

Abstract
Although gene expression profiles in peripheral blood in major depression are not likely to identify genes directly involved in the pathomechanism of affective disorders, they may serve as biomarkers for this disorder. As previous studies using baseline gene expression profiles have provided mixed results, our approach was to use an in vivo dexamethasone challenge test and to compare glucocorticoid receptor (GR)-mediated changes in gene expression between depressed patients and healthy controls. Whole genome gene expression data (baseline and following GR-stimulation with 1.5 mg dexamethasone p.o.) from two independent cohorts were analyzed to identify gene expression pattern that would predict case and control status using a training (N=18 cases/18 controls) and a test cohort (N=11/13). Dexamethasone led to reproducible regulation of 2670 genes in controls and 1151 transcripts in cases. Several genes, including FKBP5 and DUSP1, previously associated with the pathophysiology of major depression, were found to be reliable markers of GR-activation. Using random forest analyses for classification, GR-stimulated gene expression outperformed baseline gene expression as a classifier for case and control status with a correct classification of 79.1 vs 41.6% in the test cohort. GR-stimulated gene expression performed best in dexamethasone non-suppressor patients (88.7% correctly classified with 100% sensitivity), but also correctly classified 77.3% of the suppressor patients (76.7% sensitivity), when using a refined set of 19 genes. Our study suggests that in vivo stimulated gene expression in peripheral blood cells could be a promising molecular marker of altered GR-functioning, an important component of the underlying pathology, in patients suffering from depressive episodes.
Keywords: dexamethasone, FKBP5, gene-expression, glucocorticoid receptor, major depression, RNA
INTRODUCTION
Affective disorders are predicted to become a leading cause of morbidity worldwide (Ustun et al, 2004). A number of approaches have been undertaken to identify biomarkers for these disorders to optimize diagnostic and treatment algorithms (Holsboer, 2008). One of them has focused on gene expression to integrate genomic activity and environmental effects (Gerhold et al, 2002). Most studies on transcriptional profiling in affective disorders have explored RNA derived from postmortem brain tissue (Mehta et al, 2010). However, agonal and postmortem factors may influence gene expression, and lacking access to brain tissue contradicts its use as biomarker (Tomita et al, 2004). Peripheral blood cells on the other hand are readily accessible. Although gene expression profiles in peripheral blood cells are not likely to reflect profiles in neuronal cell populations, there is an overlap between gene activity measures in brain and peripheral blood. Sullivan et al (2006) found a median correlation of 0.5 between transcripts expressed in both whole blood and the central nervous system and a study by (Rollins et al (2010) showed that over 4000 transcripts are co-expressed in blood cells and postmortem brain tissue. Additionally, glucocorticoids have effects on peripheral blood cells and the hypothalamus–pituitary adrenal (HPA) axis, which is perturbed during depressive episodes of many patients (Gladkevich et al, 2004). Transcriptional profiling in whole blood has been used in the search for biomarkers for patients with neurological and other psychiatric disorders including patients with Parkinson's (Scherzer et al, 2007) and Huntington's disease (Borovecki et al, 2005), post-traumatic stress disorder (Mehta et al, 2011; Segman et al, 2005; Yehuda et al, 2009), schizophrenia (Kurian et al, 2011), bipolar disorder (Le-Niculescu et al, 2009; Padmos et al, 2008) and major depression (Segman et al, 2010; Spijker et al, 2010). Most of these studies have analyzed baseline gene expression profiles.
Evidence from HPA axis activity measures in depression indicates the advantage of in vivo challenge tests such as the dexamethasone-suppression test (DST) or the combined dexamethasone/corticotropin-releasing hormone (dex/CRH) test over single baseline measurements of peripheral cortisol or ACTH concentration to discriminate between depressed patients and healthy controls (Holsboer, 2000). Although not a specific test to identify patients that fulfilll diagnostic criteria according to current algorithms, it is of interest that the HPA axis dysregulation, including glucocorticoid receptor (GR) resistance is one of the endocrine hallmarks of mood disorders (Holsboer, 2000; Pariante and Miller, 2001b). The GR resistance proposed to underly this phenomenon has also been observed at the level of peripheral blood cells. Reduced GR function in leukocytes of depressed patients, as exemplified by a decreased nuclear translocation following dexamethasone exposure (Gormley et al, 1985) as well as a decreased capacity of dexamethasone to inhibit the ability of peripheral blood mononuclear cells to proliferate in response to mitogens were found (Lowy et al, 1988). In fact, the partial glucocorticoid resistance found in peripheral immune cells correlate with the partial glucocorticoid resistance found in the DST (Wodarz et al, 1992).
GR resistance is thus apparent in peripheral blood cells of depressed patients and glucocorticoids lead to profound and reproducible gene expression changes (Czock et al, 2005). We therefore hypothesized that GR-stimulated gene expression would be different in in-patients with moderate to severe depressive symptoms as compared with healthy controls. A recent study by Spijker et al (2010) showing gene expression differences between cases and controls using an ex vivo lipopolysaccharide (LPS) challenge test supports the notion that stimulated gene expression measures are better at discriminating between patients and controls than baseline measures.
To test whether reliable case/control differences can be identified following GR activation, we compared gene expression differences in whole blood and serum cortisol and ACTH in two independent cohorts of 18 and 11 depressed patients and 18 and 13 controls before and 3 h after ingestion of 1.5 mg of dexamethasone. Only male subjects were used to reduce the known confound of sex-steroids on GR activation (Young and Korszun, 2010).
MATERIALS AND METHODS
Patient Recruitment
We recruited 29 male patients aged 18–65 years who were admitted as inpatients to the Max Planck Institute of Psychiatry (MPI), Munich, Germany, for treatment of a depressive episode. According to the time of enrollment patients were assigned to two separate cohorts, with the discovery and the replication cohort (18 out of 11 patients). All patients were Caucasian. They were part of the Munich-Antidepressant-Response-Signature (MARS) project (www.mars-depression.de) (Ising et al, 2009) and were treated with antidepressant medications according to doctor's choice. As carbamazepine is associated with HPA-axis alterations patients treated with carbamazepine were excluded (Kunzel et al, 2003). Severity of depressive symptoms was assessed at admission by trained raters using the 21-item Hamilton Depression Rating Scale (HAM-D). Patients fulfilling the criteria for at least a moderate depressive episode (HAM-D⩾14) were eligible. Patients with severe neurological or general medical conditions were excluded. Blood was taken to measure inflammation markers, liver enzymes, renal function and coagulation, vital signs as blood pressure and pulse were monitored. The study was approved by the ethics committee of the Ludwig-Maximilans-University in Munich, and written informed consent was obtained from all subjects (Nr. 244/01).
Recruitment of Controls
The control samples consisted of 18 and 13 male subjects respectively, matched for age and ethnicity and were recruited at the MPI and were and screened for the presence of any axis I disorder using the Composite International Diagnostic Interview (CIDI) (Wittchen et al, 1999). Only individuals free of a history of psychiatric disorders as well as major neurological and general medical disorders were included in the sample, which was ensured by examination of the same laboratory measures and monitoring of vital signs as in patients.
Study Design and Blood Collection
Unstimulated peripheral blood samples were obtained at 1800 h after 2 h of fasting and abstention from coffee and physical activity, followed by oral administration of 1.5 mg dexamethasone. Additional blood samples were drawn at 2100 h. At each timepoint we collected whole blood RNA using a PAXgene (PreAnalytiX GmbH, Hombrechtikon, Switzerland) whole blood RNA collection tube, as well as serum and whole blood to analyze differential blood count, cortisol and ACTH levels as well as liver enzymes. PAXgene tubes were stored for 2.5 h at room temperature, and then transferred to -20 °C. For additional cortisol and ACTH measurements, blood was drawn at 1500 h the next day. Dexamethasone-suppressor status was defined by the cortisol value at 1500 h the day after cortisol and values below 50 ng/ml were considered suppressors and value ⩾50 ng/ml non-suppressors (Ising et al, 2007). In the same samples plasma dexamethasone concentration were assessed using LC-MSMS on an API4000 (AB Sciex).
RNA Extraction and Quality Assessment
Total RNA was isolated from PAXgene whole blood samples according to the manufacturer's instructions. The samples were processed using the PAXgene Blood RNA Kit using the Qiagen method for column purification of nucleic acids (PreAnalytiX GmbH). The quality of the total RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). The concentration and purity of total RNA was also assessed by 260 nm UV absorption and by 260/280 ratios, respectively (Nanophotometer, Implen, Munich, Germany). All samples had a RNA integrity number ⩾7 (7.0–8.9, mean 8.0±SD 0.4). Globin reduction was not applied as a technical note by Illumina suggests no benefits of globin reduction when using Illumina BeadChip expression microarrays (Expression Analysis, 2007). We could confirm this in our own pilot study comparing globin depleted and non-depleted samples from the same RNAs (Menke et al, 2011). Moreover, globin reduction protocols may lead to a loss of reproducibility and introduce other artefacts, which may interfere with gene expression data analysis (Dumeaux et al, 2008; Mehta, 2008).
Processing of Microarrays
Sample labeling was performed using the Illumina TotalPrep RNA Amplification Kit (Ambion, TX, USA, catalog number AMIL1791). Biotin-labeled cRNA (1.5 μg) was hybridized to HumanHT-12 v3 Expression BeadChips (Illumina, San Diego, CA, USA), representing a single batch of arrays. Microarrays were scanned with the Bead Array Reader (Illumina). All processing was done according to the manufacturer's instructions.
Validation and Replication with Quantitative Real-Time PCR
Total RNA was reverse-transcribed to cDNA using random primers and the Superscript II reverse transcriptase (Invitrogen) for quantitative real-time PCR (qPCR) to validate microarray results in cohorts 1 and 2 FKBP5, BEST1 and CEACAM4 were chosen for RT-PCR validation with PLA and TBP as the endogenous control genes. The first two target genes show both regulation with dexamethasone as well as differences between cases and controls and the last gene a consistent regulation by dexamethasone in all experiments. qPCR experiments were performed using the Roche 480 LightCycler (Roche Applied Science). qPCR assays were designed using the Roche universal probe library (http://qpcr.probefinder.com/organism.jsp) (Supplementary Table 1). All samples were run in duplicates and duplicates discordant in crossing points by more than 0.4 cycles, were excluded from the analysis.
Statistical Analysis
Descriptive statistics of the sample as well as the discriminant analysis using post-dexamethasone cortisol values were run using SPSS (release 16, SPSS, Chicago, Illinois, USA). For the repeated measures ANOVAs, the statistics are reported for the Greenhouse-Geisser test.
Microarray expression analysis was performed in R(R Development Core Team, 2007), making use of the packages: beadarray (providing routines to handle Illumina BeadStudio data)(Dunning et al, 2007) and vsn (for normalization, available from BioConductor (http://www.bioconductor.org/) (Huber et al, 2002).
Raw microarray scan files were exported using the Illumina Beadstudio program and loaded into R for downstream analysis. The data were first filtered by a detection P-value of 0.01 in at least five individuals and secondly, transformed and normalized using the variance stabilizing normalization method. From a total of 48 750 transcripts 15 573 remained in the analysis having significant expression according to these criteria. Pre- and post-dex samples from the training and the test cohort were processed within a single run each (that is, RNA amplification, hybridization and scanning). To further reduce the described batch effects for Illumina arrays (Kitchen et al, 2010) pre- and post-dex RNAs were hybridized to the same chip for each individual, and cases and controls were randomized across arrays and arrays positions.
For differential expression analysis linear regression models implemented in R were used on the normalized log2-transformed expression values at 1800 h, 2100 h as well as their difference comparing cases and controls.
The expression data were displayed as a heatmap using the gplots package implemented in R.
Classifications were performed with the Random Forest (RF) classification technique (Liaw and Wiener, 2002), which is a combination of tree predictors. A main advantage in RF is that there is no need for cross-validation because the training data is divided into two independent partitions. About two-thirds of the training set are used to create an in-bag partition to construct the tree and one-third, which is not used in the construction of the tree, is used as an out-of-bag (OOB) partition to test the constructed tree to evaluate the performance. All the trees vote to determine the prediction results and an OOB estimate of error rate is calculated.
The RF provides the mean decrease Gini as an importance measure that calculates the quality of a split for every node of a tree by means of the Gini Index.
Each time a split of a node is made on a variable, the Gini criterion for the two descendent nodes is less than the parent node. A higher mean decrease Gini represents a higher variable importance (Breiman, 2001; Ozcift, 2011).
RESULTS
Sociodemographic and Clinical Differences Between Groups
Sociodemographic and clinical variables of the two cohorts are listed in Table 1. All clinical variables including severity of depression, measured with the HAM-D, number of previous episodes, previous suicide attempts or family history of depression did not show significant differences between patients of cohorts 1 and 2. All patients were medicated and there were no significant differences in the type of medication between the two cohorts.
Table 1. Sociodemographic and Clinical Characteristics of Cohort 1 and 2.
|
Cohort 1 |
Cohort 2 |
P-value | |||
|---|---|---|---|---|---|
| Cases | Controls | Cases | Controls | ||
| n=18 | n=18 | n=11 | n=13 | ||
| Age (mean, SD) | 44.2 (14.3) | 43.2 (10.8) | 50.8 (15.0) | 37.7 (10.2) | 0.075 |
| BMI | 24.5 (3.0) | 25.3 (3.5) | 26.1 (2.5) | 24.7 (4.5) | 0.612 |
| Age at onset | 33.3 (13.3) | 30.1 (13.2) | 0.548 | ||
| HAMD | 25.8 (7.9) | 23.9 (9.8) | 0.573 | ||
| Bipolar disorder | 5 (28%) | 2 (18%) | 0.649 | ||
| Previous episodes | 2.7 (3.7) | 4.6 (9.6) | 0.486 | ||
| Recurrent depression | 13 (72%) | 6 (55%) | 0.331 | ||
| Previous suicide attempts | 3 (17%) | 4 (36%) | 0.172 | ||
| Family history | 9 (50%) | 7 (64%) | 0.306 | ||
| Response, 5 weeks | 9 (50%) | 5 (45%) | 0.306 | ||
| Remission, 5 weeks | 7 (38%) | 5 (45%) | 0.125 | ||
| Medication at RNA withdrawal | |||||
| TCA | 4 (22%) | 1 (9%) | 0.418 | ||
| SSRI | 2 (11%) | 4 (36%) | 0.074 | ||
| SNRI | 7 (39%) | 6 (55%) | 0.283 | ||
| NaSSA | 6 (33%) | 2 (18%) | 0.454 | ||
| SSRE | 1 (6%) | 0 | 0.448 | ||
| Antipsychotics | 6 (33%) | 3 (27%) | 0.856 | ||
| Mood stabilizer | 5 (28%) | 2 (18%) | 0.649 | ||
| Lithium | 2 (11%) | 1 (9%) | 0.927 | ||
| Benzodiazepine | 8 (44%) | 8 (73%) | 0.069 | ||
Abbrevations: HAMD, Hamilton Depression Rating Scale; NaSSA, noradrenergic and specific serotonergic antidepressant; SNRI, serotonin and noradrenaline reuptake inhibitor; SSRE, selective serotonin reuptake enhancer; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressants.
P-values of Pearson χ2 tests (qualitative data) and univariate analysis of variance (quantitative data) are reported.
Dexamethasone Effect on Cortisol and ACTH Levels
Cortisol levels were significantly suppressed in healthy controls (n=31; 75.3 ng/ml±4.8 SEM; 1800 h vs 25.4 ng/ml±1.2 SEM; 2100 h; P<0.001) and in depressed patients (n=29; 80.7 ng/ml±5.6 SEM vs 31.5 ng/ml±2.3 SEM; P<0.001) 3 h after dexamethasone challenge. At 1500 h the next day cortisol levels were significantly higher in patients compared with controls (controls: 20.9 ng/ml±3.9 SEM vs cases: 37.7 ng/ml±7.1 SEM; P=0.039) indicating relative GR resistance in depressed patients (see Figure 1a). Similar findings were observed for ACTH, with a significant reduction in ACTH levels in healthy controls (12.5 ng/ml±0.7 SEM vs 0.1 ng/ml±0.1 SEM; P<0.001) and in depressed patients (14.6 ng/ml±1.0 SEM vs 0.5 ng/ml±0.1 SEM; P<0.001) after 3 h.
Figure 1.
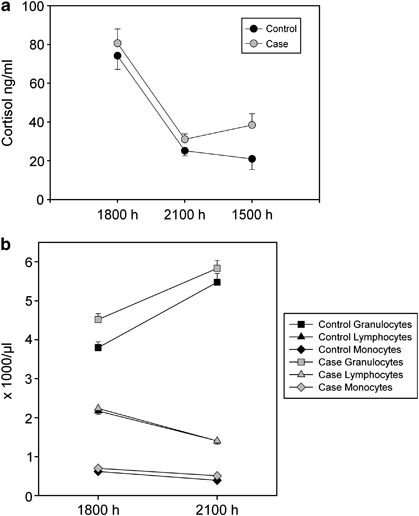
Alterations in cortisol and blood count of depressed cases and healthy volunteers: (a) dexamethasone intake resulted in a significant suppression of cortisol in healthy volunteers (n=31) and depressed patients (n=29) after 3 h (P<0.001). Cortisol levels at 1500 h the next day were significantly increased in patients compared with controls (P=0.039). (b) Dexamethasone led to a significant increase in granulocytes in cases (n=29; P<0.001) as well as controls (n=31; P<0.001). Lymphocytes and monocytes were significantly reduced by dexamethasone in both groups (P<0.001). When comparing healthy controls vs depressed cases, there was significantly less upregulation of granulocytes in cases (P=0.05) as well as baseline differences in granulocytes (P=0.067) and monocytes (P<0.001).
Differential Blood Cell Count
At baseline, patients had significantly higher levels of granulocytes and monocytes than controls (F=11.771, P<0.001; F=5.559, P=0.02, respectively). Repeated measures ANOVA showed a significant dexamethasone-induced increase in the number of granulocyte, (P<0.001) although there was a significant reduction in lymphocytes and monocytes (P<0.001; see Figure 1b) in both cases and controls. There was a significant interaction between the change in granulocyte number from 1800 h to 2100 h and disease status (F=3.933; P=0.05), indicating a smaller relative change in cases.
Dexamethasone Effect on mRNA-Expression in Patients and Controls
In controls dexamethasone led to the overlapping regulation of 2670 transcripts in both cohorts 1 and 2 at P-value ⩽0.05 of which 1132 probes or 42% were upregulated (see Supplementary Table 2). In depressed patients only 1151 transcripts were significantly regulated in both cohorts of which 507 probes or 44% were upregulated (see Supplementary Table 3). In all, 23% of the significant dexamethasone-induced transcripts level changes observed in patients were also observed in controls.
Using Gene Expression and Endocrine Measures to Classify Cases vs Controls
Classification Using Plasma Cortisol
Using the cortisol levels at 1500 h the next day, which showed the most significant difference of all the endocrine parameters between cases and controls, 60.0% could be correctly classified in a discriminant analysis, with a specificity of 83.9% and a sensitivity of only 34.5%. Discriminant analysis showed: Wilk's λ=0.929; χ2=4.250; df=1; P=0.039; canonical correlation coefficient: 0.267.
Classification Using Gene Expression Profiles
Classification was performed with the RF algorithm using cohort 1 to train the model and cohort 2 for testing.
Baseline gene expression
We first performed a feature selection by including those transcripts that showed a significant difference in baseline gene expression between cases and controls in cohort 1 at P⩽0.05 (uncorrected) and a fold change of absolute ⩾1.15. This resulted in 635 probes that were used for the RF algorithm, which was run with the best performing parameters (1000 trees (ntree) and 25 random features to build each tree (mtry)). This revealed an OOB error rate of 27.8% to classify cases vs controls in cohort 1 (with a sensitivity of 72.2% and a specificity of 72.2%). In cohort 2, however, the constructed prediction model only achieved an area under the curve (AUC) value of 0.56 with a classification rate of 10 out of 24 (41.7%).
B-GR stimulated gene expression
For feature selection for stimulated gene expression we only kept those transcripts that showed a difference in gene expression change from 1800 h to 2100 h between cases and controls at a P-value ⩽0.05 and an absolute fold change following dexamethasone in controls ⩾1.15. This resulted in 250 transcripts selected for the RF algorithm. For this analysis the best performing parameters were: ntree=1500 trees and mtry=240. Using these, RF revealed an OOB error rate of 16.7% to differentiate between cases vs controls in cohort 1 (with a sensitivity of 80% and a specificity of 87.5%). In cohort 2, an AUC value of 0.73 and a test set classification rate of 19 out of 24 (79.2%) was obtained, thus performing much better than the predictors built from baseline gene expression.
Reduction of the number of transcripts for classification
To identify the genes that contributed most to the classification in both cohorts, RF classification was performed without feature selection in all individual together, not separated by cohorts. The analysis was repeated 10 times and the importance scores (Gini index) were averaged. Out of the 206 most important features (with an average Gini index in the 10 repetitions ⩾0.02) and representing the top 1.3% transcripts, 19 transcripts also showed a significant (P⩽0.05 uncorrected) differences in dexamethasone-regulation between cases and controls in both cohorts (see Table 2 and Figure 2).
Table 2. (A) Differentially Expressed mRNA Expression at Baseline with Nominal P<0.05 in Both Cohorts and FC>1.15. (B) Differentially Regulated mRNA Expression of the Ratio Post/prestimulated mRNA with P<0.05 in Both Cohorts, FC>1.15 (in controls) and Alteration in the Same Direction of the 19 Transcripts Which Contributed Most to the Classification.
| (A) Symbol | Probe ID |
Cohort 1 |
Cohort 2 |
||
|---|---|---|---|---|---|
| FC case vs control | P-value | FC case vs control | P-value | ||
| Transcripts differentially expressed at baseline | |||||
| GPR114 | ILMN_1666902 | −1.18 | 0.036 | −1.23 | 0.02 |
| ICA1 | ILMN_2365569 | 1.18 | 0.047 | 1.18 | 0.02 |
| NCBP1 | ILMN_1777528 | −1.17 | 0.011 | −1.21 | 0.03 |
| P2RY2 | ILMN_2372915 | −1.22 | 0.035 | −1.41 | 0.00 |
| TPST1 | ILMN_1651950 | 1.26 | 0.050 | 1.39 | 0.03 |
| (B) | Cases | Controls | Cases | Controls | |||
|---|---|---|---|---|---|---|---|
| Symbol | ProbeID | FC | FC | Delta p | FC | FC | Delta p |
| Transcripts differentially expressed after dexamethasone stimulation | |||||||
| ARG1 | ILMN_1812281 | 1.70 | 2.09 | 0.03 | 1.56 | 2.04 | 0.003 |
| BEST1 | ILMN_1718982 | 1.33 | 1.62 | 0.003 | 1.43 | 1.80 | 0.01 |
| CD14 | ILMN_1740015 | −1.25 | −1.44 | 0.01 | −1.06 | −1.32 | 0.037 |
| CKLF | ILMN_1712389 | 1.15 | 1.30 | 0.039 | 1.24 | 1.48 | 0.042 |
| CPVL | ILMN_2400759 | −1.34 | −1.59 | 0.04 | −1.11 | −1.53 | 0.005 |
| FAM129B | ILMN_1661755 | −1.16 | −1.43 | 0.00 | −1.24 | −1.58 | 0.005 |
| FKBP5 | ILMN_1778444 | 4.78 | 6.32 | 0.03 | 5.16 | 6.93 | 0.031 |
| FLJ20699 | ILMN_1692464 | 1.01 | 1.19 | 0.01 | 1.02 | 1.19 | 0.019 |
| GNA15 | ILMN_1773963 | −1.11 | −1.21 | 0.02 | −1.05 | −1.25 | 0.015 |
| LAT2 | ILMN_2326953 | 1.18 | 1.33 | 0.01 | 1.37 | 1.61 | 0.031 |
| MUM1 | ILMN_1764764 | −1.09 | −1.19 | 0.04 | −1.13 | −1.27 | 0.007 |
| P2RY2 | ILMN_2372915 | −1.02 | −1.18 | 0.02 | −1.08 | −1.25 | 0.002 |
| RNF144B | ILMN_1752526 | 1.48 | 1.70 | 0.03 | 1.43 | 1.72 | 0.002 |
| RUNX1 | ILMN_1801504 | −1.08 | −1.17 | 0.03 | −1.05 | −1.20 | 0.023 |
| SELL | ILMN_1724422 | 1.22 | 1.40 | 0.02 | 1.33 | 1.68 | 0.024 |
| SOCS1 | ILMN_1774733 | 1.43 | 1.58 | 0.04 | 1.24 | 1.40 | 0.017 |
| SSH2 | ILMN_1672834 | 1.47 | 1.74 | 0.01 | 1.62 | 1.99 | 0.04 |
| TMEM176A | ILMN_1791511 | −1.08 | −1.22 | 0.04 | −1.10 | −1.27 | 0.019 |
| ILMN_1880406 | 1.42 | 1.67 | 0.05 | 1.44 | 1.65 | 0.047 | |
Abbrevations: FC, Fold change of dexamethasone induced gene expression in healthy controls and depressed patients. Probe ID, Probe ID of Illumina HumanHT-12 expression BeadChips.
Figure 2.
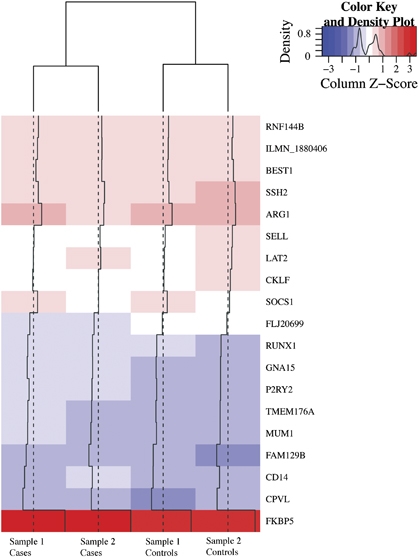
This heatmap illustrates the gene expression changes (poststimulation/prestimulation mRNA levels) of the 19 significant differentially regulated genes between cases and controls. Red indicates an upregulation following dexamethasone and blue a downregulation.
Performance of the reduced set of classifiers in dexamethasone suppressor vs dexamethasone non-suppressor patients
For this analysis, four controls who were dexamethasone non-suppressors were excluded from the analysis. Using the above-described 19 best-performing transcripts to classify controls vs dexamethasone suppressor cases (N=27/21), RF estimated an OOB error rate of 22.7% to differentiate between the groups (with a sensitivity of 76.7% and a specificity of 77.8%). To repeat the analysis with dexamethasone non-suppressor patients, we had to balance the data set as the case group only included eight individuals by randomly choosing eight controls. In this analysis the RF revealed an OOB error rate of 12.3% (with a sensitivity of 100% and specificity of 80%).
Influence of covariates
We repeated the linear regression of the change in gene expression after dexamethasone for these 19 genes adjusting for age, smoking status, body mass index and number of granulocytes, lymphocytes and monocytes. In only 3 of these 19 genes, these covariates contributed significantly (model P for the covariates ⩽0.05) to the variance in gene expression including FKBP5 and LAT2. For FKBP5 body mass index was the only significant predictor within the covariates (P=0.008) although the change in granulocytes was correlated with LAT2 expression (P=0.023). To assess a possible influence of the administrated medication on the gene expression, we correlated the duration of the antidepressant use at the day of the blood draw with the gene expression of these 19 transcripts. Mean antidepressant treatment time was 10.8 weeks (SD=16.9, ranging from 0 to 76 weeks). In patients, we found no significant effects of the duration of antidepressants treatment on the dexamethasone-induced gene expression of the 19 differentially regulated transcripts, suggesting that antidepressant treatment per se is not a very strong confound for the reported case-control effects (Supplementary Table 4). We did, however, not have sufficient power to investigate the effects of specific types of antidepressant treatments, as there were only 29 patients but five groups of different antidepressant treatments.
Next, we analyzed a possible impact of differences in dexamethasone metabolism on gene expression. The mean plasma dexamethasone concentrations in cases were 0.55 ng/ml (SD=0.33) and in controls 0.80 ng/ml (SD=0.64), t=1.897, P=0.066. Dexamethasone levels had significant effects overall on the induced expression of SOCS1 (P=0.0006) and MUM1 (P=0.0027). When adding dexamethasone levels as a covariate in analyzing the difference in dexamethasone induction of the 19 top genes, the case-status dependent difference in induction remained significant (Supplementary Table 5).
Validation of Differentially Regulated Transcripts
We used qPCR to validate mRNA expression differences in two genes regulated by dexamethasone in cases and controls in both cohorts: FKBP5, BEST1 and CEACAM4 (see Figure 3). For FKBP5 we could validate the significant interaction between the change in mRNA-expression from 1800 h and 2100 h for cohort 1 and 2 (within subject factor) and disease status for FKBP5 (cohort 1: F1,140=9.013; P=0.005; cohort 2: F1,88=8.835; P=0.007) as well as the main effect of dexamethasone stimulation (cohort 1: F1,140=39.509; P<0.001; cohort 2: F1,88=66.454; P<0.001) using a repeated measures ANOVA. For BEST1, we could validate the regulation by dexamethasone (F1,232=5.0; P=0.028) and the effect of case status when analyzing all cohorts together (F1,232=3.1; P=0.08 two-sided and 0.041 one-sided), but not separately for each cohort. However, the direction of the effect was the same in both cohorts and the observed lack of significance may reflect lack of power in the subsample analysis. For CEACAM4, we validated the significant regulation of this gene by dexamethasone in cases and controls in both cohorts (cohort 1: F1,140=12.090; P=0.001; cohort 2: F1,88=16.599; P=0.001).
Figure 3.
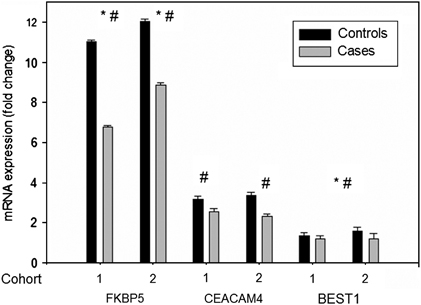
qPCR validation: mRNA expression (fold change poststimulation/prestimulation mRNA levels) of FKBP5, BEST1 and CEACAM4. Comparison of healthy controls vs depressed cases for cohort 1 and cohort 2: *significant interaction effects were observed for FKBP5 (cohort 1: P=0.005; cohort 2: P=0.007). #Significant main effects of dexamethasone were observed for FKBP5 (cohort 1: P<0.001; cohort 2: P<0.001) and CEACAM4 (cohort 1: P=0.001; cohort 2: P=0.001) and for BEST1, when analyzing the combined cohort (P=0.041 one-sided for interaction and P=0.028 for dexamethasone effects).
DISCUSSION
In this study we show that GR-stimulated gene expression changes in peripheral blood cells allow a much better discrimination between patients and controls than gene expression levels at baseline or the degree of suppression of plasma cortisol concentrations by dexamethasone.
Although there were no significant differences in the hormone levels between cases and controls in the first 3 h after dexamethasone intake, cortisol levels were significantly higher in depressed individuals 21 h later, reflecting the previously reported GR resistance in these patients (Holsboer, 2000). This endocrine measure, however, failed to reliably discriminate between cases and controls and its sensitivity was low (34.5%). In contrast, gene expression profiles of the top differentially regulated genes by GR activation showed high sensitivity and specificity in both the training as well as the test cohort (cohort 1–sensitivity of 80.0% and specificity of 87.5%) and a correct classification of 79.2% in the test cohort. The stimulated gene expression profiles outperformed baseline gene expression patterns, which only correctly classified 41.7% of individuals in the test cohort, even though a larger number of predictors was used in the analysis. This supports the previously described superiority of stimulated over baseline gene expression differences as biomarkers for depression (Spijker et al, 2010). Spijker et al (2010) investigated gene expression changes in peripheral blood monocytes following an ex vivo challenge with LPS, a strong immunogenic stimulus, which is less influenced by confounding variables than the here presented in vivo challenge test. However, the reported sensitivity of 77% and specificity of 72% is in the range of the one reported in our study. This suggests that in vivo dexamethasone-stimulated gene expression pattern, which require less hands on time in the laboratory than ex-vivo stimulation of isolated peripheral blood cells, could be further developed as potential biomarkers for depression-related GR function changes.
We found that dexamethasone-induced robust and reproducible gene expression profiles in both cases and controls. Interestingly, within the top genes regulated by dexamethasone in peripheral blood cells in controls, three (FKBP5, DUSP1 and ZBTB16) have been previously reported to be related to the development of mood and anxiety disorders or neuroprotection. Polymorphisms in FKBP5 have been associated with a number of phenotypes related to mood disorders (Binder, 2009) and gene expression differences have been reported for PTSD (Mehta et al, 2011; Segman et al, 2005; Yehuda et al, 2009). The mitogen-activated protein kinase phosphatase DUSP1 has recently been shown to be expressed more strongly in postmortem hippocampus tissue of depressed patients as compared with controls and to mediate stress-related depression-like behavior in rodents (Duric et al, 2010). ZBTB16 encodes the promyelocytic leukemia zinc finger protein and is a corticosteroid-responsive transcription factor. It is induced by stress exposure in the brain and may have a neuroprotective function (Peppi et al, 2011). Of the genes within the 19 best classifiers, FKBP5 and TMEM176A have previously been associated with mood disorders. Gene expression of the transmembrane protein 176A (TMEM176A) was previously found to be altered in the hippocampus and prefrontal cortex in a rat model of depression-like behavior (Blaveri et al, 2010).
Although our cohorts were all male and of similar age and the patients matched according to clinical and treatment parameters, patients and controls were discordant for antidepressant medication. This makes it difficult to exclude that some of the observed differences in gene expression are related to treatment and not disease status. Even if the observed differences were exclusively related to the effects of antidepressant drugs, this would not affect the observed superiority of stimulated vs baseline gene expression between the two groups. Although medication is a confound for the reported case-control differences, these differences are more likely carried by the depression than the medication status, although this can only be resolved in a new experiment comparing medicated and un-medicated patients and controls. However, the duration of the antidepressant treatment at the time of RNA collection had no significant impact on gene expression regulation in patients for the 19 genes that best differentiate between cases and controls. This suggests that antidepressant medication is not likely to have a major impact on the GR-regulated transcription of these genes. The fact that the observed differences in gene expression are more likely related to psychiatric status and not medication is also supported by a number of other studies.
It has been previously shown that at in-patient admission, markers of HPA-axis hyperreactivity are not strongly influenced by the current type of antidepressant medication (Holsboer, 2000; Kunzel et al, 2003). Moreover, antidepressants have repeatedly been shown to increase rather than decrease GR function (Carvalho and Pariante, 2008; Pariante, 2009). Antidepressants have been reported to increase GR mRNA expression (Carvalho and Pariante, 2008; Funato et al, 2006; Heiske et al, 2003; Vedder et al, 1999), GR protein expression (Carvalho and Pariante, 2008; Hery et al, 2000; Lai et al, 2003; Pepin et al, 1992) and GR function as measured using the translocation of the receptor to the nucleus (Carvalho and Pariante, 2008; Funato et al, 2006; Heiske et al, 2003; Pariante et al, 1997) or dexamethasone-induced GR-mediated gene transcription (Pariante et al, 2003; Pariante et al, 2001a; Pariante et al, 1997; Pepin et al, 1992). This pattern is opposite to the changes observed here, with less genes significantly regulated by dexamethasone in cases vs controls (1151 vs 2670) and lower fold changes in cases (see Table 2). Furthermore, two genes showing a significant case/control difference in GR regulation, FKBP5 and CD14 have both been shown not to be affected by antidepressant drug exposure alone (Anacker et al, 2011; Landmann et al, 1997).
We also tested the influence of a number of covariates, including age, BMI and immune cell fractions on the expression pattern of the 19 strongest classifiers and for only 3 of them, the model including all covariates had a significant effect on the change in gene expression. Only for one gene (LAT2), we observe a significant effect of the change in numbers of specific immune cell types on the change in gene expression. This indicates that although we (like others (Schuld et al, 2006)) observe significant differences in the baseline counts of granulocytes and monocytes and the increase of granulocyte numbers following dexamethasone in cases and controls (see Figure 1b), these are not likely confounding our main results. Experiments in separated leukocytes subtypes could further address this issue, however, such manipulations have been reported to change the gene expression profile and in addition increase hands-on time for sample processing, thus decreasing the usefulness for biomarker research.
We therefore propose that the difference in gene expression profiles between patients and controls is indeed reflecting differences in GR sensitivity in a similar, but possibly more sensitive way than clinical HPA stimulation tests (Holsboer, 2000). This is supported by the fact that FKBP5 shows less regulation after dexamethasone in patients than in controls. FKBP5 is a heatshock protein 90 associated co-chaperone of the GR and its expression is strongly induced by glucocorticoids via intronic steroid response elements as part of an intracellular short negative feedback loop for GR activity (Vermeer et al, 2003). FKBP5 induction by GR activation has been proposed as a molecular indicator of GR sensitivity (Jääskeläinen et al, 2011; Vermeer et al, 2003).
In addition, of the 19 genes that contribute most to the classification between cases and controls (see Figure 2 and Table 2), all genes are less stimulated in depressed patients than in controls, in line with the fact that the number of overall regulated genes was also less in patients than controls (N=1.151 vs N=2.670) thus likely reflecting baseline differences in GR sensitivity. Dexamethasone-induced gene expression changes also appear to be a more sensitive marker of GR-function than the DST. According to the DST, only 8 of the 29 patients fulfilled criteria for dexamethasone-non-suppression (Ising et al, 2007). In these patients, dexamethasone-stimulated genes performed best as classifiers, correctly classifying 88.7% of cases and controls with a sensitivity of 100%. Nonetheless, these transcripts also correctly classified 77.3% of the dexamethasone non-suppressor patients, indicating that they suffer from a GR-resistance that is not evident in the DST, but observable in the dexamethasone-stimulated gene expression pattern. Furthermore, dexamethasone-induced gene expression changes indicate GR-resistance much earlier than peripheral hormones. After 3 h, gene expression changes show strong differences, whereas this is not the case for serum cortisol or ACTH measures, which are only significantly different after 21 h, when gene expression changes are almost back to normal.
Our results provide support for the usefulness of dexamethasone-stimulated gene expression profiles as a biomarker for depression-related GR resistance. Studies in larger independent samples with different gender composition and different clinical settings will further explore the potential of the molecular dexamethasone-stimulation test as a biomarker helping to characterize subgroups within patient samples that fulfil current diagnostic criteria for a certain psychiatric category.
Acknowledgments
The authors would like to thank A Sangl, M Asmus, G Ernst-Jansen, E Kappelmann, M Hartung and B Siegel for their excellent technical assistance. The study is supported by a grant of the Exzellenz-Stiftung of the Max Planck Society. This work has also been funded by the Federal Ministry of Education and Research (BMBF) in the framework of the National Genome Research Network (NGFN), FKZ 01GS0481. Additionally, the study was supported by a grant of Neuronova G GmbH.
Patent applications: Menke, Binder, Holsboer inventors: Means and methods for diagnosing predisposition for treatment emergent suicidal ideation (TESI). European application number: 08016477.5 International application number: PCT/EP2009/061575. Binder, Müller-Myhsok, Uhr, Holsboer inventors: FKBP5: a novel target for antidepressant therapy. International publication number: WO 2005/054500 Polymorphisms in ABCB1 associated with a lack of clinical response to medicaments. International application number: PCT/EP2005/005194. Elisabeth B Binder currently received grant support from NIMH, Doris Duke Charitable foundation, Behrens-Weise-Stiftung and PharmaNeuroBoost. Florian Holsboer is the founder and share holder of Affectis Pharmaceuticals and HolsboerMaschmeyer NeuroChemie GmbH. The other authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Parts of the data have been presented at the Society of Biological Psychiatry, May 2010, New Orleans, Louisiana, USA and at the Annual Meeting of the American College of Neuropsychopharmacology, December 2009, Hollywood, Florida, USA.
Supplementary Material
References
- Anacker C, Zunszain PA, Cattaneo A, Carvalho LA, Garabedian MJ, Thuret S, et al. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry. 2011;16:738–750. doi: 10.1038/mp.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34 (Suppl 1:S186–S195. doi: 10.1016/j.psyneuen.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Blaveri E, Kelly F, Mallei A, Harris K, Taylor A, Reid J, et al. Expression profiling of a genetic animal model of depression reveals novel molecular pathways underlying depressive-like behaviours. PLoS One. 2010;5:e12596. doi: 10.1371/journal.pone.0012596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington's disease. Proc Natl Acad Sci USA. 2005;102:11023–11028. doi: 10.1073/pnas.0504921102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- Carvalho LA, Pariante CM. In vitro modulation of the glucocorticoid receptor by antidepressants. Stress. 2008;11:411–424. doi: 10.1080/10253890701850759. [DOI] [PubMed] [Google Scholar]
- Czock D, Keller F, Rasche FM, Haussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44:61–98. doi: 10.2165/00003088-200544010-00003. [DOI] [PubMed] [Google Scholar]
- Dumeaux V, Lund E, Borresen-Dale A-L. Comparison of globin RNA processing methods for genome-wide transcriptome analysis from whole blood. Biomark Med. 2008;2:11–21. doi: 10.2217/17520363.2.1.11. [DOI] [PubMed] [Google Scholar]
- Dunning MJ, Smith ML, Ritchie ME, Tavare S. Beadarray: R classes and methods for Illumina bead-based data. Bioinformatics. 2007;23:2183–2184. doi: 10.1093/bioinformatics/btm311. [DOI] [PubMed] [Google Scholar]
- Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, et al. A negative regulator of MAP kinase causes depressive behavior. Nat Med. 2010;16:1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Expression Analysis 2007 ). http://www.expressionanalysis.com/pdf/IlluminaBlood_TechNote_2007.pdf .
- Funato H, Kobayashi A, Watanabe Y. Differential effects of antidepressants on dexamethasone-induced nuclear translocation and expression of glucocorticoid receptor. Brain Res. 2006;1117:125–134. doi: 10.1016/j.brainres.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Gerhold DL, Jensen RV, Gullans SR. Better therapeutics through microarrays. Nat Genet. 2002;32 (Suppl:547–551. doi: 10.1038/ng1042. [DOI] [PubMed] [Google Scholar]
- Gladkevich A, Kauffman HF, Korf J. Lymphocytes as a neural probe: potential for studying psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:559–576. doi: 10.1016/j.pnpbp.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Gormley GJ, Lowy MT, Reder AT, Hospelhorn VD, Antel JP, Meltzer HY. Glucocorticoid receptors in depression: relationship to the dexamethasone suppression test. Am J Psychiatry. 1985;142:1278–1284. doi: 10.1176/ajp.142.11.1278. [DOI] [PubMed] [Google Scholar]
- Heiske A, Jesberg J, Krieg JC, Vedder H. Differential effects of antidepressants on glucocorticoid receptors in human primary blood cells and human monocytic U-937 cells. Neuropsychopharmacology. 2003;28:807–817. doi: 10.1038/sj.npp.1300056. [DOI] [PubMed] [Google Scholar]
- Hery M, Semont A, Fache MP, Faudon M, Hery F. The effects of serotonin on glucocorticoid receptor binding in rat raphe nuclei and hippocampal cells in culture. J Neurochem. 2000;74:406–413. doi: 10.1046/j.1471-4159.2000.0740406.x. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Holsboer F. How can we realize the promise of personalized antidepressant medicines. Nat Rev Neurosci. 2008;9:638–646. doi: 10.1038/nrn2453. [DOI] [PubMed] [Google Scholar]
- Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18 (Suppl 1:S96–104. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- Ising M, Horstmann S, Kloiber S, Lucae S, Binder EB, Kern N, et al. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression—a potential biomarker. Biol Psychiatry. 2007;62:47–54. doi: 10.1016/j.biopsych.2006.07.039. [DOI] [PubMed] [Google Scholar]
- Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S, et al. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;66:966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jääskeläinen T, Makkonen H, Palvimo JJ.2011Steroid up-regulation of FKBP51 and its role in hormone signaling Curr Opin Pharmacol 11326–331.(in press). [DOI] [PubMed] [Google Scholar]
- Kitchen RR, Sabine VS, Sims AH, Macaskill EJ, Renshaw L, Thomas JS, et al. Correcting for intra-experiment variation in Illumina BeadChip data is necessary to generate robust gene-expression profiles. BMC Genomics. 2010;11:134. doi: 10.1186/1471-2164-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzel HE, Binder EB, Nickel T, Ising M, Fuchs B, Majer M, et al. Pharmacological and nonpharmacological factors influencing hypothalamic-pituitary-adrenocortical axis reactivity in acutely depressed psychiatric in-patients, measured by the Dex-CRH test. Neuropsychopharmacology. 2003;28:2169–2178. doi: 10.1038/sj.npp.1300280. [DOI] [PubMed] [Google Scholar]
- Kurian SM, Le-Niculescu H, Patel SD, Bertram D, Davis J, Dike C, et al. Identification of blood biomarkers for psychosis using convergent functional genomics. Mol Psychiatry. 2011;16:37–58. doi: 10.1038/mp.2009.117. [DOI] [PubMed] [Google Scholar]
- Lai M, McCormick JA, Chapman KE, Kelly PA, Seckl JR, Yau JL. Differential regulation of corticosteroid receptors by monoamine neurotransmitters and antidepressant drugs in primary hippocampal culture. Neuroscience. 2003;118:975–984. doi: 10.1016/s0306-4522(03)00038-1. [DOI] [PubMed] [Google Scholar]
- Landmann R, Schaub B, Link S, Wacker HR. Unaltered monocyte function in patients with major depression before and after three months of antidepressive therapy. Biol Psychiatry. 1997;41:675–681. doi: 10.1016/S0006-3223(96)00176-X. [DOI] [PubMed] [Google Scholar]
- Le-Niculescu H, Kurian SM, Yehyawi N, Dike C, Patel SD, Edenberg HJ, et al. Identifying blood biomarkers for mood disorders using convergent functional genomics. Mol Psychiatry. 2009;14:156–174. doi: 10.1038/mp.2008.11. [DOI] [PubMed] [Google Scholar]
- Liaw A, Wiener M. Classification and Regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- Lowy MT, Reder AT, Gormley GJ, Meltzer HY. Comparison of in vivo and in vitro glucocorticoid sensitivity in depression: relationship to the dexamethasone suppression test. Biol Psychiatry. 1988;24:619–630. doi: 10.1016/0006-3223(88)90136-9. [DOI] [PubMed] [Google Scholar]
- Mehta D.2008Globin—to reduce or not to reducePersonal Communication.
- Mehta D, Gonik M, Klengel T, Rex-Haffner M, Menke A, Rubel J, et al. Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from Endocrine and Gene Expression Studies. Arch Gen Psychiatry. 2011;68:901–910. doi: 10.1001/archgenpsychiatry.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Menke A, Binder EB. Gene expression studies in major depression. Curr Psychiatry Rep. 2010;12:135–144. doi: 10.1007/s11920-010-0100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menke A, Rex-Haffner M, Klengel T, Binder B, Mehta D.2011Peripheral blood gene expression: it all boils down to the RNA collection tubes BMC Res Notes (in press). [DOI] [PMC free article] [PubMed]
- Ozcift A. Random forests ensemble classifier trained with data resampling strategy to improve cardiac arrhythmia diagnosis. Comput Biol Med. 2011;41:265–271. doi: 10.1016/j.compbiomed.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Padmos RC, Hillegers MH, Knijff EM, Vonk R, Bouvy A, Staal FJ, et al. A discriminating messenger RNA signature for bipolar disorder formed by an aberrant expression of inflammatory genes in monocytes. Arch Gen Psychiatry. 2008;65:395–407. doi: 10.1001/archpsyc.65.4.395. [DOI] [PubMed] [Google Scholar]
- Pariante CM. Risk factors for development of depression and psychosis. Glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci. 2009;1179:144–152. doi: 10.1111/j.1749-6632.2009.04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003;139:1111–1118. doi: 10.1038/sj.bjp.0705357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Makoff A, Lovestone S, Feroli S, Heyden A, Miller AH, et al. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol. 2001a;134:1335–1343. doi: 10.1038/sj.bjp.0704368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001b;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol. 1997;52:571–581. doi: 10.1124/mol.52.4.571. [DOI] [PubMed] [Google Scholar]
- Pepin MC, Govindan MV, Barden N. Increased glucocorticoid receptor gene promoter activity after antidepressant treatment. Mol Pharmacol. 1992;41:1016–1022. [PubMed] [Google Scholar]
- Peppi M, Kujawa SG, Sewell WF. A corticosteroid-responsive transcription factor, promyelocytic leukemia zinc finger protein, mediates protection of the cochlea from acoustic trauma. J Neurosci. 2011;31:735–741. doi: 10.1523/JNEUROSCI.3955-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team . A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna; 2007. [Google Scholar]
- Rollins B, Martin MV, Morgan L, Vawter MP. Analysis of whole genome biomarker expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:919–936. doi: 10.1002/ajmg.b.31062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer CR, Eklund AC, Morse LJ, Liao Z, Locascio JJ, Fefer D, et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc Natl Acad Sci USA. 2007;104:955–960. doi: 10.1073/pnas.0610204104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuld A, Birkmann S, Beitinger P, Haack M, Kraus T, Dalal MA, et al. Low doses of dexamethasone affect immune parameters in the absence of immunological stimulation. Exp Clin Endocrinol Diabetes. 2006;114:322–328. doi: 10.1055/s-2006-924255. [DOI] [PubMed] [Google Scholar]
- Segman RH, Goltser-Dubner T, Weiner I, Canetti L, Galili-Weisstub E, Milwidsky A, et al. 2010Blood mononuclear cell gene expression signature of postpartum depression Mol Psychiatry 1593–100.102. [DOI] [PubMed] [Google Scholar]
- Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. 2005;10:425, 500–513. doi: 10.1038/sj.mp.4001636. [DOI] [PubMed] [Google Scholar]
- Spijker S, Zanten JS, De Jong S, Penninx BW, van Dyck R, Zitman FG, et al. Stimulated gene expression profiles as a blood marker of major depressive disorder. Biol Psychiatry. 2010;68:179–186. doi: 10.1016/j.biopsych.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Fan C, Perou CM. Evaluating the comparability of gene expression in blood and brain. Am J Med Genet B Neuropsychiatr Genet. 2006;141:261–268. doi: 10.1002/ajmg.b.30272. [DOI] [PubMed] [Google Scholar]
- Tomita H, Vawter MP, Walsh DM, Evans SJ, Choudary PV, Li J, et al. Effect of agonal and postmortem factors on gene expression profile: quality control in microarray analyses of postmortem human brain. Biol Psychiatry. 2004;55:346–352. doi: 10.1016/j.biopsych.2003.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–392. doi: 10.1192/bjp.184.5.386. [DOI] [PubMed] [Google Scholar]
- Vedder H, Bening-Abu-Shach U, Lanquillon S, Krieg JC. Regulation of glucocorticoid receptor-mRNA in human blood cells by amitriptyline and dexamethasone. J Psychiatr Res. 1999;33:303–308. doi: 10.1016/s0022-3956(99)00006-0. [DOI] [PubMed] [Google Scholar]
- Vermeer H, Hendriks-Stegeman BI, van der Burg B, van Buul-Offers SC, Jansen M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: a potential marker for glucocorticoid sensitivity, potency, and bioavailability. J Clin Endocrinol Metab. 2003;88:277–284. doi: 10.1210/jc.2002-020354. [DOI] [PubMed] [Google Scholar]
- Wittchen HU, Höfler M, Gander F, Pfister H, Storz S, stün TB, et al. Screening for mental disorders: performance of the composite international diagnostic—screener (CID-S) Int J Methods Psychiatr Res. 1999;8:59–70. [Google Scholar]
- Wodarz N, Rupprecht R, Kornhuber J, Schmitz B, Wild K, Riederer P. Cell-mediated immunity and its glucocorticoid-sensitivity after clinical recovery from severe major depressive disorder. J Affect Disord. 1992;25:31–38. doi: 10.1016/0165-0327(92)90090-s. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Cai G, Golier JA, Sarapas C, Galea S, Ising M, et al. Gene expression patterns associated with posttraumatic stress disorder following exposure to the World Trade Center attacks. Biol Psychiatry. 2009;66:708–711. doi: 10.1016/j.biopsych.2009.02.034. [DOI] [PubMed] [Google Scholar]
- Young E, Korszun A. Sex, trauma, stress hormones and depression. Mol Psychiatry. 2010;15:23–28. doi: 10.1038/mp.2009.94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.