

Abstract
Mood and anxiety disorders develop in some but not all individuals following exposure to stress and psychological trauma. However, the factors underlying individual differences in risk and resilience for these disorders, including genetic variation, remain to be determined. Isogenic inbred mouse strains provide a valuable approach to elucidating these factors. Here, we performed a comprehensive examination of the extinction-impaired 129S1/SvImJ (S1) inbred mouse strain for multiple behavioral, autonomic, neuroendocrine, and corticolimbic neuronal morphology phenotypes. We found that S1 exhibited fear overgeneralization to ambiguous contexts and cues, impaired context extinction and impaired safety learning, relative to the (good-extinguishing) C57BL/6J (B6) strain. Fear overgeneralization and impaired extinction was rescued by treatment with the front-line anxiety medication fluoxetine. Telemetric measurement of electrocardiogram signals demonstrated autonomic disturbances in S1 including poor recovery of fear-induced suppression of heart rate variability. S1 with a history of chronic restraint stress displayed an attenuated corticosterone (CORT) response to a novel, swim stressor. Conversely, previously stress-naive S1 showed exaggerated CORT responses to acute restraint stress or extinction training, insensitivity to dexamethasone challenge, and reduced hippocampal CA3 glucocorticoid receptor mRNA, suggesting downregulation of negative feedback control of the hypothalamic–pituitary–adrenal axis. Analysis of neuronal morphology in key neural nodes within the fear and extinction circuit revealed enlarged dendritic arbors in basolateral amygdala neurons in S1, but normal infralimbic cortex and prelimbic cortex dendritic arborization. Collectively, these data provide convergent support for the utility of the S1 strain as a tractable model for elucidating the neural, molecular and genetic basis of persistent, excessive fear.
Keywords: gene, vmPFC, anxiety, depression, PTSD, infralimbic, stress
INTRODUCTION
Although the proportion of the US population exposed to at least one severe trauma may be as high as 75% (Breslau and Kessler, 2001), only about one-tenth of that number will meet diagnostic criterion for a clinical anxiety disorder such as post-traumatic stress disorder (PTSD) (Kessler et al, 2005). One major susceptibility factor is genetics. Twin studies estimate a significant influence of genetics in predisposing individuals to stress-related mood and anxiety disorders (Kendler, 2001). Specific gene variants influencing susceptibility, as well as resilience, interact with environmental variables to ultimately determine an individual's likelihood of developing an anxiety disorder after a trauma experience (Caspi et al, 2010; Ressler et al, 2011).
Recent human neuroimaging (Rauch et al, 2006) and preclinical (Quirk and Mueller, 2008) studies have led to important advances in understanding the neural basis of persistent, excessive fear following trauma. For example, there are a number of rodent models in which a specific rat or mouse strain (Cohen et al, 2008), sub-population (Duvarci et al, 2009), or selectively bred line (Muigg et al, 2008) exhibits features consistent with inherent susceptibility to excessive fear after an experimental trauma such as Pavlovian fear conditioning. Along these lines, we recently identified an inbred mouse strain, 129S1/SvImJ (S1), which displays a profound deficit in cued fear extinction, coupled with functional abnormalities in corticoamygdala circuitry mediating extinction (Camp et al, 2009; Hefner et al, 2008; Whittle et al, 2010).
Fear extinction is a form of inhibitory learning in which fear is reduced by repeated non-reinforced exposure to a fear-provoking stimulus (Pavlov, 1927). Given impaired extinction is an intermediate phenotype of anxiety disorders (Milad et al, 2009), the cued fear extinction deficit found in S1 raises the possibility that this strain may exhibit other fear-related abnormalities. The aim of this study was to evaluate this possibility using a combination of approaches. To this end, we tested S1 for multiple behavioral (cued and context fear generalization, context extinction, safety learning), autonomic (heart-rate and heart-rate variability during extinction), and neuroendocrine (basal, acute and chronic stress-induced, and dexamethasone (DEX)-suppression of corticosterone (CORT) responses) disturbances relevant to fear and stress. We also tested whether certain key behavioral phenotypic disturbances in S1 (cued fear extinction, context generalization) were normalized by chronic treatment with fluoxetine (FLX), a drug with efficacy in treating trauma-related anxiety. In addition, to uncover abnormalities in the fear/extinction neural circuitry, we examined the dendritic morphology and spine density of neurons in the basolateral amygdala (BLA), infralimbic cortex (IL), and prelimbic cortex (PL) of S1 mice. All analyses were made in comparison with C57BL/6J (B6) mice, an inbred strain previously shown to exhibit good cued fear extinction relative to S1 (Camp et al, 2009; Hefner et al, 2008; Whittle et al, 2010).
SUBJECTS AND METHODS
Subjects
Subjects were male S1 and B6 mice obtained at ∼8 weeks of age from the Jackson Laboratory (Bar Harbor, ME). Mice were housed by strain (2 per cage) in a temperature (72±5 °F) and humidity (45±15%) controlled vivarium under a 12-h light/dark cycle (lights on 0600 hours). The number of mice used in each experiment is given in the figure legends. Separate cohorts of naive mice were used in all experiments. All experimental procedures were approved by the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee and the Austrian Ethical Committees on Animal Care and Use (Bundesministerium für Wissenschaft und Forschung), and followed the National Institutes of Health guidelines outlined in ‘Using Animals in Intramural Research' and the local Animal Care and Use Committees.
Contextual Fear Discrimination
For the ‘ABC1' design, mice were conditioned in Cxt A as follows: after a 180-s acclimation period, there were three presentations of a 2 s, 0.6 mA scrambled footshock (the first CS at 181-s and a 60- to 90-s inter-stimulus interval between CS presentations), followed by a 120-s no-stimulus period. Cxt A was a 27 × 27 × 11 cm chamber with transparent walls and a metal rod floor, cleaned with a 79.5% water/19.5% ethanol/1% vanilla-extract solution to provide a distinctive olfactory cue. One day later, mice were exposed to Cxt A, Cxt B, and Cxt C1 for 2-min sessions, with a 4-h interval between exposures and the order counterbalanced across mice. Cxt B was designed to be readily distinguishable from Cxt A—it was a Plexiglas cylinder with black/white-checkered walls and solid-Plexiglas opaque floor, cleaned with a 99% water/1% acetic acid solution and housed in a different room from Cxt A. Cxt C1 was more ambiguous. C1 was the same as Cxt B, but contained a Cxt A metal grid floor, which has been shown to be a highly salient fear cue in rats (Gonzalez et al, 2003). The effects of strain and Cxt-type were analyzed using two-factor analysis of variance (ANOVA) (repeated measures for Cxt-type) and Newman–Keuls post hoc tests. The threshold for statistical significance for this and all analyses in this study was p<0.05.
For the ‘ABC2' design, mice were conditioned and tested as above with the exception that Cxt C1 was replaced by Cxt C2. C2 was designed to be highly ambiguous. C2 had the same size, shape and metal grid floor as A, but the same black/white-checkered walls, odor, and testing room as B. The effects of strain and Cxt-type were analyzed using two-factor ANOVA (repeated measures for Cxt-type) and Newman–Keuls post hoc tests.
For this and all other fear experiments, stimulus presentation was controlled by the Med Associates VideoFreeze and Med-PC systems (Med Associates, Burlington, VT). Freezing was measured as an index of fear (Blanchard and Blanchard, 1969) and manually scored every 5 s as no visible movement, except that required for respiration, and converted to a percentage ((number of freezing observations/total number of observations) × 100). The observer was blind to within-strain experimental conditions. We have previously shown that manual and automated scoring are equally effective in detecting and quantifying freezing differences between B6 and S1 (Hefner et al, 2008; Whittle et al, 2010).
Contextual Fear Extinction
At 24 h after context discrimination testing, mice from both the ABC1 and ABC2 discrimination experiments were tested for contextual fear extinction via non-reinforced exposure to Cxt A for 30 min. The effects of strain and time were analyzed (collapsed across the two ABC designs) using two-factor ANOVA (repeated measures for time) and Newman–Keuls post hoc tests. One day later, extinction retrieval was tested by returning the mice to Cxt A for a 3-min test. Freezing during context extinction retrieval was compared between strains via t-test, and also compared with pre-extinction freezing (ie, first time-block during extinction training) using within-subjects t-test.
Cued Fear Discrimination
Cued fear discrimination was tested as previously described (Herry et al, 2008). Mice received a single training session in Cxt A (as above). After a 120-s acclimation period, there were 3 × CS/US pairings separated by a 60- to 120-s variable interval. The CS+ was 30 s of 50 ms 80 dB pips repeated at a frequency of 0.9 Hz. For half of the mice, the frequency of the CS+ was 7.5 kHz, and for the other half it was a broadband white noise (counterbalanced across strains). The US was a 2 s, 0.6 mA scrambled footshock.
One day later, cued fear discrimination was tested in Cxt B (as above). After 120-s acclimation, there were 3 × presentations of the CS+ and 3 × presentations of whichever of the (novel) CSs not heard during training for that mouse. There was a 180-s interval between CSs and the presentation order of the CS+ and novel CS was counterbalanced across strains. A discrimination index was calculated (as freezing to the CS+ relative to total freezing to both stimuli), with higher scores indicating better discrimination. Strains were compared by t-test.
Partially Reinforced Cued Fear Discrimination
Partially reinforced cued fear discrimination was tested as previously described (Tsetsenis et al, 2007). Mice received a single training session in Cxt A (as above). After a 180-s acclimation period, there were presentations of the CS+, the CS–, in a randomized order separated by a 60- to 120-s variable interval. The CS+ and CS– was a white noise or shock, and the US was a footshock: stimulus parameters were the same as for cued fear discrimination experiment above. The CS+ was presented 6 × and always coterminated with the US, while the CS– coterminated 2 × with the US and 4 × with no US.
One day later, fear retrieval was tested in Cxt B (as above). After 180-s acclimation, there were 3 × CS+ presentations and 3 × presentations of the CS–. There was a 180-s interval between CSs and the presentation order of the CS+ and CS– was counterbalanced across strains. A discrimination index was calculated (as freezing to the CS+ relative to total freezing to both stimuli), with higher scores indicating better discrimination. Strains were compared by t-test.
Cued Fear Discrimination After Extinction
Conditioning and extinction was assessed as previously described (Gogolla et al, 2009). Mice received a single training session in Cxt A. After a 120-s acclimation period, there were 5 × CS/US pairings separated by a 20- to 180-s variable interval. The CS+ was either a tone or white noise (details the same as for cued fear discrimination). The US was a 1 s, 0.6 mA scrambled footshock.
One day, and again 2 days later, mice received extinction training sessions in Cxt B, each consisting of 12 × CS+ presentations. One day later, extinction retrieval and cued fear discrimination was tested in Cxt B. After 120-s acclimation, there were 4 × CS+ presentation and 4 × presentations of the novel CS (tone or white noise) that was not presented during training. There was a 180-s interval between CSs and the presentation order of CS-type was counterbalanced across strains. Freezing (measured with an automatic infrared beam detection system, Coulbourn Whitehall, PA) during trial 1 and trial 12 of each extinction session, and during extinction retrieval/cue discrimination, was compared between strains and between CS-type using t-tests.
Safety Learning
Safety learning was tested using an explicitly unpaired training procedure previously described for mice (Pollak et al, 2008; Rogan et al, 2005). Unpaired training took place after a 60- to 80-s acclimation period in Cxt A (as above) over three daily sessions. On each session, there was a block of 4 × US (2 s 0.6 mA scrambled footshock, 45- to 80-s variable interval between USs) and a block of 4 × CS (20 s 75 dB white noise, 70- to 90-s interval between CSs), separated by 100 s. The US-block preceded the CS-block on training sessions 1 and 3, and vice versa on training session 2. To ensure this same sequence produced good conditioned fear when the US and CS were explicitly paired, another group of mice received three daily sessions of 4 × CS-US training in which the US was presented during the last 2 s of the CS (112- to 172-s inter-pairing interval).
Successful CS safety learning was probed using two tests: conditioned inhibition of contextual fear and retarded acquisition of new fear learning (Pollak et al, 2008; Rogan et al, 2005). One day after the final training session, conditioned inhibition was tested by measuring freezing in Cxt A during a 60-s baseline followed by a 20-s CS presentation (Pollak et al, 2008). Conditioned inhibition was expressed as the reduction in freezing during the 20-s CS relative to the 20 s before the CS. The effects of strain and CS presentation on conditioned inhibition were analyzed using two-factor ANOVA (repeated measures for CS presentation).
One day later, the safety group received a session of 3 × CS-US explicitly paired training, as above. For comparison, a naive group received the same training. Fear retardation was tested, 1 day later, by measuring freezing to a 20-s CS presentation after a 60-s baseline in Cxt A. Fear retardation was expressed as the decrease in freezing to the CS in the ‘safety' group relative to the previously naive fear group. Since fear retardation can be overcome by additional training (Pollak et al, 2008; Rescorla, 1969), we conducted a second round of explicitly paired training and testing. The effects of strain and training-type (unpaired vs paired) on retardation were analyzed using two-factor ANOVA.
Chronic FLX Effects on Extinction
Mice were conditioned and extinguished as previously described (Izquierdo et al, 2006; Wellman et al, 2007). After a 180-s acclimation period to Cxt A (see discrimination experiment above), mice received three pairings (60- to 90-s variable inter-pairing interval) between a 30 s, 80 dB, white noise (CS) and a 2 s, 0.6 mA scrambled footshock (US), in which the shock was presented during the last 2 s of the CS. There was a 120-s no-stimulus period following the final CS–US pairing. For extinction training, there was a 180-s acclimation period, in Cxt B (see discrimination experiment above) followed by 50 × 30-s CS presentations (5-s no-stimulus interval). One day later, mice were tested for extinction retrieval in Cxt B via 3 × 30-s CS presentations (after a 180-s acclimation period). The effects of strain and treatment were analyzed using two-factor ANOVA and Newman–Keuls post hoc tests.
Beginning 1 day after conditioning, mice were provided with 120 mg/l FLX hydrochloride (LKT Laboratories, St Paul, MN) in (their only source of) drinking water, as previously described (Brigman et al, 2010). This concentration was chosen based on previous data from our laboratory (Karlsson et al, 2008) to attain an average self-administered daily dose of ∼10 mg/kg. Non-treated controls received water alone and solutions were refreshed weekly. FLX and water consumption was measured from bottle weights (corrected for evaporation and spillage) and converted to a mg/kg body weight daily dose. Mice remained on treatment through extinction training (day 22) and retrieval testing (day 23).
In a separate experiment, a cohort of mice was treated with FLX for 21 days before fear conditioning and remained on treatment through the completion of extinction testing (procedures as above).
Chronic FLX Effects on Context Discrimination
Mice were treated with FLX as above for 21 days before fear conditioning and remained on treatment through the completion of context discrimination testing. Testing for context discrimination followed the ABC2 design described above. The effects of strain and treatment for each context type were analyzed using two-factor ANOVA and Newman–Keuls post hoc tests.
Chronic FLX Effects on Stress-Induced Corticosterone
Mice were treated with FLX as above for 21 days and then (while still on drug) exposed to forced swim (20 cm-diameter Plexiglas cylinder filled halfway with 24±1°C water) for 6 min. Mice were then returned to the home cage and blood samples were taken 30 min later. The effects of strain and stress were analyzed using 2-factor ANOVA and Newman Keuls post hoc tests.
Telemetric Measurement of Autonomic Function
Electrocardiogram (ECG) transmitters (Data Sciences, TA10EA-F20, St Paul, MN) were implanted intraperitoneally (i.p.) with two electrodes placed subcutaneously (s.c.), as described previously (Stiedl and Spiess, 1997). We have established telemetry procedures for C57BL/6N (Charles River Laboratories, Germany) and this substrain shows similar autonomic functions as B6 (Stiedl et al, 2003). We have also previously shown that C57BL/6N show good extinction in comparison with S1 (Whittle et al, 2010). For these reasons, we used C57BL/6N rather than B6 in this experiment. After 2–3 weeks of recovery in individual home cages, mice were fear conditioned via 3 × CS (30 s, 80 dB, white noise)/US (2 s, 0.6 mA scrambled footshock) explicit-pairings. Conditioning took place, after 180-s acclimation, in a novel 26 × 30 × 32 cm chamber with transparent walls and a metal rod floor, with stimuli controlled by the Habitest operant system (Coulbourn Instruments, Whitehall, PA). ECG could not be recorded during conditioning. One day later, after a 180-s no-stimulus baseline period mice received extinction training via 50 × non-reinforced CS+ presentations (5-s inter-CS intervals) in the home cage (to avoid ECG artifacts because of handling) (Depino and Gross, 2007; Tovote et al, 2004).
The ECG signal was measured during baseline and extinction training. The signal was digitized at a rate of 2 kHz and stored for later off-line analysis using Dataquest A.R.T. 4.1 (Data Sciences). Recordings were edited for correction of unrecognized beats and exclusion of artifacts. As each mouse does not provide a good ECG signal for every CS, we increased the power of the analysis by collating useable signals for each strain during the first and second 25-CS trial blocks. We used successive R–R waves of the ECG signal to derive heart rate (HR) (defined as beats per min (b.p.m.) and calculated as 60 (s in 1 min)/successive R–R interval differences (s)) and HR variability (HRV) ((expressed (ms) as the root-mean-square of successive R–R interval differences (RMSSD)). Basal strain differences were analyzed via t-tests. Cardiovascular data during extinction data from each strain were normalized to their respective baselines, and the effects of strain and CS-block were analyzed using two-factor ANOVA (repeated measures for CS-block) and Newman–Keuls post hoc tests.
HPA-Axis Profile and Hippocampal Glucocorticoid Receptor Expression
Basal CORT
Mice were individually housed for 1 day to minimize disturbance before sampling. Basal CORT levels were measured during the early (0800–0900 hours) and late (1700–1800 hours) phase of the light cycle, when mouse serum CORT levels are expected to be low and high, respectively (Chen et al, 2006; Lechner et al, 2000). Samples for this and all other CORT analyses were collected from trunk blood, following cervical dislocation and rapid decapitation, within 30 s of disturbing the cage. Samples were centrifuged at 13 000 r.p.m. for 30 s. Serum was extracted and assayed for total CORT (bound and free) using the ImmuChem Double Antibody 125I RIA kit from MP Biomedicals, LLC (Orangeburg, NY) as previously described (Boyce-Rustay et al, 2007). The effects of strain and time of day were analyzed using two-factor ANOVA.
Acute restraint-induced CORT
Stress-induced activation and recovery of CORT was measured by placing mice in ventilated 50 ml Falcon tubes for 2 h. Mice were then returned to the home cage and blood samples were taken either 30 min (=activation) or 75 min (=recovery) later (1700–1800 hours, concomitant sampling basal levels of the late phase of the light cycle). The effects of strain and stress were analyzed using two-factor ANOVA and Newman–Keuls post hoc tests.
Extinction-induced CORT
Mice were conditioned and extinguished as described above for the FLX experiments, but immediately killed after extinction training for CORT analysis. Strains were compared using t-tests.
Acute swim-induced CORT after chronic restraint
As previously reported (Mozhui et al, 2010) and reanalyzed and redrawn here, a naive cohort was restrained daily (1000–1200 hours) for 10 days and, 2 days later, exposed to forced swim (20 cm-diameter Plexiglas cylinder filled halfway with 24±1 °C water) for 6 min. Mice were then returned to the home cage and blood samples were taken 30 min later. The effects of strain and stress were analyzed using two-factor ANOVA and Newman–Keuls post hoc tests.
DEX suppression of CORT
Stress-naive mice were injected (s.c.) with 0, 0.05 or 0.1 mg/kg DEX (in a volume of 5 ml/kg body weight). Blood samples were taken 6 h later (1700–1800 hours). Doses were based on those previously described for mice on a C57BL/6 background (Mitsukawa et al, 2006; Ridder et al, 2005). The effects of strain and treatment were analyzed using two-factor ANOVA and Newman–Keuls post hoc tests. A separate cohort of S1 mice was injected with 0, 0.075 or 0.2 mg/kg DEX and blood samples were taken 6 h later as above.
GR mRNA expression
Stress-naive mice were killed, brains were immediately extracted, snap-frozen in isopentane, maintained in dry ice and then stored at −80 °C. Glucocorticoid (GR) mRNA was measured via in situ hybridization using an oligodeoxynucleotide (cDNA) probe complementary to GR mRNA (169–213 pb access number NM_008173), labeled with a digoxigenin (DIG) oligonucleotide 3′-OH tailing kit (Roche, Molecular Biochemicals, Mannheim, Germany). The hybridization procedure was as previously described (Bravo et al, 2009). Briefly, 10 μm-thick coronal brain sections were cut and mounted on superfrost glass slides (Menzel-Glaser, Menxel GmbH Germany). Sections were post-fixed in 4% paraformaldehyde made in PBS for 30 min, permeabilized with Proteinase K (0.5 mg/100 ml in TE buffer) and then treated with acetic anhydride buffer. Next, the slides were put through a series of ethanol dilutions (70, 95 and 100%) before being dehydrated in chloroform for 5 min. The tissues were then rehydrated and placed in a humidity chamber with the hybridization solution (formamide 50%, saline and sodium citrate buffer (SSC) 4 × , sheared salmon DNA 6.25 mg/ml, tRNA 125 μg/ml, and cDNA probe at fixed concentration of 100 pmol/ml for each probe) and incubated overnight at 37 °C. Sections were then washed in ascending dilutions of SSC buffer (4 × , 2 × , 1 × , and 0.5 × ) and equilibrated with maleic acid 0.1 M buffer before blocking for unspecific protein binding with Roche's blocking reagent (Roche, Molecular Biochemicals). After 30 min of blocking, the DIG molecules attached to the hybridized probes were detected with an anti-DIG antibody, conjugated with an alkaline phosphatase (Roche, Molecular Biochemicals). Finally, a substrate for the alkaline phosphatase (NBT/BCIP, Sigma, St Louis, MO) was added until a violet/blue precipitate was apparent. Slides were then left to air-dry and cover-slips were mounted with DPX mounting medium (Fisher Scientific, Loughborough, UK). Once the mounting medium was dry, hippocampal images from both hemispheres were obtained using an Olympus DP71 digital camera attached to an Olympus BX51 microscope (Olympus, Tokyo, Japan). Specificity of the hybridization was evaluated by the use of 100-fold excess of the unlabeled oligodeoxynucleotide (see Supplementary Figure S1).
For semi-quantitative analysis, densitometric measurements of each hippocampal area (DG, CA1 and CA3) were analyzed using FujiFilm's Science Lab Multi Gauge v2.2 software (Fuji Photo Film). Images were analyzed in grey scale and the value given by the software corresponds to the intensity of pixels (the darkest staining is the highest intensity; the lightest staining the lowest intensity) in a given area (density of pixels). The hybridization signal in the stratum radiatum was considered as background and was subtracted from the pixel density values obtained in the hippocampal cell layers. Strains were compared via t-tests.
Dendritic Morphology of BLA, IL, and PL Neurons
The dendritic morphology and spine density of pyramidal neurons in BLA, IL, and PL was determined using Glaser and Van der Loos' modified Golgi stain (Glaser and Van der Loos, 1981), essentially as described previously (Izquierdo et al, 2006). Within 3 h of the completion of extinction testing, mice were overdosed with xylazine/ketamine and then transcardially perfused with 0.9% saline. Brains were removed and immersed in Golgi-Cox solution (1 : 1 solution of 5% potassium dichromate and 5% mercuric chloride diluted 4 : 10 with 5% potassium chromate) for 18 days (optimal period determined from a pilot study). Brains were dehydrated, infiltrated with a graded series of celloidins, and embedded in 8% celloidin. Coronal sections were cut at 160 μm on a sliding microtome (American Optical 860, San Marcos) and alkalinized, developed, fixed, dehydrated, cleared, mounted, and cover-slipped.
Pyramidal neurons in layers II–III of IL and PL were drawn. PL was readily identified by its position on the medial wall of rostral cortex and its location dorsal to IL, which is markedly thinner than the PL area and has fewer, less well-defined layers. PL was distinguished from anterior cingulate cortex by the curve of the apical dendrites, which is more pronounced in anterior cingulate, as the cortex wraps around the midline. Layers II–III in PL is readily identifiable in Golgi-stained material based on its characteristic cytoarchitecture. It is positioned immediately ventral deep to the relatively cell-poor layer I (which also contains the distal dendritic tufts of layers II–III pyramidals) and superficial to layer V; this boundary is pronounced because of greater cell-packing density and smaller somata of pyramidal cells in layers II–III relative to layer V (Zilles and Wree, 1995). IL was distinguished from the ventrally adjacent dorsal peduncular cortex by thinning of gray matter and by increased distance between layers II–III and the cortical surface. Morphology of pyramidal cells in dorsal penduncular peduncular cortex is also markedly different from that of the IL cortex. BLA is readily identified in Golgi-stained material, as the external capsule branches into two smaller fiber tracts that define the dorsal, medial and lateral borders of the BLA.
Pyramidal neurons in these three regions were defined by the presence of at least two basilar dendritic trees with at least third-order branches, a distinct, single apical dendrite, and dendritic spines. Neurons selected for reconstruction did not have truncated branches and were unobscured by neighboring neurons and glia, with dendrites that were easily discriminable by focusing through the depth of the tissue. In 4–6 sections evenly spaced through the rostral-caudal extent of medial prefrontal cortex, all pyramidal neurons meeting these criteria were identified. An average of 5 neurons per mouse per region (average of 2.5 from each hemisphere) were randomly selected (using a random number generator, http://www.randomizer.org) from all identified neurons and reconstructed.
Neurons were drawn in three-dimensions by an experimenter blind to strain, using a 100 × objective on an Olympus BX41 system microscope using a computer-based neuron tracing system (Neurolucida, MBF Biosciences, Williston, VT). The length and number of dendrites, as well as the length and number of terminal branches, was measured for apical and basilar arbors. Strains were compared using t-tests. In addition, to assess the overall amount and location of dendritic material, a three-dimensional version of a Sholl analysis (Larkman, 1991; Wellman et al, 2007) was performed by measuring the number of dendritic intersections within 10-μm concentric spheres radiating from the soma. The effects of strain and distance from soma were analyzed using two-factor ANOVA (repeated measures for distance) and Newman–Keuls post hoc tests.
Dendritic spines were counted on first- through third-order branches, as these make up ∼90% of the dendritic arbor of IL and PL pyramidal neurons. For each neuron, spines were counted on segments of the basilar primary, secondary, and tertiary branches, and the oblique and horizontal branches of the apical trunk. Segments averaged ∼50 μm in length that were unobscured by neighboring dendrites and glia, and were not randomly selected. Spines were counted using Neurolucida as previously described (Mozhui et al, 2010). Strains were compared for spines per μm using t-tests.
RESULTS
Context Overgeneralization
One cohort of mice was tested using an ‘ABC1' Cxt design. Fear conditioning was in Cxt A via 3 × presentations of a footshock US. One day later, mice were exposed to Cxt A, Cxt B (different from A) and Cxt C1 (same as B, but with same grid floor as A) for 3-min sessions with 4 h between exposures and the order of exposure counterbalanced. Results showed that S1 and B6 were equally able to discriminate B and C1 from A, indicated by lower freezing to B and C1 than A (Cxt-type: F2, 28=38.81, p<0.01) (Figure 1a).
Figure 1.

Fear overgeneralization and impaired safety learning. (a) S1 and B6 froze more to the conditioned context (Cxt A) than either a novel context (Cxt B) or slightly ambiguous context (Cxt C1) (**p<0.01 vs Cxt A, n=8 per strain). (b) S1 froze more to the conditioned Cxt (Cxt A) than a novel Cxt (Cxt B) but not a highly ambiguous context (Cxt C2). B6 froze more to Cxt A than either Cxt B or Cxt C2 (**p<0.01 vs Cxt A/same strain, n=8 per strain). (c) During Cxt extinction, there was a decrease in freezing from the first to the last timebin in B6 but not S1, and S1 froze more than B6 during all timebins except the first (*p<0.05 vs B6/same timepoint, n=16 per strain). During the extinction retrieval test S1 froze more than B6 (**p<0.01 vs B6), although freezing was lower than it was at the beginning of extinction training in S1 (#p<0.05) and B6 (##p<0.01). (d) S1 and B6 were equally able to discriminate a novel CS– from a conditioned CS+ (n=13–14 per strain). (e) S1 were poorer than B6 at discriminating a partially reinforced CS– from a fully reinforced CS+ (**p<0.01 vs B6, n=9–12 per strain). (f) S1 and B6 were equally able to discriminate a novel CS– from a conditioned CS+ after fear extinction, although discrimination in B6 was accounted for by a sub-population of poorly extinguishing mice (‘B6 high') (see inset) (*p<0.05, **p<0.01 vs B6, ##p<0.01 vs CS+/same strain, n=10–14 per strain). (g) Safety-trained B6, but not S1, showed inhibition of contextual fear during presentation of a CS safety signal (##p<0.01 vs Cxt/B6, n=9–10 per strain). (h) Safety-trained B6 and S1 mice showed retardation of new fear learning, as compared with previously naive controls (**p<0.01 vs previously naive, n=6–9 per strain per group). (i) Following safety training in which the CS block always preceded the US block, S1 and B6 both showed inhibition of contextual fear during presentation of the CS safety signal (**p<0.01 vs Cxt, n=7 per strain). Data are means±SEM.
A separate cohort of mice was tested using an ‘ABC2' Cxt design. Here, mice were tested as above except that C1 was replaced by C2, which was rendered more ambiguous by having multiple features of Cxt A. Here, S1 discriminated B but not C2 from A (Cxt-type: F2, 28=29.51, p<0.01; strain × Cxt-type: F2, 28=3.22, p=0.0553) (Figure 1b).
One day after context discrimination, contextual fear extinction was tested via non-reinforced exposure to Cxt A for 30 min. S1 froze more than B6 on all timebins except the first, and there was a decrease in freezing from the first to last timebin in B6 not S1 (strain × time: F14, 420=2.80, p<0.01) (Figure 1c). One day later, extinction retrieval was tested by returning mice to Cxt A for a 3-min test. S1 showed more freezing than B6 (t30=4.15, p<0.01), although freezing was lower during retrieval than the corresponding time point before extinction training in S1 (t15=2.52, p<0.05) and B6 (t15=7.00, p<0.01) (Figure 1c).
Cue Overgeneralization
We first tested for the ability of mice to discriminate a novel stimulus from a fully conditioned stimulus. Mice were conditioned via 3 × pairings of a CS+ (7.5 kHz pure tone, or white noise pips) with footshock US in Cxt A. One day later, the CS+ and a novel CS (order counterbalanced) were presented in Cxt B. A discrimination index was calculated as freezing to the CS+ relative to total freezing to both stimuli, with higher scores indicating better discrimination. S1 and B6 showed high (>0.75) and similar cue discrimination (Figure 1d).
Next, we tested the ability to discriminate a fully conditioned CS+ from a CS– rendered ambiguously predictive of danger by being occasionally paired with footshock. Here, mice were conditioned via 6 × pairings of a CS+ with the US and 6 × presentations of a CS– paired only 2 × with the US. One day later, mice were presented with the CS+ and the ambiguous CS– (order of CS-type counterbalanced). S1 had a lower discrimination index than B6 (t19=3.57, p<0.01) (Figure 1e).
In a third experiment, we tested for discrimination of a novel stimulus from a fully conditioned CS+ after mice had undergone extinction to the CS+. Mice were conditioned via 5 × pairings of a CS+ (7.5 kHz or white noise pips) with footshock US. Over 2 days, mice were given 12 per day non-reinforced presentations of the CS+ in Cxt B. During the first (strain × trial: F1, 22=33.42, p<0.01) and second (strain: F1, 22=15.38, p<0.01; trial: F1, 22=9.53, p<0.01) extinction sessions, B6, not S1, reduced freezing (Figure 1f). Freezing was higher in S1 than B6 by the final trial of both sessions and on first trial of session 2, confirming impaired extinction. One day later, mice were presented with the CS+ and novel CS– (order counterbalanced). Both B6 and S1 showed more freezing to the CS+ than CS– (F1, 22=5.87, p<0.05, followed by post hoc tests), and S1 froze more than B6 regardless of CS-type (F1, 22=12.69, p<0.01; strain × CS-type interaction: p>0.05) (Figure 1f). As it has previously found that B6 segregate into good and poor extinguishing sub-populations (Herry and Mons, 2004), we also analyzed discrimination in B6 as a function of high (S1-like) or low CS+ freezing after extinction. This revealed that, as with S1, poor extinguishing B6 still retain good discrimination (t4=7.52, p<0.01) (Figure 1f, inset).
Impaired Safety Learning
As a measure of safety learning, mice were tested for contextual fear inhibition during presentation of a CS safety signal. B6, but not S1, showed fear inhibition to a safety signal (strain × CS: F1, 17=6.14, p<0.05) (Figure 1g). Both strains tested with explicit CS/US pairing showed robust and equivalent freezing to CS during a post-conditioning fear probe (S1=75.0±6.7%, B6=71.4±3.6, n=6–7).
In a second test for safety learning, both strains exhibited significant retardation of new fear learning (training-type: F1, 26=49.81, p<0.01), although there was a nonsignificant trend for lesser retardation in S1 than B6 (Figure 1h). Retardation was overcome after this second round of training in both strains (S1 naive=100±5%, S1 safety=77±10%, B6 naive=100±14, B6 safety=86±10).
In a separate cohort of mice, we tested for contextual fear inhibition after a safety training protocol in which the CS block always preceded the US block. Here, both strains exhibited contextual fear inhibition during presentation of the CS safety signal (CS effect: F1, 12=28.67, p<0.01) (Figure 1i).
FLX Rescue of Impaired Extinction and Context Overgeneralization
In mice given FLX before fear conditioning, freezing increased across conditioning trials (F3, 72=221.60, p<0.01), but FLX-treated S1 tended (post hoc: p=0.059) to freeze less by the final CS-trial than water-treated S1 counterparts (trials × strain interaction: F3, 174=3.09, p<0.01, trials × drug: F3, 174=4.72, p<0.01) (Figure 2b). During extinction training, B6 showed a greater decrease in freezing across trial-blocks than S1, although S1 again tended (post hoc: p=0.12) to freeze less after FLX treatment, relative to water treatment, during the first trial-block (strain × trial-block × drug: F9, 522=3.26, p<0.01) (Figure 2b). On the extinction retrieval test, water-treated S1 had higher freezing than B6, but this strain difference was absent in FLX-treated mice because of a FLX-induced reduction in S1 freezing (strain × drug: F1, 24=9.58, p<0.01, Figure 2c).
Figure 2.

FLX recue of impaired fear extinction and context overgeneralization. (a) Schematic of experimental design in which mice were FLX treated for 3 weeks before conditioning (Con), and remained on treatment through extinction training (Ext) and retrieval testing (Test). (b) S1 showed more freezing than B6 over extinction trials, regardless of treatment, although there was a nonsignificant trend for lesser freezing during the first trial-block in FLX-treated S1 mice (n=8 per strain per treatment). (c) S1 had higher freezing than B6 during extinction retrieval after water but not FLX treatment (**p<0.01 vs B6/water, ##p<0.01 vs S1/water, n=8 per strain per treatment). (d) Schematic of experimental design in which mice were FLX treated for 3 weeks starting after conditioning, and remained on treatment through testing. (e) S1 showed more freezing than B6 over extinction trials, regardless of treatment, although decreases in freezing were evident in all groups during the first four trial-blocks. (f) S1 had higher freezing than B6 during extinction retrieval after water but not (post-conditioning) FLX treatment (**p<0.01 vs B6/water, ##p<0.01 vs S1/water, n=7 per strain per treatment). (g) Schematic of experimental design in which mice were FLX treated for 3 weeks before context conditioning, and remained on treatment through the context generalization test. (h) S1 froze more to an ambiguous context than B6 after water but not FLX treatment (**p<0.01 vs B6/water, #p<0.05 vs S1/water, n=6–8 per strain per treatment). (i) Schematic of experimental design in which mice were FLX treated for 3 weeks before acute forced swim stress. (j) S1 had higher acute swim stress-induced CORT than B6 after water but not FLX treatment. CORT was higher in B6 after FLX treatment relative to water (**p<0.01 vs B6/water, ##p<0.05 vs B6/water, n=7–8 per strain per treatment). Data are means±SEM.
A separate cohort of mice was not given FLX until after fear conditioning. Freezing increased across conditioning trials (F3, 72=139.43, p<0.01), and strains and treatment groups did not differ (effects of strain/treatment and treatment × strain/trial: ns) (Figure 2e). During extinction training, B6 showed a greater decrease in freezing across trial-blocks than S1 (strain × trial-block: F9, 216=5.65, p<0.01), regardless of treatment (effect of treatment and treatment × strain/trial-block: ns) (Figure 2e). However, on the extinction retrieval test, water-treated S1 had higher freezing than similarly treated B6, and FLX treatment significantly reduced freezing in S1 to B6 levels (strain × drug: F1, 25=5.89, p<0.05) (Figure 2f).
We next tested the effects of FLX, given before conditioning, on context discrimination (ABC2 design above). FLX-treated S1 froze less than water-treated S1 to the ambiguous context, such that S1 froze more than B6 after water (as seen in Figure 1b) but not FLX (Figure 2h) (strain: F1, 34=11.03, p<0.01, drug: F1, 34=4.63, p<0.05, strain × drug: F1, 34=3.19, p=0.083, after exclusion of 1 S1 and 1 B6 with >2 SDs outlying scores).
Finally, we treated mice for 21 days with FLX and then measured CORT responses to acute swim stress. Results showed that water-treated S1 showed higher stress-induced CORT than B6, but not after FLX (strain × drug: F1, 25=5.27, p<0.05) because of a significant increase in CORT in FLX-treated B6 relative to water-treated B6 (Figure 2j).
Autonomic Abnormalities
Mice received explicitly paired CS/US (noise/footshock) training in Cxt A and, 1 day later, 50 × CS extinction training in the home cage (tested in this familiar environment to avoid ECG handling artifacts). S1 had higher basal HR (t100=6.35, p<0.01; S1=618.3±4.7, B6=566.8±6.6 heart b.p.m.) and lower HRV (t100=2.58, p<0.05; S1=3.30±.24, B6=4.60±.44 RMSSD index) relative to B6. Moreover, both strains showed increased HR (strain × CS-block: F2, 100=14.90, p<0.01) and decreased HRV from baseline to the first (CS1-25) block (strain × CS-block: F2, 100=3.24, p<0.01), but changes across these blocks were significantly greater in B6 than S1 (Figure 3a and b). Then, from the first to second extinction block, HRV (not HR) increased in B6 but did not change, and remained below baseline, in S1 (Figure 3b).
Figure 3.
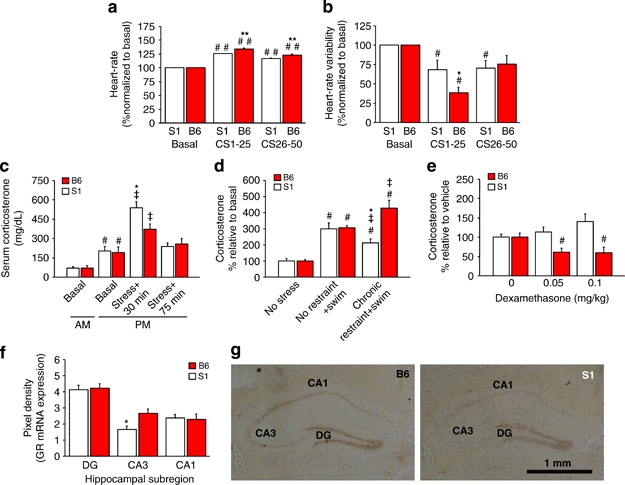
Autonomic and neuroendocrine abnormalities. (a) S1 and B6 showed an increase in HR from baseline to the first-half and second-half blocks of extinction training, although this increase was greater in B6 than S1 (##p<0.01 vs basal/same strain, **p<0.01 vs S1, n=8 per strain). (b) Both strains showed a reduction in HRV from baseline to the first-half block of extinction training, with a greater reduction in B6 than S1 and a recovery to baseline in B6, but not S1, on the second-half block (##p<0.05 vs basal/same strain, *p<0.05 vs S1, n=8 per strain). (c) AM or PM basal CORT did not differ between strains, and PM CORT was generally higher than AM CORT (#p<0.05 vs AM/same strain). CORT was increased 30 min after acute 2-h restraint stress, relative to basal PM (‡p<0.05 same strain) but was elevated more so in S1 than B6 (*p<0.05 vs B6). CORT returned to PM basal levels by 75 min (n=7–9 per strain per condition). (d) CORT was increased by acute swim stress (#p<0.05 vs basal/same strain) in previously stress-naive S1 and B6. In mice previously subjected to chronic restraint stress, acute swim produced an augmented CORT response in B6, but an attenuated response in S1, relative to the no restraint+swim groups (‡p<0.05 vs no restraint+swim, *p<0.05 vs B6, 5–14 per strain per group) (from Mozhui et al, 2010 with permission). (e) Injection with DEX (0.05 and 0.1 mg/kg) reduced CORT relative to vehicle in B6, but not in S1 (#p<0.05 vs 0 dose/B6, n=10–12 per strain per dose). (f) S1 had lower GR mRNA expression than B6 in the CA3, not dentate gyrus (DG) or CA1, hippocampal subregions (*p<0.05 vs B6/same region, n=5–6 per strain). (g) Representative in situ hybridization images of GR mRNA from S1 and B6 (scale bar=1 mm). Data are means±SEM.
Neuroendocrine Abnormalities
CORT levels were higher in the p.m. than a.m. but did not differ between strains (time: F3, 52=2.79, p<0.05) (Figure 3c). After a 2-h acute restraint stress, CORT levels were elevated in both strains at 30 min, but to a significantly greater extent in S1 than B6 (strain × stress: F1, 24=4.79, p<0.05) (Figure 3c). By 75-min post-stress, CORT levels had recovered to baseline in both strains (Figure 3c).
After exposure to 50 × CS extinction training, CORT levels were significantly higher in S1 than B6 (t20=3.40, p<0.01; S1=421±66, B6=208±29 mg/dl).
The CORT response to an acute swim stress was significantly lesser in S1 than B6 with a history of chronic restraint stress, but similar between strains that had no prior restraint stress (strain × stress: F2, 42=10.21, p<0.01) (Figure 3d).
Injection of DEX significantly suppressed CORT levels relative to vehicle in B6 but not S1 (strain × treatment: F2, 59=3.69, p<0.05) (Figure 3e). Both strains did, however, show similar CORT suppression after challenge with a very high dose of DEX (vehicle as %vehicle=100.0±10.5, 0.2 mg/kg=22.6±7.3, F2, 19=23.26, p<0.01).
S1 had significantly lower GR mRNA expression than B6 in the CA3, but not DG or CA1, hippocampal subregion (t9=2.76, p<0.05) (Figures 3f and g) (levels of unlabeled oligo probe were no different).
Increased BLA Dendritic Arborization
In BLA neurons, apical, but not basilar (S1=5.16±0.43 μm, B6=5.26±0.13) dendrites were longer in S1 than B6 (t14=2.18, p<0.05) (Figures 4a and b). Spine density was not different between strains (Figure 4c). Sholl analysis indicated that S1 had more apical dendritic material than B6, particularly at distances relatively distal from the soma (strain × distance-from-soma: F11, 154=1.87, p<0.05) (Figure 4d).
Figure 4.

Increased dendritic arborization of BLA neurons. (a) Examples of reconstructed BLA neurons in S1 and B6. (b) Apical dendrites in BLA were on average longer in S1 than B6. (c) BLA spine density was no different between strains. (d) Sholl analysis showing greater apical dendritic material relatively distal from the soma in S1 as compared with B6. n=6–8 neurons per mouse, n=6–8 mice per strain. Data are means±SEM. *p<0.05 vs B6.
In PL neurons, apical dendritic length did not differ between strains as measured by overall averages (Figure 5a and b) or Sholl analysis (Figure 5c). PL spine density was similar in S1 and B6 (Figure 5d).
Figure 5.

Normal PL and IL dendritic morphology and spine density. (a) Examples of reconstructed PL neurons. Apical dendritic length of PL neurons did not differ between strains as measured by overall averages (b) or Sholl analysis (c). (d) PL spine density was similar in S1 and B6. (e) Examples of reconstructed IL neurons. Apical dendritic length of IL neurons did not differ between strains as measured by overall averages (f) or Sholl analysis (g). (h) IL spine density was similar in S1 and B6. n=6–8 mice per strain per region. Data are means±SEM.
Finally, strains did not differ in apical dendritic length of IL neurons, measured either by overall averages (Figures 5e and f) or Sholl analysis (Figure 5g). IL spine density was similar across strains (Figure 5h).
DISCUSSION
Anxiety patients perceive ambiguous stimuli and places ‘that resemble or symbolize an aspect of the traumatic event' as threatening (Bonne et al, 2004; DSM-IV, 1994). We tested for a similar bias in S1 by assessing discrimination of contexts and cues ambiguously associated with trauma. S1 and B6 were equally able to discriminate between a novel context and slightly ambiguous, but S1 generalized fear to an ambiguous context with multiple features of the conditioned context. In addition, extinction of fear to the conditioned context was impaired in S1, relative to B6, extending the previous observation that the strain exhibits profoundly deficient cued fear extinction (Hefner et al, 2008; Whittle et al, 2010). We also found that while S1 and B6 equally discriminated a novel from a fully conditioned auditory cue (after either conditioning or extinction), S1 froze more than B6 to an auditory cue ambiguously predictive of danger. Thus, S1 exhibited a bias to overgeneralize fear to both ambiguous contexts and discrete environmental cues. Whether this reflects a broader tendency of S1 to generalize learning remains to be determined, for example, by testing for generalization of positively reinforced stimuli, such as drug and food rewards. Although it is relevant to note here that, in contrast to impaired fear extinction, extinction of an instrumental response for reward is normal in S1 (Hefner et al, 2008).
Impaired fear extinction and fear generalization in S1 could be a reflection of a more general deficit in the ability to form or express memories that inhibit fear. We tested for a form of ‘safety learning' that results from explicit CS–US unpairing and produces conditioned fear inhibition to contexts and retardation of new fear learning (Rescorla, 1969). Unlike B6, S1 failed to show inhibition of a contextual response, demonstrative of impaired safety learning. Interestingly, however, both strains exhibited retardation of new fear learning following safety training. This suggests that S1 acquired a safety signal but failed to express it to overcome a contextual fear response. Moreover, we found that S1 were in fact able to successfully demonstrate contextual inhibition using a training protocol in which the CS safety signal was consistently presented before footshock during safety training. In other words, the order in which safety and trauma signals are experienced is a critical determinant of the ability of S1 to show safety learning. Although the precise nature of this deficit will need to be delineated in future studies, these data demonstrate that the S1 fear phenotype goes beyond the cued extinction deficit we originally observed to encompass multiple impairments in inhibitory fear learning.
Another novel finding was that the S1 fear phenotype was rescued by FLX, a frontline pharmacotherapeutic for trauma-related anxiety disorders such as PTSD. Specifically, chronic FLX normalized long-term cued fear extinction in S1 to B6 levels, without producing any observable facilitation of extinction in B6 mice. This effect was evident either when FLX was given before conditioning or between conditioning and extinction. Furthermore, chronic FLX treatment also resolved strain differences in contextual fear generalization. Interestingly, FLX normalized S1 fear to B6 levels on the post-extinction retrieval test without producing reductions in fear during extinction training. The most parsimonious account of this pattern of drug effects is that FLX selectively promoted the consolidation of the extinction memory, and not its acquisition. This contrasts with other manipulations, such as dietary Zinc depletion, which strongly facilitates extinction acquisition in S1 (Whittle et al, 2010) and suggest that FLX treatment might selectively targets processes, such as IL neuronal bursting (Burgos-Robles et al, 2007), which are implicated in extinction-consolidation. This could be interrogated further by, for example, measuring IL bursting in FLX-treated S1 or testing whether the ability of FLX to reduce fear on extinction–retrieval is contingent upon extinction training (ie, consolidation dependent). Further studies could also extend the current data set by testing the effects of FLX on other fear abnormalities in S1, including contextual fear extinction and safety learning. In lieu of these additional studies, these rescue experiments are of importance to the S1 as a potential model with clinical relevance because SSRIs such as FLX and sertraline remain first-line treatments for anxiety disorders (Brady et al, 2000). By extension, S1 could be used as a tractable model to screen for novel fear inhibiting treatments (Holmes and Quirk, 2010).
Providing further support for the face validity of the S1 as a clinical model, we found perturbations in various autonomic and neuroendocrine parameters. Behavioral abnormalities in mood and anxiety disorders are accompanied by autonomic and neuroendocrine disturbances, including reduced HRV in at least some anxiety patients (Cohen and Benjamin, 2006). Measuring HR and HRV at baseline and during extinction learning via i.p. implanted ECG transmitter revealed that S1 had higher basal HR and lower HRV relative to B6. As HR and HRV regulation are strongly modulated by blood pressure (Fazan et al, 2005), these low HRV values might reflect higher (around 10% vs B6) basal systolic blood pressure in S1 (Barrick et al, 2007).
During extinction, both strains showed increased HR and decreased HRV from baseline to the first half of extinction training (as previously seen in Gaburro et al, 2011), but these changes were greater in B6 than S1. Furthermore, HRV (not HR) recovered in B6 from the first to second half of extinction training, but remained depressed below baseline in S1. These data indicate a relative lack of plasticity in the S1 HRV response during fear retrieval and extinction, and echoes slow recovery of HRV seen after trauma recall in human subjects who experienced relatively severe trauma (Arditi-Babchuk et al, 2009). Autonomic disturbances of this kind could reflect aberrant sympathovagal balance, mediated via brainstem nuclei as well as higher corticolimbic regions (Neafsey, 1990). Indeed, recent human fMRI work has shown an association between reduced HRV, increased anxiety, and abnormal corticolimbic activity (Mujica-Parodi et al, 2009).
Corticolimbic dysfunction is also implicated in the neuroendocrine alterations found in depression and anxiety disorders, such as abnormal circulating cortisol levels and altered sensitivity to low-dose DEX cortisol suppression (Muller and Holsboer, 2006; Yehuda and LeDoux, 2007). In this context, we found that CORT levels were higher in S1 than B6 30 min after 2-h restraint, then recovered to an equivalent baseline in both strains by 75 min. CORT was also higher in S1 than B6 after cued extinction training, indicating an augmented HPA-axis response to various acute stressors in S1. A notably different pattern of strain differences emerged after chronic (10-day restraint) stress. Specifically, we observed that a novel swim stressor produced equivalent CORT increases in restraint-naive S1 and B6, but that the CORT response was further augmented in B6 with a history of chronic restraint, and, conversely, attenuated in S1. Thus, B6 were hypersensitive to novel stressor after chronic stress, while S1 became hyposensitive.
We also found that treatment with the synthetic glucocorticoid agonist DEX suppressed p.m. CORT relative to vehicle in B6 but not S1. S1 were not completely insensitive to DEX, as demonstrated by challenge with a very high dose. This relative resistance to DEX (and exaggerated stress-induced CORT) resembles the effect of constitutive GR haploinsufficiency or forebrain-specific GR knockout (Boyle et al, 2005; Ridder et al, 2005). Indeed, in situ hybridization revealed lower GR mRNA expression than B6 in the CA3, but not DG or CA1, hippocampal subregion of S1, relative to B6, mice. Collectively, these data suggest downregulation of GR-mediated negative feedback in S1 under stress-naive conditions that may revert to upregulation after a history of chronic stress. Precisely how this HPA-axis dysregulation might be a diathesis variable contributing to excessive fear in S1 remains a key question for future studies. For example, given recent evidence that elevating glucocorticoids promotes negative bias in rats (Enkel et al, 2010), it will be interesting to determine whether glucocorticoid abnormalities contribute to the fear generalization phenotype in S1.
There is compelling evidence implicating a PFC-amygdala neural circuit in excessive fear in humans and rodents (Quirk and Mueller, 2008). Previous studies have demonstrated that impaired fear extinction in S1 is associated with abnormal activation of this circuit, consistent with a failure to properly engage regions of PFC and amygdala, and a resultant bias toward sustained amygdala output (Hefner et al, 2008; Whittle et al, 2010). Via analysis of the dendritic arborization of BLA pyramidal neurons, we found that S1 had longer dendrites than B6, and more apical dendritic material than B6, particularly at distances relatively distal from the soma. Spine density was not different between strains, although the overall number of synapses on the apical dendrites was likely greater in S1 given their longer arbors. In contrast to these changes in BLA neurons, we did not find any significant differences between S1 and B6 in the morphology or spine density of neurons in either IL or PL. This does not exclude the possibility of more subtle structural changes in these neurons that we were unable to detect. However, the more parsimonious conclusion is that the clear functional alterations we have found in these prefrontal areas, including IL hypoactivation and PL hyperactivation during extinction (Hefner et al, 2008; Whittle et al, 2010), are not associated with major neuronal morphological alterations. These may, instead, likely be driven by to be determined molecular abnormalities or abnormal interactions with other regions (eg, the amygdala).
One key question is how the enlarged apical arbors of BLA neurons relate to the fear abnormalities in S1 mice. Enlarged dendritic arborization in S1 is highly reminiscent of the finding that stress causes dendritic hypertrophy and spinogenesis in BLA pyramidal neurons and produces associated increases in anxiety-like behavior (Vyas et al, 2002) (stress causes dendritic retraction of prefrontal neurons, by contrast, eg, Holmes and Wellman, 2009; Roozendaal et al, 2009; Shansky et al, 2009). One hypothesis is that once encoded, fear signaled by these larger neurons may be resistant to extinction. Combined with the failure to recruit extinction processes mediated by other brain regions, such as IL, in S1 (Hefner et al, 2008; Whittle et al, 2010), this could provide a neural substrate for the rigid maintenance and overgeneralization of fear in S1. More generally, these observations provide novel evidence of an important link between amygdala dysmorphology and excessive fear and anxiety.
In this study, we found that the S1 inbred mouse strain exhibits a tendency to overgeneralize fear to ambiguous environmental cues and contexts and has difficulty using cues that signal safety to inhibit fear—two processes posited to underlie the intractable nature of excessive anxiety in the clinic. We also found that S1 display a depression of resting HRV, and persistent autonomic disturbances during extinction training. HPA-axis abnormalities in S1 were consistent with a loss of GR-mediated negative feedback regulation. Supporting the utility of the model as a tool for drug discovery, we found that behavioral abnormalities in S1 were rescued by the front-line anxiety medication, FLX. It also provides further insight into the neural basis of the aberrant phenotype in S1, we found enlarged apical dendritic arbors in the pyramidal neurons of a key brain region within the fear circuit, the BLA.
Collectively, these data provide convergent support for the validity of the S1 strain as a model of excessive fear. This model could prove valuable for future studies aimed at elucidating the neural and molecular basis of fear-related disorders such as PTSD, with a view to developing novel behavioral and pharmacological approaches to treating the disorder. Finally, because inbred mouse strains offer a powerful approach to finding gene–phenotype associations, the S1 strain provides a unique model for identifying genes underlying individual differences in trait fear and risk for fear disorders.
Acknowledgments
We thank Daniela Pollak, Fred Helmstetter, and O Stiedl for valuable discussions and methods advice, and Heather Cameron for RIA equipment. This study was supported by the NIAAA Intramural Research Program and the Austrian Science Fund (F4410-B19).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary Material
References
- Arditi-Babchuk H, Feldman R, Gilboa-Schechtman E. Parasympathetic reactivity to recalled traumatic and pleasant events in trauma-exposed individuals. J Trauma Stress. 2009;22:254–257. doi: 10.1002/jts.20417. [DOI] [PubMed] [Google Scholar]
- Barrick CJ, Rojas M, Schoonhoven R, Smyth SS, Threadgill DW. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am J Physiol Heart Circ Physiol. 2007;292:H2119–H2130. doi: 10.1152/ajpheart.00816.2006. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- Bonne O, Grillon C, Vythilingam M, Neumeister A, Charney DS. Adaptive and maladaptive psychobiological responses to severe psychological stress: implications for the discovery of novel pharmacotherapy. Neurosci Biobehav Rev. 2004;28:65–94. doi: 10.1016/j.neubiorev.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Cameron HA, Holmes A. Chronic swim stress alters sensitivity to acute behavioral effects of ethanol in mice. Physiol Behav. 2007;91:77–86. doi: 10.1016/j.physbeh.2007.01.024. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, et al. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci USA. 2005;102:473–478. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder: a randomized controlled trial. JAMA. 2000;283:1837–1844. doi: 10.1001/jama.283.14.1837. [DOI] [PubMed] [Google Scholar]
- Bravo JA, Diaz-Veliz G, Mora S, Ulloa JL, Berthoud VM, Morales P, et al. Desipramine prevents stress-induced changes in depressive-like behavior and hippocampal markers of neuroprotection. Behav Pharmacol. 2009;20:273–285. doi: 10.1097/FBP.0b013e32832c70d9. [DOI] [PubMed] [Google Scholar]
- Breslau N, Kessler RC. The stressor criterion in DSM-IV posttraumatic stress disorder: an empirical investigation. Biol Psychiatry. 2001;50:699–704. doi: 10.1016/s0006-3223(01)01167-2. [DOI] [PubMed] [Google Scholar]
- Brigman JL, Mathur P, Harvey-White J, Izquierdo A, Saksida LM, Bussey TJ, et al. Pharmacological or genetic inactivation of the serotonin transporter improves reversal learning in mice. Cereb Cortex. 2010;20:1955–1963. doi: 10.1093/cercor/bhp266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Camp M, Norcross M, Whittle N, Feyder M, D'Hanis W, Yilmazer-Hanke D, et al. Impaired Pavlovian fear extinction is a common phenotype across genetic lineages of the 129 inbred mouse strain. Genes Brain Behav. 2009;8:744–752. doi: 10.1111/j.1601-183X.2009.00519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Hariri AR, Holmes A, Uher R, Moffitt TE. Genetic sensitivity to the environment: the case of the serotonin transporter gene and its implications for studying complex diseases and traits. Am J Psychiatry. 2010;167:509–527. doi: 10.1176/appi.ajp.2010.09101452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Zorrilla E, Smith S, Rousso D, Levy C, Vaughan J, et al. Urocortin 2-deficient mice exhibit gender-specific alterations in circadian hypothalamus-pituitary-adrenal axis and depressive-like behavior. J Neurosci. 2006;26:5500–5510. doi: 10.1523/JNEUROSCI.3955-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen H, Benjamin J. Power spectrum analysis and cardiovascular morbidity in anxiety disorders. Auton Neurosci. 2006;128:1–8. doi: 10.1016/j.autneu.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Cohen H, Geva AB, Matar MA, Zohar J, Kaplan Z. Post-traumatic stress behavioural responses in inbred mouse strains: can genetic predisposition explain phenotypic vulnerability. Int J Neuropsychopharmacol. 2008;11:331–349. doi: 10.1017/S1461145707007912. [DOI] [PubMed] [Google Scholar]
- Depino AM, Gross C. Simultaneous assessment of autonomic function and anxiety-related behavior in BALB/c and C57BL/6 mice. Behav Brain Res. 2007;177:254–260. doi: 10.1016/j.bbr.2006.11.025. [DOI] [PubMed] [Google Scholar]
- DSM-IV 1994Diagnostic and Statistical Manual of Mental Disorders4th edn.APA Press: Washington, DC [Google Scholar]
- Duvarci S, Bauer EP, Pare D. The bed nucleus of the stria terminalis mediates inter-individual variations in anxiety and fear. J Neurosci. 2009;29:10357–10361. doi: 10.1523/JNEUROSCI.2119-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkel T, Gholizadeh D, von Bohlen Und Halbach O, Sanchis-Segura C, Hurlemann R, Spanagel R, et al. Ambiguous-cue interpretation is biased under stress- and depression-like states in rats. Neuropsychopharmacology. 2010;35:1008–1015. doi: 10.1038/npp.2009.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazan R, Jr, de Oliveira M, da Silva VJ, Joaquim LF, Montano N, Porta A, et al. Frequency-dependent baroreflex modulation of blood pressure and heart rate variability in conscious mice. Am J Physiol Heart Circ Physiol. 2005;289:H1968–H1975. doi: 10.1152/ajpheart.01224.2004. [DOI] [PubMed] [Google Scholar]
- Gaburro S, Stiedl O, Giusti P, Sartori SB, Landgraf R, Singewald N. A mouse model of high trait anxiety shows reduced heart rate variability that can be reversed by anxiolytic drug treatment. Int J Neuropsychopharmacol: 2011;14:1–15. doi: 10.1017/S1461145711000058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–125. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Luthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science. 2009;325:1258–1261. doi: 10.1126/science.1174146. [DOI] [PubMed] [Google Scholar]
- Gonzalez F, Quinn JJ, Fanselow MS. Differential effects of adding and removing components of a context on the generalization of conditional freezing. J Exp Psychol Anim Behav Process. 2003;29:78–83. [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, et al. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Herry C, Mons N. Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur J Neurosci. 2004;20:781–790. doi: 10.1111/j.1460-9568.2004.03542.x. [DOI] [PubMed] [Google Scholar]
- Holmes A, Quirk GJ. Pharmacological facilitation of fear extinction and the search for adjunct treatments for anxiety disorders – the case of yohimbine. Trends Pharmacol Sci. 2010;31:2–7. doi: 10.1016/j.tips.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Wellman CL. Stress-induced prefrontal reorganization and executive dysfunction in rodents. Neurosci Biobehav Rev. 2009;33:773–783. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26:5733–5738. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson RM, Choe JS, Cameron HA, Thorsell A, Crawley JN, Holmes A, et al. The neuropeptide Y Y1 receptor subtype is necessary for the anxiolytic-like effects of neuropeptide Y, but not the antidepressant-like effects of fluoxetine, in mice. Psychopharmacology (Berl) 2008;195:547–557. doi: 10.1007/s00213-007-0945-2. [DOI] [PubMed] [Google Scholar]
- Kendler KS. Twin studies of psychiatric illness: an update. Arch Gen Psychiatry. 2001;58:1005–1014. doi: 10.1001/archpsyc.58.11.1005. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617–627. doi: 10.1001/archpsyc.62.6.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman AU. Dendritic morphology of pyramidal neurones of the visual cortex of the rat: I. Branching patterns. J Comp Neurol. 1991;306:307–319. doi: 10.1002/cne.903060207. [DOI] [PubMed] [Google Scholar]
- Lechner O, Dietrich H, Oliveira dos Santos A, Wiegers GJ, Schwarz S, Harbutz M, et al. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J Autoimmun. 2000;14:325–333. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsukawa K, Mombereau C, Lotscher E, Uzunov DP, van der Putten H, Flor PJ, et al. Metabotropic glutamate receptor subtype 7 ablation causes dysregulation of the HPA axis and increases hippocampal BDNF protein levels: implications for stress-related psychiatric disorders. Neuropsychopharmacology. 2006;31:1112–1122. doi: 10.1038/sj.npp.1300926. [DOI] [PubMed] [Google Scholar]
- Mozhui K, Karlsson RM, Kash TL, Ihne J, Norcross M, Patel S, et al. Strain differences in stress responsivity are associated with divergent amygdala gene expression and glutamate-mediated neuronal excitability. J Neurosci. 2010;30:5357–5367. doi: 10.1523/JNEUROSCI.5017-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muigg P, Hetzenauer A, Hauer G, Hauschild M, Gaburro S, Frank E, et al. Impaired extinction of learned fear in rats selectively bred for high anxiety – evidence of altered neuronal processing in prefrontal-amygdala pathways. Eur J Neurosci. 2008;28:2299–2309. doi: 10.1111/j.1460-9568.2008.06511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujica-Parodi LR, Korgaonkar M, Ravindranath B, Greenberg T, Tomasi D, Wagshul M, et al. Limbic dysregulation is associated with lowered heart rate variability and increased trait anxiety in healthy adults. Hum Brain Mapp. 2009;30:47–58. doi: 10.1002/hbm.20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller MB, Holsboer F. Mice with mutations in the HPA-system as models for symptoms of depression. Biol Psychiatry. 2006;59:1104–1115. doi: 10.1016/j.biopsych.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Neafsey EJ.1990Prefrontal cortical control of the autonomic nervous system: anatomical and physiological observations Prog Brain Res 85147–165.discussion 165-6. [DOI] [PubMed] [Google Scholar]
- Pavlov IP. Conditioned Reflexes. Oxford University Press: London; 1927. [Google Scholar]
- Pollak DD, Monje FJ, Zuckerman L, Denny CA, Drew MR, Kandel ER. An animal model of a behavioral intervention for depression. Neuron. 2008;60:149–161. doi: 10.1016/j.neuron.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research – past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rescorla RA. Conditioned inhibition of fear resulting from negative CS-US contingencies. J Comp Physiol Psychol. 1969;67:504–509. doi: 10.1037/h0027313. [DOI] [PubMed] [Google Scholar]
- Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, et al. Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature. 2011;470:492–497. doi: 10.1038/nature09856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, et al. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25:6243–6250. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Leon KS, Perez DL, Kandel ER. Distinct neural signatures for safety and danger in the amygdala and striatum of the mouse. Neuron. 2005;46:309–320. doi: 10.1016/j.neuron.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McEwen BS, Chattarji S. Stress, memory and the amygdala. Nat Rev Neurosci. 2009;10:423–433. doi: 10.1038/nrn2651. [DOI] [PubMed] [Google Scholar]
- Shansky RM, Hamo C, Hof PR, McEwen BS, Morrison JH. Stress-induced dendritic remodeling in the prefrontal cortex is circuit specific. Cereb Cortex. 2009;19:2479–2484. doi: 10.1093/cercor/bhp003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiedl O, Meyer M, Kishimoto T, Rosenfeld MG, Spiess J. Stress-mediated heart rate dynamics after deletion of the gene encoding corticotropin-releasing factor receptor 2. Eur J Neurosci. 2003;17:2231–2235. doi: 10.1046/j.1460-9568.2003.02659.x. [DOI] [PubMed] [Google Scholar]
- Stiedl O, Spiess J. Effect of tone-dependent fear conditioning on heart rate and behavior of C57BL/6N mice. Behav Neurosci. 1997;111:703–711. doi: 10.1037//0735-7044.111.4.703. [DOI] [PubMed] [Google Scholar]
- Tovote P, Meyer M, Beck-Sickinger AG, von Horsten S, Ove Ogren S, Spiess J, et al. Central NPY receptor-mediated alteration of heart rate dynamics in mice during expression of fear conditioned to an auditory cue. Regul Pept. 2004;120:205–214. doi: 10.1016/j.regpep.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Tsetsenis T, Ma XH, Lo Iacono L, Beck SG, Gross C. Suppression of conditioning to ambiguous cues by pharmacogenetic inhibition of the dentate gyrus. Nat Neurosci. 2007;10:896–902. doi: 10.1038/nn1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman CL, Izquierdo A, Garret JE, Martin KP, Carroll J, Millstein R, et al. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J Neurosci. 2007;27:684–691. doi: 10.1523/JNEUROSCI.4595-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J Neurosci. 2010;30:13586–13596. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. 2007;56:19–32. doi: 10.1016/j.neuron.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Zilles K, Wree A. Cortex: Areal and Laminar Structure. Academic Press: San Diego; 1995. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.