cai S, Wang X, et al. Functional consequence of protein kinase A-dependent phosphorylation of the cardiac ryanodine receptor: sensitization of store overload-induced Ca2+ release. J Biol Chem. 2007 Oct 12;282(41):30256–64. doi: 10.1074/jbc.M703510200. [DOI] [PubMed] [Google Scholar]
- 36.Soltis AR, Saucerman JJ. Synergy between CaMKII Substrates and beta-Adrenergic Signaling in Regulation of Cardiac Myocyte Ca(2+) Handling. Biophys J. 2010 Oct 6;99(7):2038–47. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Benitah JP, Alvarez JL, Gomez AM. L-type Ca(2+) current in ventricular cardiomyocytes. J Mol Cell Cardiol. 2010 Jan;48(1):26–36. doi: 10.1016/j.yjmcc.2009.07.026. [DOI] [PubMed] [Google Scholar]
- 38.Song LS, Wang SQ, Xiao RP, Spurgeon H, Lakatta EG, Cheng H. beta-Adrenergic stimulation synchronizes intracellular Ca(2+) release during excitation-contraction coupling in cardiac myocytes. Circ Res. 2001 Apr 27;88(8):794–801. doi: 10.1161/hh0801.090461. [DOI] [PubMed] [Google Scholar]
- 39.Alon U. Network motifs: theory and experimental approaches. Nat Rev Genet. 2007 Jun;8(6):450–61. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- 40.Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, et al. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci U S A. 2006 Aug 22;103(34):12923–8. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frace AM, Mery PF, Fischmeister R, Hartzell HC. Rate-limiting steps in the beta-adrenergic stimulation of cardiac calcium current. J Gen Physiol. 1993 Mar;101(3):337–53. doi: 10.1085/jgp.101.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, et al. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001 May 25;88(10):1059–65. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 43.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006 Oct 13;99(8):884–90. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 44.Bers DM, Despa S. Na/K-ATPase--an integral player in the adrenergic fight-or-flight response. Trends Cardiovasc Med. 2009 May;19(4):111–8. doi: 10.1016/j.tcm.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang JH, Saucerman JJ. Computational models reduce complexity and accelerate insight into cardiac signaling networks. Circ Res. 2011 Jan 7;108(1):85–97. doi: 10.1161/CIRCRESAHA.110.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenstein JL, Winslow RL. Integrative systems models of cardiac excitation-contraction coupling. Circ Res. 2011 Jan 7;108(1):70–84. doi: 10.1161/CIRCRESAHA.110.223578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang LJ, Sobie EA. Mathematical model of the neonatal mouse ventricular action potential. Am J Physiol Heart Circ Physiol. 2008 Jun;294(6):H2565–75. doi: 10.1152/ajpheart.01376.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin SY, Choo SM, Woo SH, Cho KH. Cardiac systems biology and parameter sensitivity analysis: intracellular Ca2+ regulatory mechanisms in mouse ventricular myocytes. Adv Biochem Eng Biotechnol. 2008;110:25–45. doi: 10.1007/10_2007_093. [DOI] [PubMed] [Google Scholar]
- 49.Koivumaki JT, Korhonen T, Takalo J, Weckstrom M, Tavi P. Regulation of excitation-contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modelling. BMC Physiol. 2009;9:16. doi: 10.1186/1472-6793-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L, Niederer SA, Idigo W, Zhang YH, Swietach P, Casadei B, et al. A mathematical model of the murine ventricular myocyte: a data-driven biophysically based approach applied to mice overexpressing the canine NCX isoform. Am J Physiol Heart Circ Physiol. 2010 Oct;299(4):H1045–63. doi: 10.1152/ajpheart.00219.2010. [DOI] [PubMed] [Google Scholar]
- 51.Li L, Louch WE, Niederer SA, Andersson KB, Christensen G, Sejersted OM, et al. Calcium dynamics in the ventricular myocytes of SERCA2 knockout mice: A modeling study. Biophys J. 2011 Jan 19;100(2):322–31. doi: 10.1016/j.bpj.2010.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huke S, Bers DM. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J Mol Cell Cardiol. 2007 Mar;42(3):590–9. doi: 10.1016/j.yjmcc.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002 Oct;82(4):893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.