

Abstract
Acute ethanol intoxication increases the production of reactive oxygen species (ROS). Hemorrhagic shock with subsequent resuscitation (H/R) also induces ROS resulting in cellular and hepatic damage in vivo. We examined the role of acute ethanol intoxication upon oxidative stress and subsequent hepatic cell death after H/R. 14 h before H/R, rats were gavaged with single dose of ethanol or saline (5 g/kg, EtOH and ctrl; H/R_EtOH or H/R_ctrl, resp.). Then, rats were hemorrhaged to a mean arterial blood pressure of 30 ± 2 mmHg for 60 min and resuscitated. Two control groups underwent surgical procedures without H/R (sham_ctrl and sham_EtOH, resp.). Liver tissues were harvested at 2, 24, and 72 h after resuscitation. EtOH-gavage induced histological picture of acute fatty liver. Hepatic oxidative (4-hydroxynonenal, 4-HNE) and nitrosative (3-nitrotyrosine, 3-NT) stress were significantly reduced in EtOH-gavaged rats compared to controls after H/R. Proapoptotic caspase-8 and Bax expressions were markedly diminished in EtOH-gavaged animals compared with controls 2 h after resuscitation. EtOH-gavage increased antiapoptotic Bcl-2 gene expression compared with controls 2 h after resuscitation. iNOS protein expression increased following H/R but was attenuated in EtOH-gavaged animals after H/R. Taken together, the data suggest that acute EtOH-gavage may attenuate H/R-induced oxidative stress thereby reducing cellular injury in rat liver.
1. Introduction
Excessive ethanol consumption is one of the leading causes of mortality in the United States [1]. It is associated with one-third of all traumatic injury deaths per year [2, 3]. About 47% of trauma patients have positive blood alcohol concentrations (BACs) at the time of injury, of those up to 15% have legal intoxication BAC levels [4–6]. Ethanol consumption is associated with increased risk of inflammatory and infectios complications in trauma patients such as pneumonia, sepsis, and multiple organ failure (MOF) and higher morbidity and mortality [7–13]. Controversy results reported from others show that acute ethanol intoxication does not affect the outcome after trauma [13, 14]. Our recent findings show that acute ethanol gavage improves survival after hemorrhagic shock with subsequent resuscitation (H/R) in vivo [15]. Other clinical results show that acute ethanol intoxication is associated with decreased 24 h mortality after trauma (own unpublished data).
Acute ethanol abuse suppressed innate immunity and production of the proinflammatory cytokines as well as their release in human and animal studies [7, 8, 16–18]. Previously, we demonstrated an anti-inflammatory effect of an acute ethanol gavage, which was associated with reduced NF-κB activity and improved H/R-induced liver injury and a reduction in 72 h mortality in rats after massive blood loss [15]. However, excessive ethanol consumption is linked to increased production of reactive oxygen and nitrogen species (RONS) and oxidative stress due to the metabolism of ethanol resulting in liver injury [19–21]. Chronic ethanol use is described to increase inducible nitric oxide synthase (iNOS) expression which leads to high amounts of nitric oxide and its oxidation product peroxynitrite in the liver [22–24]. Peroxynitrite represents an important DNA-damaging reactive nitrogen species after an ethanol binge [22, 23, 25]. Increased formation of RONS by hepatocytes, activated macrophages, and the infiltrating neutrophils damages hepatic proteins, lipids, and DNA [26–28].
Moreover, ROS can initiate both apoptosis and necrosis [29–31]. An alternative pathway in which ROS may damage the liver cells is mitochondrial damage via the multidomain Bcl-2 family of proteins, including Bax and Bcl-2. Increased expression of proapoptotic Bax has been observed in patients with alcoholic liver disease [32]. Oxidative stress can contribute to liver dysfunction after H/R [33]. Therefore, RONS may play a crucial role in the induction and progression of hepatic injury after H/R.
Finally, the harmful pathophysiological changes after H/R can lead to MOF, with increased mortality after trauma up to 60% [34–37]. Hemorrhagic shock initiates an inflammatory response characterized by enhanced activation of immune cells (monocytes, polymorphonuclear leukocytes, PMNL), production and release of proinflammatory cytokines, accumulation of neutrophils in the liver, and enhanced production of RONS [38–40]. These changes also result in damaged cell membranes, DNA, and proteins, with subsequent organ damage induced by hemorrhagic shock [27, 28, 41–43].
To address this important issue, we analyzed hepatic oxidative and nitrosative stress and apoptosis in rats after acute ethanol gavage prior H/R.
2. Material and Methods
2.1. Animals and Experimental Model
Female LEWIS rats (180–250 g) were obtained from Harlan (Borchen, Germany). Fourteen hours before H/R, rats were gavaged with single dose of ethanol or saline (5 g/kg, 30% EtOH, H/R_EtOH or H/R_ctrl, resp.). After an overnight fast, rats were anesthetized with isoflurane (1.5%), and the right carotid artery, the right femoral artery, and the left jugular vein were cannulated with polyethylene tubing. Then, hemorrhagic shock was induced over 5 min by withdrawing blood from the right carotid artery into a heparinized syringe (10 U) to a mean blood pressure of 30 ± 2 mmHg. Systemic blood pressure was monitored in the right femoral artery using a blood pressure analyzer (BPA 400, Digi-Med, Louisville, KY). Constant pressure was maintained by further withdrawal of small volumes of blood as necessary for 60 min. Then, rats were resuscitated by transfusion of 60% of the shed blood plus a volume of lactated Ringer's solution corresponding to 50% of the shed blood volume with a syringe pump over 30 min via the left jugular vein [39]. At the end of resuscitation, catheters were removed, the vessels were occluded and the wounds were closed. Control groups underwent all surgical procedures without H/R (sham_ctrl and sham_EtOH, resp.). Animals were reanesthetized at 2 h, 24 h and 72 h after the end of resuscitation and liver was harvested. For each rat, the two right dorsal liver lobes were snap-frozen in liquid nitrogen. The remaining liver was flushed with normal saline, infused and fixed with 10% buffered formalin through the portal vein, embedded in paraffin, and subsequently sectioned and stained. Body temperature was measured in the colon and maintained at 37°C throughout the experiment with a heating pad. Animal protocols were approved by the Veterinary Department of the Regional Council in Darmstadt, Germany.
2.2. Group Allocation
Twenty-four animals were randomly allocated to 4 groups. Each group included 6 rats. 2 groups of sham-operated animals underwent surgical procedures, but H/R was not carried out. Two groups of shock animals underwent hemorrhage followed by resuscitation. In one sham and one shock group, respectively, rats were gavaged either with EtOH or saline in the same way as described above. Tissue was harvested at 2 h after resuscitation. Twelve animals were also randomly allocated to 4 groups (n = 3 per each group) and euthanised at 24 h after resuscitation. Twenty-four animals were allocated to 4 groups as described above and euthanised at 72 h after resuscitation (n = 6).
2.3. Immunohistochemistry
Paraffin-embedded liver tissue was sectioned, deparaffinized, and rehydrated. Then endogenous peroxidase activity was blocked with hydrogen peroxide (2% for 10 min). Hepatic sections were incubated with blocking solution (10% Tween 20 and 2% bovine serum albumin in phosphate-buffered saline) for 1 h at RT. Rabbit antibody against 4-hydroxynonenal (4-HNE, Alpha Diagnostics International, Biotrend, Cologne, Germany) diluted 1 : 500 in phosphate-buffered saline (pH 7.4) containing 10% Tween 20 and 1% bovine serum albumin was used as primary antibody. Anti-rabbit horseradish peroxidase-linked secondary antibody and diaminobenzidine (Peroxidase EnVision Kit, DakoCytomation, Hamburg, Germany) were used to detect specific binding. Sections were counterstained with hematoxylin. The immunostained tissue sections were captured at 400x and analyzed in a blinded manner. The extent of labeling in the liver lobule was defined as the percentage of the field area within a preset color range determined by the software (Adobe Photoshop 7.0). Data from each tissue section (10 fields per section) were pooled to determine mean values, as described previously [44].
After blocking as described above, the following staining protocol was used for the detection of 3-nitrotyrosine. A mouse antibody against nitrotyrosine (HyCult Biotechnology, Uden, the Netherlands) diluted 1 : 500 in phosphate-buffered saline (pH 7.4) containing 10% Tween 20 and 1% bovine serum albumin was used as primary antibody. A kit system consisting of anti-mouse horseradish peroxidase-linked secondary antibody (Simple Stain Rat MAX PO MULTI, Histofine, Nichirei Biosciences Inc., Tokyo, Japan) and diaminobenzidine (Detection UltraVision Plus Detection System DAB, Lab Vision, Fremont, USA) were used to detect specific binding. Sections were counterstained with hematoxylin, captured at 400x, and analyzed in a blinded manner. The extent of labeling in the liver lobule was defined as the percentage of the field area within a preset color range determined by the software (Adobe Photoshop 7.0). Data from each tissue section (10 fields per section) were pooled to determine mean values, as described previously [40].
2.4. Western Blotting for Intracellular Signalling
Liver tissue was homogenized in lysis buffer at 4°C, followed by centrifugation for 30 min at 4°C at 20.000 g. Supernatants were stored at −80°C for later analysis. Lysates (40 μg protein) were separated by electrophoresis on 12% polyacrylamide SDS gels and transferred to nitrocellulose membranes (Amersham-Buchler, Braunschweig, Germany). iNOS was detected using rabbit Inducible Nitric Oxide Synthase 2 (iNOS/NOS-2) antibody (Alpha Diagnostic International, BioTrend, Cologne, Germany). Determination of β-actin with anti-β-actin antibody (Sigma, Taufkirchen, Germany) served as a loading control. Blots were blocked (10% nonfat dry milk in 1 mM Tris, 150 mM NaCl, pH 7.4) for 1 h, incubated 1 h at RT with primary antibody (diluted according to manufacturer's instructions in blocking buffer with 0.5% tween 20 and 0.5% BSA) and then incubated 1 h with horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1 : 1000 in blocking buffer with 0.5% tween 20 and 0.5% bovine serum albumin at RT. Proteins were detected with ECL western blotting detection reagents (GE Healthcare, Munich, Germany). Films were digitized, and the integrated density of individual bands was determined using the software Multianalyst (Bio-rad, Munich, Germany). By densitometric measurements using the same software the amount of protein expression was normalized to β-actin.
2.5. Ribonucleic Acid (RNA) Isolation, Quantitative Reverse-Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA of snap-frozen liver lobes was isolated using the RNeasy-system (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The residual amounts of DNA remaining were removed using the RNase-Free DNase Set according to the manufacturer's instructions (Qiagen, Hilden, Germany). The RNA was stored immediately at −80°C. Quality and amount of the RNA were determined photometrically using the NanoDrop ND-1000 device (NanoDrop Technologies, Wilmington, DE, USA).
RNA was subsequently used for qRT-PCR. In brief, 100 ng of total hepatic RNA was reversely transcribed using the Affinity script QPCR-cDNA synthesis kit (Stratagene, La Jolla, CA, USA) following the manufacturer's instructions. To determine the mRNA expression of Bcl-2, BAX, and Caspase 8, qRT-PCR was carried out on a Stratagene MX3005p QPCR system (Stratagene) using gene-specific primers for rat Bcl-2 (NM_016993, UniGene#: Rn.9996, Cat#: PPR06577A), rat BAX (NM_017059, UniGene#: Rn.10668, Cat#: PPR06496B), and rat Casp8 (NM_022277, UniGene#: Rn.54474, Cat#: PPR06555A) purchased from SABiosciences (SuperArray, Frederick, MD, USA). As reference gene, the expression of GAPDH with rat Gapdh (NM_017008, UniGene#: Rn.91450, Cat#: PPR06557A, SABiosciences, SuperArray, Frederick, MD, USA) was measured. Sequences of these primers are not available. PCR reaction was set up with 1x RT2 SYBR Green/Rox qPCR Master mix (SABiosciences) in a 25 μL volume according to manufacturer's instructions. A two-step amplification protocol consisting of initial denaturation at 95°C for 10 min followed by 40 cycles with 15 s denaturation at 95°C and 60 s annealing/extension at 60°C was chosen. A melting-curve analysis was applied to control the specificity of amplification products.
Relative expression of target gene mRNA level was then calculated using the comparative threshold cycle (CT) method (2-ΔΔCT method). In brief, the amount of target mRNA in each sample was first normalized to the amount of GAPDH mRNA, to give ΔCT and then to a calibrator consisting of samples obtained from the sham_ctrl group. The relative mRNA expression of target genes is presented as fold change calculated in relation to sham_ctrl after normalization to GAPDH.
2.6. Statistical Analysis
Differences between groups were determined by one-way analysis of variance (ANOVA) using a multiple comparison procedure (Student-Newman-Keuls). Changes in target gene expression were analyzed by Wilcoxon matched-pair analysis followed by Bonferroni correction. A P value of less than 0.05 was considered significant. Data are given as mean ± standard error of the mean. All statistical analyses were performed employing GraphPad Prism 5 (Graphpad Software, Inc., San Diego, CA).
3. Results
3.1. Oxidative and Nitrosative Stress after Hemorrhage and Resuscitation
Lipid peroxidation and protein nitrosylation occur after H/R. Hepatic oxidative stress was evaluated by immunohistochemical staining of 4-hydroxynonenal (4-HNE), indicating lipid peroxidation. The amount of 4-HNE following H/R increased significantly at 2 h after resuscitation (38 ± 2%), with further increase at 24 h after resuscitation (49 ± 2%) and a decline at 72 h after resuscitation (30 ± 3%) compared with sham-operated rats (25 ± 2%, P < 0.05, Figures 1(a)–1(d) and 3(a)). EtOH gavage significantly reduced hepatic 4-HNE at 2 h (31 ± 1%) and 24 h (32 ± 3%) after resuscitation compared to the corresponding control groups after H/R (Figures 1(e)–1(h) and 3(a)).
Figure 1.
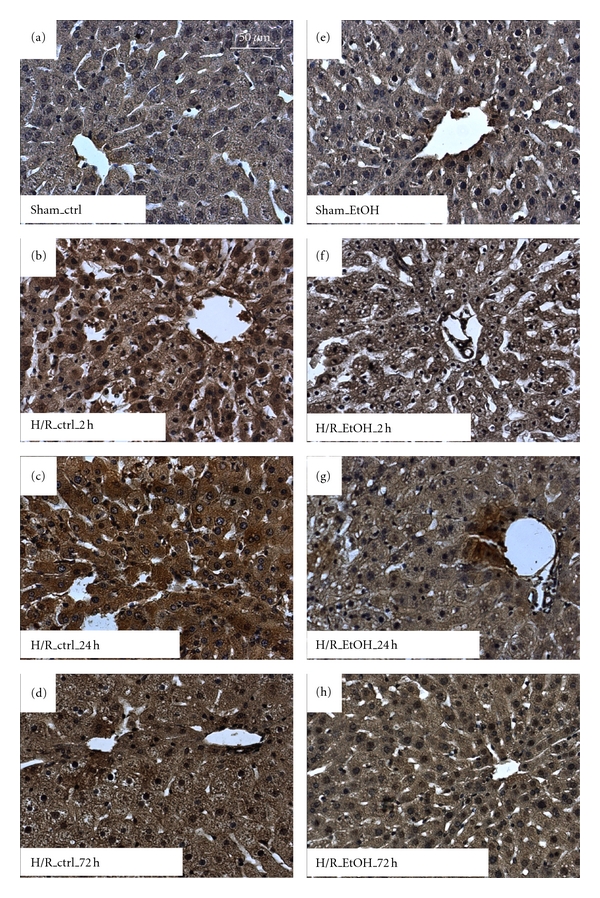
Acute ethanol gavage decreases lipid peroxidation after hemorrhage and resuscitation. Rats were gavaged with saline (ctrl) or ethanol (5 g/kg, 30% EtOH) 12 h before hemorrhagic shock and resuscitation (H/R). Sham-operated animals underwent the same surgical procedures but H/R was not carried out. Immunohistochemical staining for 4-hydroxynonenal (4-HNE) reveals areas of lipid peroxidation. 4-HNE-stained, representative liver sections from sham-operated rats are given in (a) (ctrl) and (e) (EtOH). 4-HNE-stained, representative liver sections from rats at 2 h, 24 h, and 72 h after H/R are given in (c)-(d) (ctrl) and (f)–(h) (EtOH); bar is 50 μm.
Figure 3.
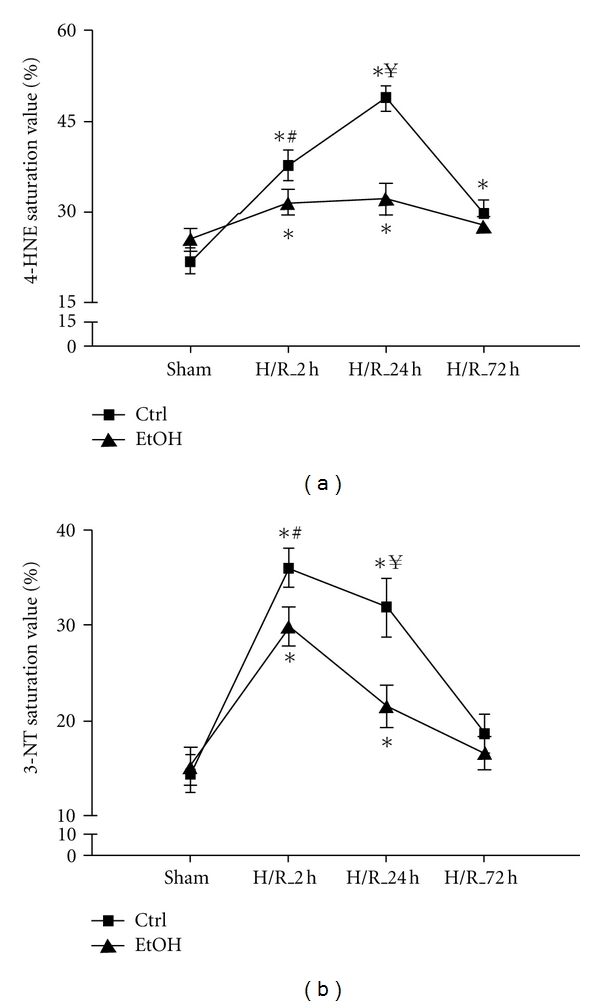
Semiquantitative analysis of lipid peroxidation and protein nitrosylation after acute ethanol gavage after hemorrhage and resuscitation in a time course. The percentage of 4-HNE (a) and 3-NT (b) positive cells (staining) was semiquantified in liver sections 2 h, 24 h, and 72 h after H/R in a blinded manner. Ctrl: saline gavage, EtOH: ethanol gavage, sham: surgical procedure without the onset of H/R and H/R: rats undergoing hemorrhagic shock with subsequent resuscitation (*P < 0.05 versus both sham groups, # P < 0.05 ctrl_H/R_2 h versus EtOH_H/R_2 h group, ¥ P < 0.05 ctrl_H/R_24 h versus EtOH_H/R_24 h group, n = 3 for 24 h groups, and n = 6 for other groups).
Hepatic nitrosative stress was evaluated by immunhistochemical staining of 3-nitrotyrosine (3-NT) indicating peroxynitrite-dependent nitrosylation of protein tyrosine residues. 3-NT was significantly increased in saline-gavaged rats at 2 h and 24 h after resuscitation when compared to sham-operated rats (36 ± 2, 33 ± 3% and 14 ± 2%, resp., P < 0.05, Figures 2(a)–2(d) and 3(b)). EtOH gavage markedly reduced 3-NT-stained areas at 2 h (30 ± 2%) and 24 h (21 ± 3%) after resuscitation as compared to the corresponding control groups after H/R (P < 0.05, Figures 1(e)–1(h) and 3(b)). 72 h after H/R, 3-NT levels declined almost to basal levels in both groups. Both hepatic oxidative and nitrosative stress following H/R were largely diminished by EtOH administration.
Figure 2.
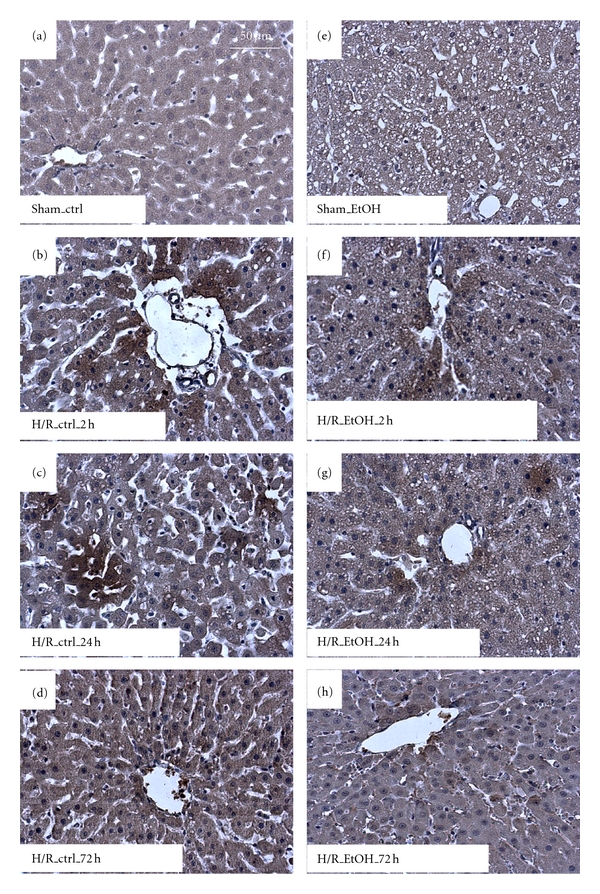
Acute ethanol gavage decreases protein nitrosylation after hemorrhage and resuscitation. Immunohistochemical staining for 3-Nitrotyrosine (3-NT) reveals areas of protein nitrosylation. 3-NT-stained, representative liver sections from sham-operated rats are given in (a) (saline control, ctrl) and (e) (acute ethanol gavage, EtOH). 3-NT-stained, representative liver sections from rats at 2 h, 24 h, and 72 h after H/R are given in (c)-(d) (saline control treatment, ctrl) and (f)–(h) (acute ethanol gavage, EtOH); bar is 50 μm.
3.2. Analysis of Hepatic Gene Expression of Apoptosis Relevant Genes after Hemorrhage and Resuscitation
The semi-quantitative real-time PCR showed a decrease in Bcl-2 : BAX ratio at 2 h after resuscitation in liver samples obtained from H/R ctrl animals as compared to all other groups (P < 0.05, Figure 4(a)). The Bcl-2 : BAX ratio was significantly enhanced in the H/R group of animals that received EtOH gavage compared with H/R ctrl group (2 h: 1.9 ± 0.3 and 0.3 ± 0.1, resp.). In both groups, this trend was also found at 24 h (H/R_EtOH: 1.4 ± 0.2 and H/R_ctrl: 0.6 ± 0.2, resp.) and 72 h (72 h: 1.2 ± 0.2 and 0.5 ± 0.2) after resuscitation, with significant differences between the 24 h H/R_EtOH group versus sham (P < 0.05). In parallel, caspase-8 mRNA expression enhanced significantly 2 h after H/R compared to other groups (2.5 ± 0.6, 0.8 ± 0.2 in the sham EtOH group and 0.6 ± 0.1 in the H/R EtOH group, resp., P > 0.05, Figure 4(b)). Caspase-8 mRNA was not increased in livers from rats receiving EtOH before H/R.
Figure 4.
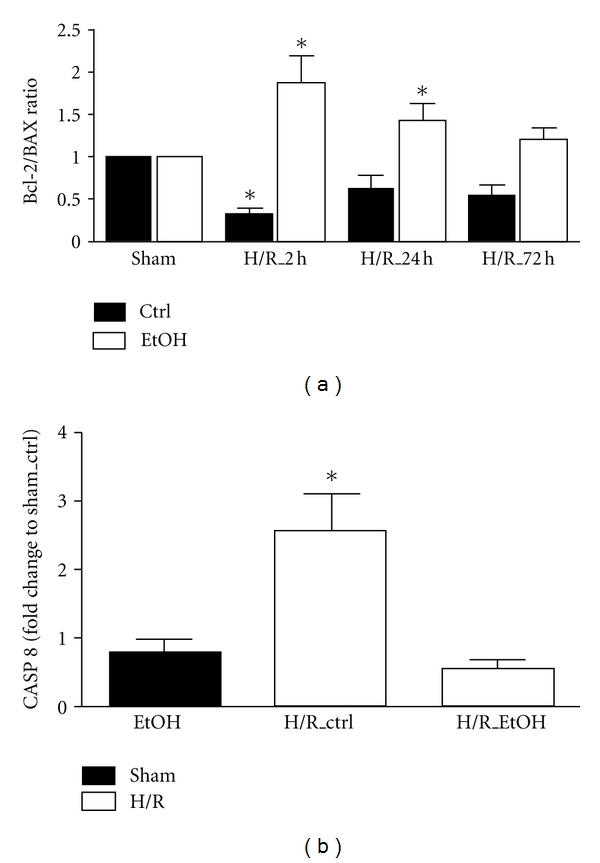
Effects of acute ethanol gavage on hepatic gene expression of BAX, Bcl-2, and caspase 8 at 2 h, 24 h, and 72 h after H/R. Hepatic gene expression of Bax, Bcl-2 (a), and caspase 8 (b) was analyzed. After normalization to GAPDH expression, the ratio of Bcl-2 : Bax gene expression was determined and caspase 8 gene expression was measured as fold change compared to sham-operated control group. Ctrl: saline gavage, EtOH: ethanol gavage, sham: surgical procedure without the onset of H/R and H/R: rats undergoing hemorrhagic shock with subsequent resuscitation (*P < 0.05 versus other groups, n = 3 for 24 h groups, and n = 6 for other groups).
These results indicate that EtOH reduces early apoptotic changes at the gene expression level after H/R.
3.3. Analysis of the iNOS Expression after Hemorrhagic Shock and Resuscitation
To analyze the mechanism influenced by EtOH after H/R, we evaluated the iNOS expression in a time course by western blotting in liver homogenates collected at 2 h, 24 h, and 72 h after resuscitation. In parallel to previous work, hepatic iNOS expression increased during the time course after H/R compared with sham group. Densitometric analysis of protein content related to β-actin content showed an increase in iNOS at 2 h after H/R to 37 ± 7%, 24 h after H/R to 41 ± 7%, and 72 h after H/R to 28 ± 5%, respectively, compared to 5 ± 2% in sham-operated rats (P < 0.05, Figures 5(a) and 5(b)). EtOH prevented the increase in the iNOS expression at 2 h after H/R to 17 ± 5%, 24 h after H/R to 20 ± 5%, and 72 h after H/R to 14 ± 4%, respectively, compared to ctrl rats after H/R (7 ± 2%; P < 0.05, Figures 5(a) and 5(b)).
Figure 5.
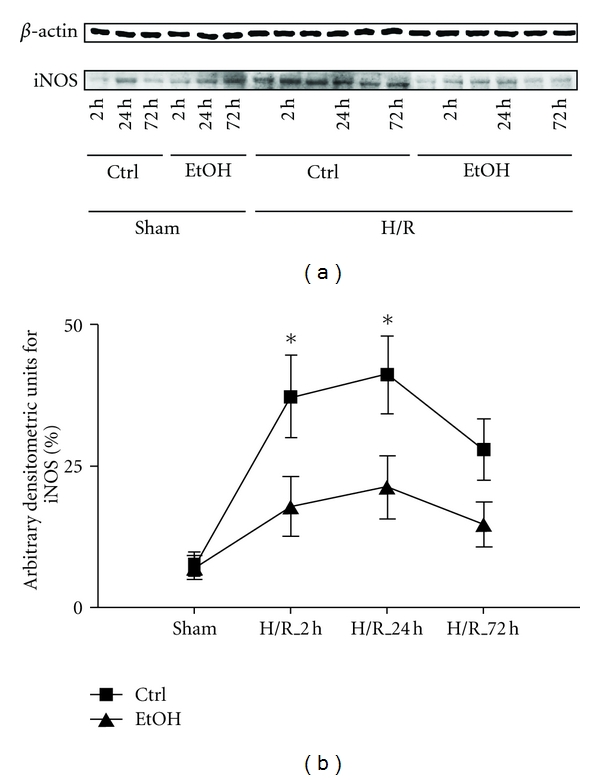
Acute ethanol exposure reduces hepatic iNOS gene expression early after H/R. Saline (Ctrl) or ethanol (EtOH) gavaged rats were subjected to H/R or sham operation. 2 h, 24 h, and 72 h after the end of resuscitation, liver tissue was harvested and western blotting for iNOS and β-actin was performed. (a): lanes 1–6 depict liver protein extracts from rats after sham operation (sham, lanes 1–3: ctrl time course, lane 4–6: EtOH gavage time course) or H/R (lanes 7–12: ctrl time course, lanes 13–18: EtOH gavage time course). In (b), densitometric measurement after normalization to β-actin staining is plotted (*P < 0.05 versus sham and corresponding H/R_EtOH group, n = 3 for 24 h groups, and n = 6 for other groups, representative gel from 3 experiments is shown).
4. Discussion
Oxidative and nitrosative stress cause liver injury in several hypoxia models and were associated with higher apoptosis and necrosis rates [31]. Liver injury after ischemia/reperfusion and H/R is closely associated with oxidative and nitrosative stress and characterized by lipid peroxidation, thereby damaging cell membranes, proteins, and DNA [31, 38, 45, 46]. Acute ethanol gavage induces an acute fatty liver. After H/R, acute ethanol gavage reduces proinflammatory changes, hepatic injury as well as mortality rates, effects that may be associated and explained with downregulated NF-κB activation by ethanol in the H/R in vivo model [15]. To further extend these results, we hypothesized that generation of RONS may be influenced by acute ethanol exposure. Accordingly, we now demonstrated that both oxidative and nitrosative stress were strongly diminished by ethanol in our model of H/R. This effect was at least partly brought about by a reduced expression of iNOS, caspase 8, and proapoptotic Bax and increased antiapoptotic Bcl-2 protein after acute ethanol gavage prior the onset of H/R.
Positive BACs are frequently detected in trauma victims [4–6]. Due to anti-inflammatory effects of ethanol, numerous studies associate ethanol intoxication with increased risk of complications, such as secondary infections, MOF, and higher morbidity and mortality after trauma [7, 10, 12, 47, 48]. Previously, we have shown that acute ethanol gavage reduces mortality and hepatic injury after H/R in vivo [15]. As suggested, we expected an additive and negative effect of acute ethanol gavage and H/R on liver function, leading to increased risk for hepatic failure or damage in our previous study [49]. However, only early hepatic injury was increased after H/R, whereas harmful pathophysiological changes were decreased [15]. Elevated levels of RONS can cause oxidative, and nitrosative cell stress [50]. Hepatic damage after ethanol exposure is associated with oxidative stress in different experimental models [51–53]. An ethanol-dependent increase in generation of RONS initiates lipid peroxidation, oxidative, and nitrosative stress as well as decreased hepatic antioxidant defense [54–57].
NF-κB induction after H/R is involved in subsequent activation of the inflammatory cascade, finally culminating in cellular activation and organ damage [38, 58, 59]. The oxidative state of the cell influences the induction of NF-κB [60]. Reactive oxygen intermediates may induce IkappaB phosphorylation via tyrosine kinases, thereby activating NF-κB [61]. Moreover, antioxidants, such as vitamin E, its derivative pentamethyl-hydroxychromane, and green tea catechins inhibit the NF-κB activation [38, 62]. In our previous studies, we found that acute ethanol inhibits NF-κB activation in our model of H/R, but the mechanism was not clearly evaluated [15]. Here, taken into account the dependence of NF-κB activation upon RONS production, we detect an ethanol-induced reduction of oxidative and nitrosative stress after H/R (Figures 1–3).
H/R-induced liver injury is associated with enhanced lipid peroxidation and peroxynitrite anion and nitrotyrosine adducts formation, which result from interaction between superoxide anion and NO. Increases in NO occur when iNOS is induced. H/R upregulates iNOS, an NF-κB-dependent protein [63, 64]. To further evaluate the mechanism that contributes to the production of RONS in our model, we examined the expression of iNOS after ethanol gavage and H/R. Previously, it has been reported that, after 4 weeks of ethanol feeding, iNOS knockout mice and wild-type mice treated with the iNOS inhibitor W1400 revealed blunted liver damage, an effect that was associated with a reduced production of NO-derived prooxidants such as peroxynitrite [65]. Furthermore, iNOS is upregulated after H/R, and iNOS knockout mice are protected from lung and liver injury produced by hemorrhagic shock [66]. Our previous study demonstrates that alcohol gavage prior to H/R blunts NF-κB activation as described above [15]. Consistent with these results we now demonstrate a reduced iNOS expression in acute ethanol gavaged rats after H/R, an effect that was associated with reduced oxidative and nitrosative stress (Figures 1, 2, and 5). These results further highlight the central role of iNOS in the pathophysiology of resuscitated blood loss.
Liver apoptosis after H/R is associated with enhanced formation of RONS [38, 67]. Prevention of caspase activation reduced cellular injury after H/R [68, 69]. Chronic ethanol feeding induces hepatic cell death by mechanisms including Fas ligand expression, increased cytokine levels, and/or oxidative stress [70–73]. Low concentrations of ethanol were associated with caspase 8 activation in HepG2 cells in vitro [74]. Due to well-described H/R-induced hepatocellular apoptosis and necrosis, we hypothesized that ethanol exposure prior to hypoxia (H/R) sensitizes the liver to oxidative stress and potentiates hepatic injury. 2 h after H/R the mRNA expression ratio of hepatic Bcl-2 and Bax was reduced indicating ongoing apoptosis. Proapoptotic Bax is involved in perforation of membranes facilitating Ca2+ efflux with subsequent activation of the caspase cascade [75]. Bcl-2 is able to counteract the proapoptotic changes and plays a role in the antioxidant pathway and the relative levels of these two proteins determine cell death and survival [76]. Interestingly, we found enhanced ratio of Bcl-2 : Bax at 2 h and 24 h after resuscitation in animals that received an acute ethanol gavage before H/R (Figure 4(a)). This early antiapoptotic phenotype was confirmed by reduced gene expression of caspase 8 in the ethanol-gavaged group, whereas caspase 8 was strongly upregulated in the control group after H/R (Figure 4(b)). Our findings support other findings that H/R induces apoptosis [77]. However, H/R-induced apoptosis can be reduced by acute ethanol gavage in our model.
Acute ethanol intoxication before the H/R decreases mortality and ameliorates tissue damage [15]. Taken together, this study describes that iNOS, an important NF-κB-dependent proinflammatory mediator, is associated with increased oxidative and nitrosative stress and apoptosis after H/R. These harmful H/R-induced effects are blunted after acute ethanol gavage before H/R and may help to explain the beneficial effects of an acute ethanol gavage prior to a hemorrhagic insult.
Acknowledgments
The study was supported by DFG MA 1119/3-3 and DFG LE 1346/2-1.
References
- 1.Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. Journal of the American Medical Association. 2004;291(10):1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- 2.Li G, Keyl PM, Smith GS, Baker SP. Alcohol and injury severity: reappraisal of the continuing controversy. Journal of Trauma. 1997;42(3):562–569. doi: 10.1097/00005373-199703000-00032. [DOI] [PubMed] [Google Scholar]
- 3.Rehm J, Room R, Graham K, Monteiro M, Gmel G, Sempos CT. The relationship of average volume of alcohol consumption and patterns of drinking to burden of disease: an overview. Addiction. 2003;98(9):1209–1228. doi: 10.1046/j.1360-0443.2003.00467.x. [DOI] [PubMed] [Google Scholar]
- 4.Rivara FP, Jurkovich GJ, Gurney JG, et al. The magnitude of acute and chronic alcohol abuse in trauma patients. Archives of Surgery. 1993;128(8):907–913. doi: 10.1001/archsurg.1993.01420200081015. [DOI] [PubMed] [Google Scholar]
- 5.Gentilello LM, Ebel BE, Wickizer TM, Salkever DS, Rivara FP. Alcohol interventions for trauma patients treated in emergency departments and hospitals: a cost benefit analysis. Annals of Surgery. 2005;241(4):541–550. doi: 10.1097/01.sla.0000157133.80396.1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.London JA, Battistella FD. Testing for substance use in trauma patients: are we doing enough? Archives of Surgery. 2007;142(7):633–638. doi: 10.1001/archsurg.142.7.633. [DOI] [PubMed] [Google Scholar]
- 7.Boé DM, Nelson S, Zhang P, Quinton L, Bagby GJ. Alcohol-induced suppression of lung chemokine production and the host defense response to Streptococcus pneumoniae. Alcoholism: Clinical and Experimental Research. 2003;27(11):1838–1845. doi: 10.1097/01.ALC.0000095634.82310.53. [DOI] [PubMed] [Google Scholar]
- 8.Boé DM, Nelson S, Zhang P, Bagby GJ. Acute ethanol intoxication suppresses lung chemokine production following infection with Streptococcus pneumoniae. Journal of Infectious Diseases. 2001;184(9):1134–1142. doi: 10.1086/323661. [DOI] [PubMed] [Google Scholar]
- 9.Ruiz M, Ewig S, Torres A, et al. Severe community-acquired pneumonia: risk factors and follow-up epidemiology. American Journal of Respiratory and Critical Care Medicine. 1999;160(3):923–929. doi: 10.1164/ajrccm.160.3.9901107. [DOI] [PubMed] [Google Scholar]
- 10.Messingham KAN, Faunce DE, Kovacs EJ. Alcohol, injury, and cellular immunity. Alcohol. 2002;28(3):137–149. doi: 10.1016/s0741-8329(02)00278-1. [DOI] [PubMed] [Google Scholar]
- 11.Bagby GJ, Zhang P, Stoltz DA, Nelson S. Suppression of the granulocyte colony-stimulating factor response to Escherichia coli challenge by alcohol intoxication. Alcoholism: Clinical and Experimental Research. 1998;22(8):1740–1745. [PubMed] [Google Scholar]
- 12.Zhang P, Bagby GJ, Boé DM, et al. Acute alcohol intoxication suppresses the CXC chemokine response during endotoxemia. Alcoholism: Clinical and Experimental Research. 2002;26(1):65–73. [PubMed] [Google Scholar]
- 13.Cowperthwaite MC, Burnett MG. Treatment course and outcomes following drug and alcohol-related traumatic injuries. Journal of Trauma Management and Outcomes. 2011;5(1, article 3) doi: 10.1186/1752-2897-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jurkovich GJ, Rivara FP, Gurney JG, et al. The effect of acute alcohol intoxication and chronic alcohol abuse on outcome from trauma. Journal of the American Medical Association. 1993;270(1):51–56. [PubMed] [Google Scholar]
- 15.Relja B, Höhn C, Bormann F, et al. Acute alcohol intoxication reduces mortality, inflammatory responses and hepatic injury after haemorrhage and resuscitation in vivo. British Journal of Pharmacology. 2012;165(4):1188–1199. doi: 10.1111/j.1476-5381.2011.01595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gluckman SJ, MacGregor RR. Effect of acute alcohol intoxication on granulocyte mobilization and Kinetics. Blood. 1978;52(3):551–559. [PubMed] [Google Scholar]
- 17.Szabo G, Chavan S, Mandrekar P, Catalano D. Acute alcohol consumption attenuates interleukin-8 (IL-8) and monocyte chemoattractant peptide-1 (MCP-1) induction in response to ex vivo stimulation. Journal of Clinical Immunology. 1999;19(1):67–76. doi: 10.1023/a:1020518703050. [DOI] [PubMed] [Google Scholar]
- 18.Nelson S, Bagby GJ, Bainton BG, Summer WR. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. Journal of Infectious Diseases. 1989;160(3):422–429. doi: 10.1093/infdis/160.3.422. [DOI] [PubMed] [Google Scholar]
- 19.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB Journal. 2001;15(8):1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 20.Hoek JB, Pastorino JG. Cellular signaling mechanisms in alcohol-induced liver damage. Seminars in Liver Disease. 2004;24(3):257–272. doi: 10.1055/s-2004-832939. [DOI] [PubMed] [Google Scholar]
- 21.Cahill A, Cunningham CC, Adachi M, et al. Effects of alcohol and oxidative stress on liver pathology: the role of the mitochondrion. Alcoholism: Clinical and Experimental Research. 2002;26(6):907–915. [PMC free article] [PubMed] [Google Scholar]
- 22.Epe B, Ballmaier D, Roussyn I, Briviba K, Sies H. DNA damage by peroxynitrite characterized with DNA repair enzymes. Nucleic Acids Research. 1996;24(21):4105–4110. doi: 10.1093/nar/24.21.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radical Biology and Medicine. 2002;33(11):1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- 24.Yuan GJ, Zhou XR, Gong ZJ, Zhang P, Sun XM, Zheng SH. Expression and activity of inducible nitric oxide synthase and endothelial nitric oxide synthase correlate with ethanol-induced liver injury. World Journal of Gastroenterology. 2006;12(15):2375–2381. doi: 10.3748/wjg.v12.i15.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larosche I, Lettéron P, Berson A, et al. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. Journal of Pharmacology and Experimental Therapeutics. 2010;332(3):886–897. doi: 10.1124/jpet.109.160879. [DOI] [PubMed] [Google Scholar]
- 26.Wieland P, Lauterburg BH. Oxidation of mitochondrial proteins and DNA following administration of ethanol. Biochemical and Biophysical Research Communications. 1995;213(3):815–819. doi: 10.1006/bbrc.1995.2202. [DOI] [PubMed] [Google Scholar]
- 27.Passos JF, Saretzki G, Von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Research. 2007;35(22):7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akgür FM, Brown MF, Zibari GB, et al. Role of superoxide in hemorrhagic shock-induced P-selectin expression. American Journal of Physiology. 2000;279(2):H791–H797. doi: 10.1152/ajpheart.2000.279.2.H791. [DOI] [PubMed] [Google Scholar]
- 29.Davies KJ. Oxidative stress: the paradox of aerobic life. Biochemical Society symposium. 1995;61:1–31. doi: 10.1042/bss0610001. [DOI] [PubMed] [Google Scholar]
- 30.Korenaga M, Wang T, Li Y, et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. Journal of Biological Chemistry. 2005;280(45):37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- 31.Jaeschke H. Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. Journal of Gastroenterology and Hepatology. 2011;26(supplement 1):173–179. doi: 10.1111/j.1440-1746.2010.06592.x. [DOI] [PubMed] [Google Scholar]
- 32.Hong F, Kim WH, Tian Z, et al. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: Involvement of induction of Bcl-2 and Bcl-xL proteins. Oncogene. 2002;21(1):32–43. doi: 10.1038/sj.onc.1205016. [DOI] [PubMed] [Google Scholar]
- 33.Mela L. Altered mitochondrial metabolism in circulatory shock. Advances in Experimental Medicine and Biology. 1977;78:371–373. doi: 10.1007/978-1-4615-9035-4_31. [DOI] [PubMed] [Google Scholar]
- 34.Baue AE, Durham R, Faist E. Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock. 1998;10(2):79–89. doi: 10.1097/00024382-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Moore FA, Sauaia A, Moore EE, Haenel JB, Burch JM, Lezotte DC. Postinjury multiple organ failure: a bimodal phenomenon. Journal of Trauma. 1996;40(4):501–510. doi: 10.1097/00005373-199604000-00001. [DOI] [PubMed] [Google Scholar]
- 36.Sauaia A, Moore FA, Moore EE, et al. Epidemiology of trauma deaths: a reassessment. Journal of Trauma. 1995;38(2):185–193. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 37.Treggiari MM, Hudson LD, Martin DP, Weiss NS, Caldwell E, Rubenfeld G. Effect of acute lung injury and acute respiratory distress syndrome on outcome in critically ill trauma patients. Critical Care Medicine. 2004;32(2):327–331. doi: 10.1097/01.CCM.0000108870.09693.42. [DOI] [PubMed] [Google Scholar]
- 38.Relja B, Töttel E, Breig L, et al. Plant polyphenols attenuate hepatic injury after hemorrhage/resuscitation by inhibition of apoptosis, oxidative stress, and inflammation via NF-kappaB in rats. European Journal of Nutrition. 2012;51(3):311–321. doi: 10.1007/s00394-011-0216-1. [DOI] [PubMed] [Google Scholar]
- 39.Relja B, Lehnert M, Seyboth K, et al. Simvastatin reduces mortality and hepatic injury after hemorrhage/resuscitation in rats. Shock. 2010;34(1):46–54. doi: 10.1097/SHK.0b013e3181cd8d05. [DOI] [PubMed] [Google Scholar]
- 40.Relja B, Schwestka B, Lee VSY, et al. Inhibition of c-jun n-terminal kinase after hemorrhage but before resuscitation mitigates hepatic damage and inflammatory response in male rats. Shock. 2009;32(5):509–516. doi: 10.1097/SHK.0b013e3181a2530d. [DOI] [PubMed] [Google Scholar]
- 41.Partrick DA, Moore FA, Moore EE, Barnett CC, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horizons. 1996;4(2):194–210. [PubMed] [Google Scholar]
- 42.Redl H, Gasser H, Schlag G, Marzi I. Involvement of oxygen radicals in shock related cell injury. British Medical Bulletin. 1993;49(3):556–565. doi: 10.1093/oxfordjournals.bmb.a072630. [DOI] [PubMed] [Google Scholar]
- 43.Meng ZH, Dyer K, Billiar TR, Tweardy DJ. Essential role for IL-6 in postresuscitation inflammation in hemorrhagic shock. American Journal of Physiology. 2001;280(2):C343–C351. doi: 10.1152/ajpcell.2001.280.2.C343. [DOI] [PubMed] [Google Scholar]
- 44.Lehnert M, Uehara T, Bradford BU, et al. Lipopolysaccharide-binding protein modulates hepatic damage and the inflammatory response after hemorrhagic shock and resuscitation. American Journal of Physiology. 2006;291(3):G456–G463. doi: 10.1152/ajpgi.00480.2005. [DOI] [PubMed] [Google Scholar]
- 45.Zhong Z, Froh M, Connor HD, et al. Prevention of hepatic ischemia-reperfusion injury by green tea extract. American Journal of Physiology. 2002;283(4):G957–G964. doi: 10.1152/ajpgi.00216.2001. [DOI] [PubMed] [Google Scholar]
- 46.Farber JL. Mechanisms of cell injury by activated oxygen species. Environmental Health Perspectives. 1994;102(10):17–24. doi: 10.1289/ehp.94102s1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zambell KL, Phelan H, Stouwe CV, Zhang P, Shellito JE, Molina PE. Acute alcohol intoxication during hemorrhagic shock: impact on host defense from infection. Alcoholism: Clinical and Experimental Research. 2004;28(4):635–642. doi: 10.1097/01.alc.0000122104.85971.55. [DOI] [PubMed] [Google Scholar]
- 48.Greiffenstein P, Mathis KW, Stouwe CV, Molina PE. Alcohol binge before trauma/hemorrhage impairs integrity of host defense mechanisms during recovery. Alcoholism: Clinical and Experimental Research. 2007;31(4):704–715. doi: 10.1111/j.1530-0277.2007.00355.x. [DOI] [PubMed] [Google Scholar]
- 49.Phelan H, Stahls P, Hunt J, Bagby GJ, Molina PE. Impact of alcohol intoxication on hemodynamic, metabolic, and cytokine responses to hemorrhagic shock. Journal of Trauma. 2002;52(4):675–682. doi: 10.1097/00005373-200204000-00010. [DOI] [PubMed] [Google Scholar]
- 50.Yokouchi M, Hiramatsu N, Hayakawa K, et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. Journal of Biological Chemistry. 2008;283(7):4252–4260. doi: 10.1074/jbc.M705951200. [DOI] [PubMed] [Google Scholar]
- 51.Kasdallah-Grissa A, Mornagui B, Aouani E, et al. Resveratrol, a red wine polyphenol, attenuates ethanol-induced oxidative stress in rat liver. Life Sciences. 2007;80(11):1033–1039. doi: 10.1016/j.lfs.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 52.Jurczuk M, Brzóska MM, Moniuszko-Jakoniuk J, Gałazyn-Sidorczuk M, Kulikowska-Karpińska E. Antioxidant enzymes activity and lipid peroxidation in liver and kidney of rats exposed to cadmium and ethanol. Food and Chemical Toxicology. 2004;42(3):429–438. doi: 10.1016/j.fct.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Husain K, Mejia J, Lalla J, Kazim S. Dose response of alcohol-induced changes in BP, nitric oxide and antioxidants in rat plasma. Pharmacological Research. 2005;51(4):337–343. doi: 10.1016/j.phrs.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 54.Polavarapu R, Spitz DR, Sim JE, et al. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology. 1998;27(5):1317–1323. doi: 10.1002/hep.510270518. [DOI] [PubMed] [Google Scholar]
- 55.Lieber CS. Alcohol and the liver: metabolism of alcohol and its role in hepatic and extrahepatic diseases. Mount Sinai Journal of Medicine. 2000;67(1):84–94. [PubMed] [Google Scholar]
- 56.French SW, Wong K, Jui L, Albano E, Hagbjork AL, Ingelman-Sundberg M. Effect of ethanol on cytochrome P450 2E1 (CYP2E1), lipid peroxidation, and serum protein adduct formation in relation to liver pathology pathogenesis. Experimental and Molecular Pathology. 1993;58(1):61–75. doi: 10.1006/exmp.1993.1006. [DOI] [PubMed] [Google Scholar]
- 57.Kaur J, Shalini S, Bansal MP. Influence of vitamin e on alcohol-induced changes in antioxidant defenses in mice liver. Toxicology Mechanisms and Methods. 2010;20(2):82–89. doi: 10.3109/15376510903559950. [DOI] [PubMed] [Google Scholar]
- 58.Rushing GD, Britt LD. Reperfusion injury after hemorrhage: a collective review. Annals of Surgery. 2008;247(6):929–937. doi: 10.1097/SLA.0b013e31816757f7. [DOI] [PubMed] [Google Scholar]
- 59.Papia G, Fan J, Kapus A, et al. Altered inhibitory κb alpha expression in LPS-stimulated alveolar macrophages following resuscitated hemorrhagic shock. Shock. 2011;35(2):171–177. doi: 10.1097/SHK.0b013e3181f21d2d. [DOI] [PubMed] [Google Scholar]
- 60.Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO Journal. 1991;10(8):2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee JI, Burckart GJ. Nuclear factor kappa B: important transcription factor and therapeutic target. Journal of Clinical Pharmacology. 1998;38(11):981–993. doi: 10.1177/009127009803801101. [DOI] [PubMed] [Google Scholar]
- 62.Hattori S, Hattori Y, Banba N, Kasai K, Shimoda SI. Pentamethyl-hydroxychromane, vitamin E derivative, inhibits induction of nitric oxide synthase by bacterial lipopolysaccharide. Biochemistry and Molecular Biology International. 1995;35(1):177–183. [PubMed] [Google Scholar]
- 63.Kiang JG, Bowman PD, Lu X, et al. Geldanamycin inhibits hemorrhage-induced increases in caspase-3 activity: role of inducible nitric oxide synthase. Journal of Applied Physiology. 2007;103(3):1045–1055. doi: 10.1152/japplphysiol.00100.2007. [DOI] [PubMed] [Google Scholar]
- 64.Kiang JG, Krishnan S, Lu X, Li Y. Inhibition of inducible nitric-oxide synthase protects human T cells from hypoxia-induced apoptosis. Molecular Pharmacology. 2008;73(3):738–747. doi: 10.1124/mol.107.041079. [DOI] [PubMed] [Google Scholar]
- 65.McKim SE, Gäbele E, Isayama F, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125(6):1834–1844. doi: 10.1053/j.gastro.2003.08.030. [DOI] [PubMed] [Google Scholar]
- 66.Hierholzer C, Harbrecht B, Menezes JM, et al. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. Journal of Experimental Medicine. 1998;187(6):917–928. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayuste EC, Chen H, Koustova E, et al. Hepatic and pulmonary apoptosis after hemorrhagic shock in swine can be reduced through modifications of conventional Ringer’s solution. The Journal of trauma. 2006;60(1):52–63. doi: 10.1097/01.ta.0000200156.05397.0b. [DOI] [PubMed] [Google Scholar]
- 68.Yang R, Lisa MH, Woodall C, et al. Administration of glutamine after hemorrhagic shock restores cellular energy, reduces cell apoptosis and damage, and increases survival. Journal of Parenteral and Enteral Nutrition. 2007;31(2):94–100. doi: 10.1177/014860710703100294. [DOI] [PubMed] [Google Scholar]
- 69.Maitra SR, Bhaduri S, El-Maghrabi R, Shapiro MJ. Inhibition of matrix metalloproteinase on hepatic transforming growth factor β1 and caspase-3 activation in hemorrhage. Academic Emergency Medicine. 2005;12(9):797–803. doi: 10.1197/j.aem.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 70.Benedetti A, Brunelli E, Risicato R, Cilluffo T, Jezequel AM, Orlandi F. Subcellular changes and apoptosis induced by ethanol in rat liver. Journal of Hepatology. 1988;6(2):137–143. doi: 10.1016/s0168-8278(88)80024-2. [DOI] [PubMed] [Google Scholar]
- 71.Galle PR, Hofmann WJ, Walczak H, et al. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. Journal of Experimental Medicine. 1995;182(5):1223–1230. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kurose I, Higuchi H, Miura S, et al. Oxidative stress-mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology. 1997;25(2):368–378. doi: 10.1053/jhep.1997.v25.pm0009021949. [DOI] [PubMed] [Google Scholar]
- 73.Miller AM, Horiguchi N, Jeong W-I, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcoholism: Clinical and Experimental Research. 2011;35(5):787–793. doi: 10.1111/j.1530-0277.2010.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castañeda F, Kinne RKH. Apoptosis induced in HepG2 cells by short exposure to millimolar concentrations of ethanol involves the Fas-receptor pathway. Journal of Cancer Research and Clinical Oncology. 2001;127(7):418–424. doi: 10.1007/s004320000227. [DOI] [PubMed] [Google Scholar]
- 75.Schendel SL, Montal M, Reed JC. Bcl-2 family proteins as ion-channels. Cell Death and Differentiation. 1998;5(5):372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- 76.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes and Development. 1999;13(15):1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 77.Duvigneau JC, Kozlov AV, Zifko C, et al. Reperfusion does not induce oxidative stress but sustained endoplasmic reticulum stress in livers of rats subjected to traumatic-hemorrhagic shock. Shock. 2010;33(3):289–298. doi: 10.1097/SHK.0b013e3181aef322. [DOI] [PubMed] [Google Scholar]