

Abstract
Although p120-catenin (p120) is crucial for E-cadherin function, ablation experiments in epithelial tissues from different organ systems reveal markedly different effects. Here, we examine for the first time the consequences of p120 knockout during mouse mammary gland development. An MMTV-Cre driver was used to target knockout to the epithelium at the onset of puberty. p120 ablation was detected in approximately one-quarter of the nascent epithelium at the forth week post-partum. However, p120 null cells were essentially nonadherent, excluded from the process of terminal end bud (TEB) morphogenesis and lost altogether by week six. This elimination process caused a delay in TEB outgrowth, after which the gland developed normally from cells that had retained p120. Mechanistic studies in vitro indicate that TEB dysfunction is likely to stem from striking E-cadherin loss, failure of cell-cell adhesion and near total exclusion from the collective migration process. Our findings reveal an essential role for p120 in mammary morphogenesis.
Keywords: p120 catenin (catenin delta 1), Cadherin, Mammary development, Morphogenesis, Mouse
INTRODUCTION
Cell-cell adhesion plays a key role in development, tissue maintenance and cancer (Birchmeier, 1995; Gumbiner, 2005; Takeichi, 1995; Yap, 1998). In vertebrates, the classical cadherins (i.e. type I and type II cadherins) comprise a large family (26 members) of transmembrane glycoproteins found in essentially all adhesive tissues (Gallin, 1998; Hulpiau and van Roy, 2009). Epithelial cadherin (E-cadherin, or cadherin 1) is the main cadherin in epithelial tissues and plays an important role in morphogenesis and homeostasis in most glandular tissues, including the mammary gland. Although the importance of cadherins in mammary morphogenesis is widely accepted, the role of p120-catenin (also known as catenin delta 1) in this process remains to be investigated.
The extracellular domains of cadherins connect adjacent cells via homophilic interaction, while the cytoplasmic domains form a complex with a group of proteins known as catenins (Gumbiner, 2005; Takeichi, 1991). p120-catenin (hereafter p120) and β-catenin are armadillo repeat domain proteins that bind directly to distinct regions of the cytoplasmic domain (Davis et al., 2003; Hulsken et al., 1994; Ireton et al., 2002; McCrea and Gumbiner, 1991; Reynolds et al., 1996; Takeichi et al., 1989; Thoreson et al., 2000; Yap et al., 1998). β-catenin connects the cadherins physically and/or functionally to the actin cytoskeleton through a mechanism involving α-catenin (Herrenknecht et al., 1991; Nagafuchi et al., 1991; Rimm et al., 1995; Yamada et al., 2005). By contrast, p120 appears to regulate the strength of cell-cell adhesion by modulating cadherin retention at the cell surface (Davis et al., 2003; Ireton et al., 2002; Xiao et al., 2003). In its absence, cadherins are internalized and degraded, thus defining p120 as a master regulator of cadherin stability (Davis et al., 2003). p120 is also thought to modulate actin dynamics via Rho-GTPases, -GEFs and -GAPs (Anastasiadis et al., 2000; Noren et al., 2000; Wildenberg et al., 2006). Together, these observations suggest that the catenins play a central role in regulating functional interactions between cadherins and the actin cytoskeleton.
Phenotypes associated with p120 ablation in vivo appear to be largely tissue dependent and surprisingly unpredictable (Bartlett et al., 2010; Davis and Reynolds, 2006; Elia et al., 2006; Marciano et al., 2011; Oas et al., 2010; Perez-Moreno et al., 2006; Smalley-Freed et al., 2010; Stairs et al., 2011). For example, in the developing salivary gland, p120 ablation completely blocks acini formation (Davis and Reynolds, 2006). Ducts are grossly distorted and characterized by cell-cell adhesion defects reminiscent of those observed in intraepithelial neoplasia. By contrast, p120 knockout (KO) in the epidermis induces a massive inflammatory response despite essentially normal adhesion and barrier function (Perez-Moreno et al., 2006). In the intestine, p120 KO causes a prominent barrier defect along with cell-cell adhesion abnormalities and inflammation (Smalley-Freed et al., 2010). These animals die from gastrointestinal bleeding within 3 weeks of birth. Other p120 KO-associated defects include reduced vessel density and anomalies in dendritic spine and synapse development in hippocampal neurons (Elia et al., 2006; Oas et al., 2010). Surprisingly, p120 KO in the prostate has no detectable effect on either cell morphology or adhesion despite near complete loss of E-cadherin expression (A.B.R., unpublished). These studies, for the most part, reflect dramatic phenotypes, although the consequences of p120 ablation differ markedly from one organ system to the next. However, the effects of p120 loss in the mammary gland have not been formally addressed.
The mammary gland provides an outstanding in vivo system for studying morphogenetic events (e.g. invasion and differentiation), as the majority of the development of this non-vital organ occurs after birth. Prior to puberty, the mammary gland exists as a rudimentary ductal tree. At the onset of puberty at ∼3 weeks of age, proliferative structures at the tips of ducts, known as terminal end buds (TEBs), develop and begin to invade the surrounding stroma (Hinck and Silberstein, 2005). TEBs comprise a dynamic mass of E-cadherin-positive luminal body cells surrounded by a motile cap cell layer expressing P-cadherin (cadherin 3) (Daniel et al., 1995; Ewald et al., 2008; Hinck and Silberstein, 2005). The TEBs bifurcate repeatedly to form the ductal tree and, ultimately, the mature gland. This process, termed branching morphogenesis, concludes at ∼10-12 weeks, when the TEBs have traversed the length of the fat pad and a fully developed ductal tree has formed (Cardiff and Wellings, 1999; Hennighausen and Robinson, 2005; Richert et al., 2000; Sternlicht, 2006). Disruption of TEBs is often associated with delayed ductal outgrowth and impaired branching morphogenesis, thus suggesting an essential function of TEBs in the overall development of the mammary gland (Jackson-Fisher et al., 2004; Kouros-Mehr et al., 2006; Lu et al., 2008; Parsa et al., 2008; Srinivasan et al., 2003; Sternlicht et al., 2006).
Here, we examine the role of p120 in the developing mammary epithelium. MMTV promoter-driven Cre recombinase expression in p120f/f mice was used to induce p120 ablation at the onset of puberty. In week 4, developing epithelial structures exhibited mosaic p120 ablation, the extent of which varied widely between mice. p120 loss in nascent ducts caused severe morphological defects (e.g. cell rounding and sloughing into the lumen), despite the presence of p120 family members, which were unable to compensate for p120 loss. p120 null cells were observed less frequently in the TEB itself owing to rapid shedding from TEBs. In vitro two- and three-dimensional modeling suggest that TEB function is compromised in the absence of p120, most likely owing to defects in cell-cell adhesion and collective cell migration. At the whole organ level, the phenotype manifested as a transient delay in ductal outgrowth due to selective loss of p120 null cells and preferential outgrowth of the p120-positive cell population. Reconstitution with pure populations of p120-depleted cells blocked mammary gland formation completely. These data reveal an essential, non-redundant role for p120 in mammary gland development.
MATERIALS AND METHODS
Animals
p120f/f mice were backcrossed onto an FVB/NJ background and crossed with MMTV-Cre #7 obtained from Dr W. J. Muller on an FVB background (Andrechek et al., 2000; Andrechek et al., 2005; Davis and Reynolds, 2006). Genotyping was performed as previously described (Andrechek et al., 2000; Davis and Reynolds, 2006). All experiments involving animals were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Whole-mount mammary gland analysis
Inguinal mammary glands were fixed overnight in Carnoy’s II (1:3:6 glacial acetic acid:chloroform:ethanol) fixative and gradually rehydrated. Glands were stained with carmine alum, washed and dehydrated. Clearing was performed using Histoclear (National Diagnostics, Atlanta, GA, USA). Whole-mount images were acquired using an Olympus QColor 3TM digital camera and assessed using MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). To quantify outgrowth, the average distance of the three longest ducts was measured relative to a line tangential to the nipple-proximal face of the lymph node. Outgrowths beyond and before the lymph node were quantified with positive and negative values, respectively. To quantify TEB area, the average area of the six largest TEBs per whole-mount was obtained. For both assessments, glands from at least five mice per genotype were analyzed.
Immunofluorescence/immunohistochemistry
Immunostaining on tissue was performed as previously described (Davis and Reynolds, 2006). Briefly, tissues were fixed in 10% formalin. Paraffin-embedded tissue sections were deparaffinized and rehydrated. Antigen retrieval was performed by boiling slides in 10 mM sodium citrate pH 6.0 for 10 minutes. After blocking, slides were incubated in primary and secondary antibody overnight and for 2 hours, respectively. Sections were mounted with Prolong Gold Antifade Mounting Medium (Invitrogen, Carlsbad, CA, USA). Tissue processing and H&E staining were performed by the Vanderbilt Translational Pathology Shared Resource Core using standard techniques. TUNEL staining was performed as per the manufacturer’s instructions (Millipore, Danvers, MA, USA) with the following modification: antigen retrieval was for 10 minutes in proteinase K (Clontech, Mountain View, CA, USA). Staining was visualized using an Axioplan 2 microscope (Zeiss, Oberkochen, Germany). Images were collected with either an Olympus QColor 3TM digital camera or a Hamamatsu Orca ER fluorescence camera and processed using MetaMorph software.
Antibodies for immunohistochemistry
The following primary antibodies were used for fluorescent immunohistochemistry: anti-p120 (F1aSH, 0.8 μg/ml); anti-Arvcf (1:100) (Walter et al., 2008); anti-∂-catenin (1 μg/ml, EMD Millipore 07-295); anti-p0071 (1:300) (Hofmann et al., 2009); anti-p120/pp120 (0.6 μg/ml, BD Bioscience, Franklin Lakes, NJ, USA); anti-E-cadherin (0.5 μg/ml, BD Biosciences); anti-β-catenin (1:800, Sigma-Aldrich, St Louis, MO, USA); anti-crumbs 3 (1:500, gift from Ben Margolis, Ann Arbor, MI, USA); anti-phosphorylated histone H3 (2 μg/ml, EMD Millipore 06-570); anti-cleaved caspase 3 (1:200, Cell Signaling, Danvers, MA, USA); anti-SMA (0.2 μg/ml, Sigma-Aldrich); anti-ZO-1 (1.25 μg/ml, Invitrogen); anti-desmoglein (1 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-p63 (1 μg/ml, Santa Cruz Biotechnology); TROMAI/anti-keratin 8 (0.2 μg/ml, University of Iowa Hybridoma Core). Secondary antibodies were conjugated to 488, 594 or 647 Alexa-Fluor dyes (1:500, Invitrogen).
Cell culture and generation of cell lines
CommaD-beta (CDβ) cells, a gift from Dr Medina at Baylor University, were cultured as previously described (Zhan et al., 2008). Primary mammary epithelial cells were isolated and cultured as previously described (Vaught et al., 2009). NMuMG and Phoenix293 cells were cultured in DMEM supplemented with antibiotics and 10% heat-inactivated fetal bovine serum. MCF10A cells were cultured as previously described (Debnath et al., 2003). pRetroSuper-puromycin vectors expressing shRNA against human p120 and pLZRS (neomycin) vectors expressing mouse p120 isoforms 1A or 3A were utilized to deplete or add back p120, respectively (Davis et al., 2003). pLZRS-neomycin vector expressing mouse p120 isoform 3A Δarm1.CAAX was generated as previously described (Wildenberg et al., 2006). Production of virus for protein expression and shRNA expression was conducted in Phoenix293 cells as previously described (Ireton et al., 2002). MCF10A cells were selected for expression of pRetroSuper and pLZRS constructs by addition of 2 μg/ml puromycin and 500 μg/ml G418, respectively. After transduction and selection, monoclonal cell lines of MCF10A p120 shRNA cells were generated using limiting dilution. For rescue experiments, clonal cell lines were transduced with empty vector or vectors expressing mouse p120 isoform 1A, 3A or 3A Δarm1.CAAX.
Wound-healing assays
MCF10A cells were plated to confluence and scratched with a P200 tip to generate the wound. Cells were rinsed with PBS, covered in growth media, and imaged at six regions per scratch every 6 hours. Cell migration was calculated as percentage closure of the original wound. For time-lapse microscopy, the above procedure was performed and cells were imaged every 10 minutes. Images were acquired using an Axiovert 200M microscope (Zeiss) and processed using MetaMorph software.
Western blot analysis
Protein was isolated as previously described (Mariner et al., 2004). Briefly, cells were washed with PBS, lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Nonidet P40, 0.5% deoxycholic acid, 0.1% SDS) containing inhibitors (1 mM phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mM sodium orthovanadate, 1 mM EDTA, 50 mM NaF, 40 mM β-glycerophosphate) and spun at 14,000 g at 4°C for 5 minutes. Cleared total protein was quantified using a bicinchoninic acid assay (Pierce, Rockford, IL, USA). Then 20 μg protein per sample was boiled in 2× Laemmli sample buffer and separated by SDS-PAGE. Proteins were transferred to nitrocellulose (PerkinElmer, Waltham, MA, USA). Non-specific binding was blocked by incubating membranes in 3% nonfat milk in Tris-buffered saline and Odyssey blocking buffer (LI-COR, Lincoln, NE, USA) prior to addition of primary and secondary antibodies, respectively. Anti-p120/pp120 (0.1 μg/ml, BD Biosciences), anti-E-cadherin (0.1 μg/ml, BD Biosciences), anti-tubulin/DM1α (1:1000, Sigma-Aldrich), anti-N-cadherin (0.8 μg/ml, 13A9, Millipore) and anti-P-cadherin (1:250, BD Biosciences) antibodies were used. The Odyssey system was used for detection of secondary goat anti-mouse IgG IRDye 800CW antibodies (1:10,000, LI-COR).
Three-dimensional branching assays and mammary transplants
Primary mammary epithelial cells (PMECs) were isolated as previously described (McCaffrey and Macara, 2009). Lentivirus was generated by transfecting HEK293T cells with pLL5.0-GFP expressing shRNA against human (control) or mouse p120 (p120i) and psPAX2 and pMD2.G (Addgene, Cambridge, MA, USA). PMECs were infected with lentivirus (MOI=100) for 3 hours during centrifugation at 300 g. Cells were grown in suspension on low-adhesion plates (Corning, NY, USA) for 5-7 days in mammosphere media (DMEM:F12, 20 ng/ml EGF, 20 ng/ml FGF2, 2% B27 supplement) (Dontu et al., 2003). For 3D branching assays, 100 mammospheres were suspended in 50 μl Matrigel (BD Bioscences) per well of a 96-well plate and equilibrated in minimal media [DMEM:F12, 1% (v/v) insulin/transferrin/selenium, 1% penicillin/streptomycin] (Ewald et al., 2008). Branching was induced with 2.5 nM FGF2 in minimal media changed twice. Quantification of percent branched and number and fluorescence status of branches were performed after 7 days (>30 mammospheres per experiment). For mammary transplantation assays, PMECs were infected and grown in suspension. After 7 days, cells were trypsinized and flow sorted for GFP by the Vanderbilt Flow Cytometry Laboratory. Then 1×105 cells in 10 μl PBS with 10% Matrigel expressing control GFP or p120i GFP virus were injected into contralateral cleared fat pads of 3-week-old FVB mice. After 6 weeks, glands were removed and analyzed for GFP-positive outgrowth relative to gland size using a Nikon AZ 100M fluorescence wide-field microscope.
Statistical analysis
Statistical analyses were preformed using Prism (GraphPad La Jolla, CA, USA) as described in the figure legends. For assays with or without normal distribution, two-tailed Student’s t-tests or Mann-Whitney tests were performed, respectively.
RESULTS
Characterization of p120 expression in the developing mammary gland
To characterize baseline p120 expression patterns in the developing mammary gland, sections of glands from 4-week-old control mice were co-immunostained with antibodies to p120 along with the basal and luminal cell markers smooth muscle actin (SMA) and keratin 8 (K8), respectively (Fig. 1A). Fig. 1A illustrates diffuse p120 staining of stromal cells (arrows) and sharp junctional staining in the epithelium of ducts (top panels) and TEBs (bottom panels). Note that p120 staining in the basal compartment of the TEB is markedly reduced relative to the very strong staining in the luminal compartment. These patterns of p120 localization were the same throughout all stages of pubertal development (data not shown).
Fig. 1.

p120 is ubiquitously expressed in the mouse mammary gland. (A) Immunostaining for p120, SMA and K8 on sections from glands of 4-week-old females. SMA and K8 mark the cap and body cells, respectively. Representative images for ducts and terminal end buds (TEBs) are shown. Arrow indicates diffuse stromal p120 staining. Dashed line indicates the division between body and cap cells. Scale bar: 50 μm. (B) Immunoblots for p120 and tubulin in a panel of normal mammary epithelial cell types. (C) The mammary TEB and duct.
Typically, epithelial cells predominantly express p120 isoform 3, whereas fibroblasts express isoform 1. However, Fig. 1B illustrates biochemically that primary mammary epithelial cells (PMECs) and the untransformed mouse mammary cell lines CDβ and NMuMG express both p120 isoforms 1 and 3. Furthermore, both layers of the mammary epithelium demonstrated positive immunostaining using an antibody that only recognizes p120 isoforms 1 and 2, suggesting that isoforms other than 3 are also expressed in the epithelium in vivo (data not shown). Collectively, these results demonstrate expression of p120 in the mature basal and luminal mammary epithelium, as well as in the body and cap layers of the TEB.
Mosaic p120 knockout at puberty induces transient delay of ductal outgrowth
To target p120 KO to the mammary gland, MMTV-Cre;p120fl/fl mice were generated by crossing MMTV-Cre #7 mice on an FVB background (Andrechek et al., 2000; Andrechek et al., 2005) to p120 floxed mice, which were backcrossed to an FVB background (Davis and Reynolds, 2006). Effects of p120 ablation were examined initially by whole-mount analysis at time points spanning pubertal development (Fig. 2). At 3 weeks, KO and control rudimentary mammary trees were grossly indistinguishable (Fig. 2A). By contrast, ductal outgrowth was significantly reduced at weeks 4, 5 and 6 in the p120 KO mammary gland (Fig. 2A,B). By week 9, however, control and KO glands were again indistinguishable. Thus, p120 ablation induces a transient delay in ductal outgrowth that is ultimately resolved by week 9.
Fig. 2.

p120 ablation in the developing mammary gland delays ductal outgrowth. (A) Virgin mammary gland whole-mounts from control and p120 knockout (KO) animals. LN, lymph node. (B) Quantitative comparisons of ductal outgrowth. (C) Immunostaining for p120 in 4-week-old glands. (D) Quantification of p120 ablation in glands from mice at the ages indicated. (B,D) Mean with s.e.m. *P<0.05, Mann-Whitney test. (E) p120 and K8 immunostaining of glands at indicated ages. Scale bars: 1 mm in A; 50 μm in C,E.
Further analysis of the glands by immunostaining revealed mosaic, epithelium-specific p120 ablation in all experimental animals starting at week 3. Significant p120 KO was observed at week 4 (Fig. 2C). The overall percentage of KO cells varied widely (6.7-38%, n=6), but averaged 22% of the nascent epithelium following puberty-induced expression of Cre (Fig. 2D,E). Thereafter, p120 null cells were increasingly scarce and almost completely absent by week 6 (0-0.9%, n=5) (Fig. 2D,E). Thus, from week 6 on, glands were essentially p120 positive (despite the MMTV-Cre;p120fl/fl genotype) and further development (including pregnancy and lactation) was indistinguishable from that of p120fl/fl controls.
The rapid loss of p120 null cells between weeks 3 and 6 suggested the possibility of reduced cell proliferation or elevated cell death. We broadly assessed cell death by TUNEL staining, which marks cells undergoing apoptosis, necrosis or lysosome-mediated death (Grasl-Kraupp et al., 1995; McIlroy et al., 2000; Overholtzer et al., 2007). There was no statistically significant change in global cell death in p120 KO ducts or TEBs (Fig. 3Aa,b,Ba,b). However, the percentage of TUNEL-positive cells increased 3-fold when analyzing only those detached from the body of the TEB in KO mice (Fig. 3Bb, arrows). Similarly, detached cells demonstrated a 4-fold increase in the percentage positive for cleaved caspase 3, further suggesting that p120 null cells are dying by anoikis, i.e. detachment-induced cell death (Fig. 3Bc,d, arrows). This cell detachment was rarely seen in control TEBs. Cell proliferation in ducts and TEBs was unaffected by p120 ablation as monitored by phosphorylated histone H3 staining (Fig. 3Ae,f,Be,f).
Fig. 3.

Analysis of proliferation and cell death in the absence of p120. (A,B) Analysis of cell death and proliferation performed on ducts (A) and TEBs (B). (Aa,b,Ba,b) Representative TUNEL stained sections from glands of 4-week-old control and p120 KO mice. Arrows indicate detached TUNEL-positive cells. (Ac-f,Bc-f) Analysis of apoptosis and proliferation performed by immunostaining for cleaved caspase 3 (Ac,d,Bc,d) and phosphorylated histone H3 (Ae,f,Be,f), respectively. Arrows indicate detached cleaved caspase 3-positive cells. Mean ± s.e.m is shown; n, number of animals. D denotes analysis of detached cells. *P<0.05, Student’s t-test. Scale bars: 50 μm.
We also examined the possibility that p120 null cells might be removed or engulfed by elements of the immune system. In several other organ systems (e.g. intestine, esophagus, epidermis), p120 ablation is associated with significant immune cell infiltration, which could facilitate rapid clearance of malfunctioning cells. However, immunostaining for macrophage/eosinophil [anti-F4/80 (Emr1)] and neutrophil (anti-Ly6B.2) markers showed little, if any, evidence for unusual recruitment of these cell types (data not shown).
Collectively, these experiments suggest that p120 null cells are being selectively lost by anoikis and subsequent clearance by mechanisms that do not involve obvious inflammation.
p120 is required and non-redundant for ductal architecture
To determine the immediate consequences of p120 loss on ductal architecture, sections from week 4 glands were analyzed by Hematoxylin and Eosin (H&E) staining and immunofluorescence microscopy (Fig. 4). H&E analyses revealed cell-cell adhesion defects manifested by dramatic cell sloughing and frequent partial occlusions of the lumens (Fig. 4A). Immunostaining for the apical marker crumbs 3 revealed obvious rounding of the apical cell surface, presumably reflecting poor basolateral cell-cell adhesions (Fig. 4B). By contrast, basal cells [marked by p63 (Trp63)] were never displaced from their normal position at the basement membrane, but appeared more sparse than in the wild-type glands (Fig. 4C).
Fig. 4.
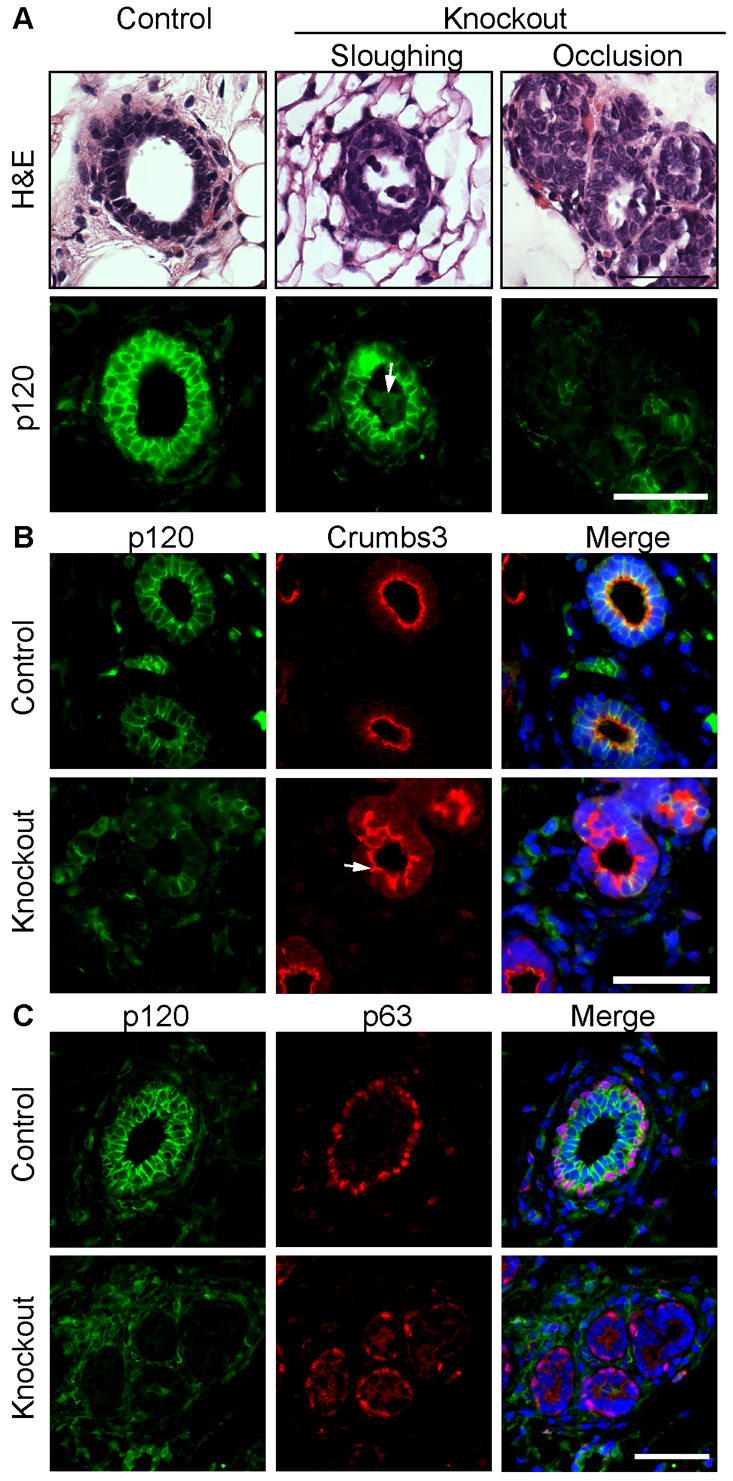
p120 ablation disrupts ductal architecture. (A) Serial sections of mammary glands from 4-week-old mice stained with H&E and with antibodies against p120. Representative images are shown of the two phenotypes observed: luminal sloughing and partial occlusions. Arrow indicates sloughed KO cells. (B) Four-week-old glands were co-immunostained for p120 and crumbs 3. Arrow indicates the mislocalized apical marker and severe disruption of ductal morphology in areas of p120 ablation. (C) Four-week-old glands were co-immunostained for p120 and p63. Scale bars: 50 μm.
As observed in several other organ systems, p120 ablation selectively affected the adherens junction as shown by decreased expression of E-cadherin and β-catenin in p120 null cells (Fig. 5A,B). Loss of p120 did not affect ZO-1 (Tjp1)-expressing tight junctions (Fig. 5C) or desmosomes (data not shown). Since immunostaining of E-cadherin and β-catenin was dramatically decreased in the absence of p120, these data suggest that p120 family members, if present, are unable to compensate in this tissue. Therefore, we analyzed the expression of the p120 family members Arvcf, p0071 (plakophilin 4) and ∂-catenin using well-characterized antibodies (Hofmann et al., 2009; Marciano et al., 2011; Walter et al., 2008; Walter et al., 2010). Family members demonstrated membranous and cytoplasmic localization in the mammary epithelium, which was not altered in the absence of p120 (Fig. 6). Thus, p120 provides a non-redundant function in cell-cell adhesion of mammary ductal cells.
Fig. 5.
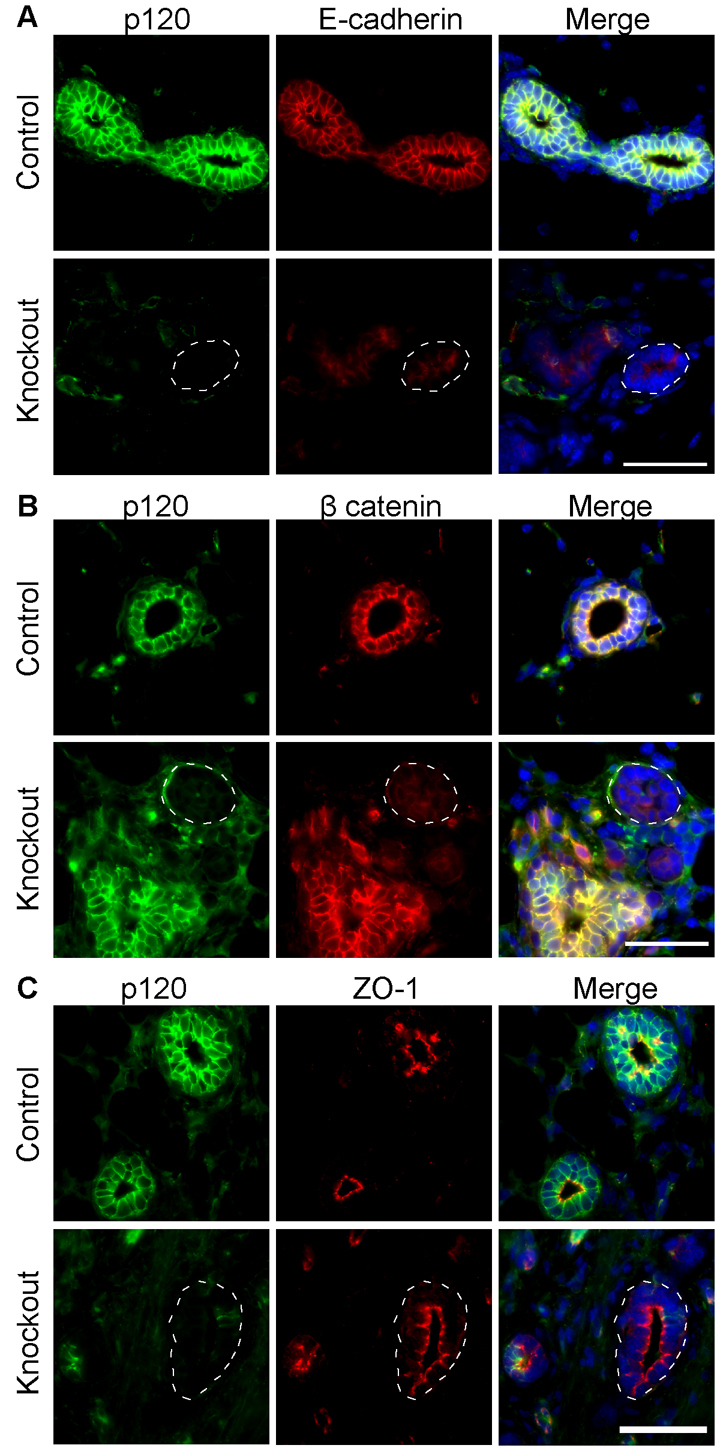
Dysregulation of the mammary gland cadherin complexes in the absence of p120. Sections of mammary glands from control and p120 KO mice were co-immunostained for (A) p120 and E-cadherin, (B) p120 and β-catenin and (C) p120 and ZO-1. Dashed circles indicate areas of p120 ablation and consequent downregulation of E-cadherin and β-catenin, but not ZO-1. Scale bars: 50 μm.
Fig. 6.
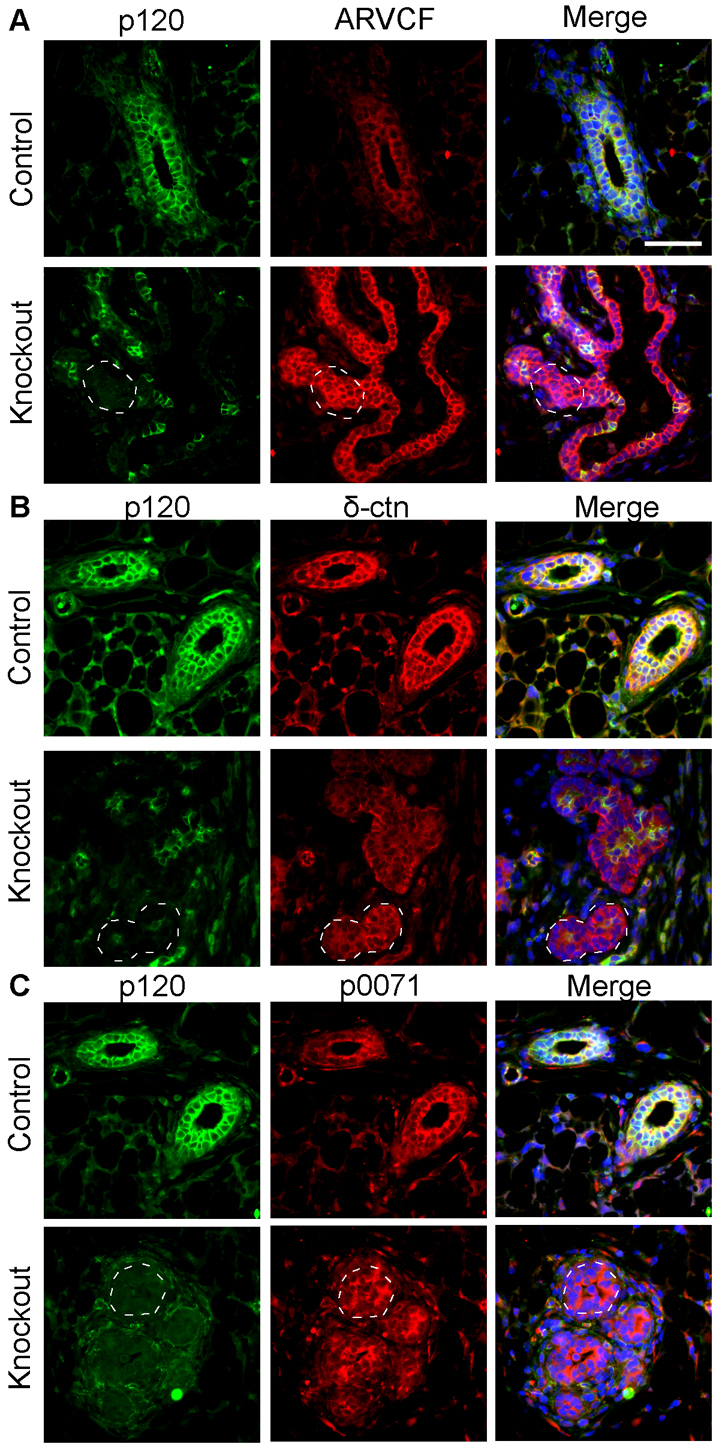
p120 family members cannot compensate for p120 loss. Sections from mammary glands of 4-week-old control and p120 KO mice co-immunostained for (A) p120 and Arvcf, (B) p120 and ∂-catenin or (C) p120 and p0071. Dashed regions indicate areas of p120 ablation. Scale bar: 50 μm.
p120 null cells are rapidly sorted and eliminated from nascent TEBs
The driving force behind the development of the mammary gland during puberty is the TEB, where the vast majority of cell growth, death and invasion occur. To determine the role of p120 in the TEB, we examined the effects of p120 ablation on TEB size and morphology (Fig. 7). Whereas total TEB number was unaffected in p120 KO mice (data not shown), average TEB size was significantly reduced at week 4 (Fig. 7A,B). However, TEB size normalized by week 5, well before the KO gland growth caught up with that of the wild-type gland (Fig. 7A,B and Fig. 2). Thus, the early delay in ductal elongation due to p120 ablation might occur in response to events taking place within the TEBs, which rely on p120 expression.
Fig. 7.

p120 null cells are rapidly shed and fail to participate in TEB development. (A) Whole-mount images of control and p120 KO mammary glands. Dashed circles highlight representative TEBs. (B) Analysis of TEB size. KO mice exhibited a statistically significant decrease in TEB size at 4 weeks but not at 5 weeks of age. Mean with s.e.m. *P<0.05, Student’s t-test. (C) Transverse sections of TEBs from inguinal mammary glands harvested at 4 weeks. Serial sections were stained with H&E (a,d,g) or immunostained for p120, SMA and K8. Nuclei were co-stained with Hoechst dye. Examples of luminal (d-f) and subcapsular (g-i) sloughing are shown. (D) Longitudinal sections of TEBs from inguinal mammary glands harvested at 4 weeks. Sections were immunostained for p120 and E-cadherin. Examples show luminal (top) and subcapsular (bottom) cell sloughing. Arrows, body cells; arrowheads, cap cells. The TEB is outlined. Scale bars: 1 mm in A; 50 μm in C,D.
To understand the nature of the defect at week 4, the histological morphology of TEBs from 4-week-old mice was examined. Fig. 7C,D show examples of typical TEB phenotypes in transverse and longitudinal sections, respectively. Interestingly, the majority of TEBs from KO mice lacked p120 null cells altogether, suggesting that the early size discrepancy might reflect very rapid clearing and/or loss of p120 null cells from these structures. In TEBs retaining significant numbers of p120 null cells, the cells were invariably rounded, non-adhesive and unlikely to be able to participate in end bud activity. In general, such structures segregated into one of two distinct scenarios based on where the p120 null cells accumulated. Fig. 7Cd-f illustrates sloughing of p120 null cells into the lumen. Alternatively, TEBs from p120 KO mice frequently contained aberrantly large subcapsular spaces, and these were also found to accumulate significant numbers of p120 null cells (Fig. 7Cg-i). The presence of p120 family members was insufficient to support TEB morphology in the absence of p120 (data not shown).
To identify the origin of the sloughed p120 null cells (i.e. cap versus body cells), we analyzed the images for basal (SMA) or luminal (K8) markers (Fig. 7C). Transverse sections of TEBs from control mice demonstrated a multilayered K8+ p120+ body surrounded by a single-cell SMA+ cap cell layer that also expressed p120, albeit at lower levels (Fig. 7C). p120 null cells shed into the lumen were K8+ SMA– (Fig. 7Cf, arrow), whereas p120 null cells accumulating in the subcapsular compartment were predominantly K8– SMA+, suggesting that they were derived from the cap layer or myoepithelial cells (Fig. 7Ci, arrowhead). Occasional examples of K8+ SMA– p120– cells were detected in the subcapsular region (Fig. 7Ci, arrow). Note that p120-expressing cells were rarely seen in the lumen or in the subcapsular space. Similar results were observed in longitudinal TEB sections stained with antibodies against p120 and E-cadherin (Fig. 7D).
Together, these observations show that p120 null mammary epithelial cells generated at puberty in the nascent TEBs are derived from both body and cap cell components, underscoring the requirement for p120 in both epithelial cell populations for adhesion and TEB morphogenesis.
p120 loss in vitro disables the collective migration required for branching morphogenesis in vivo
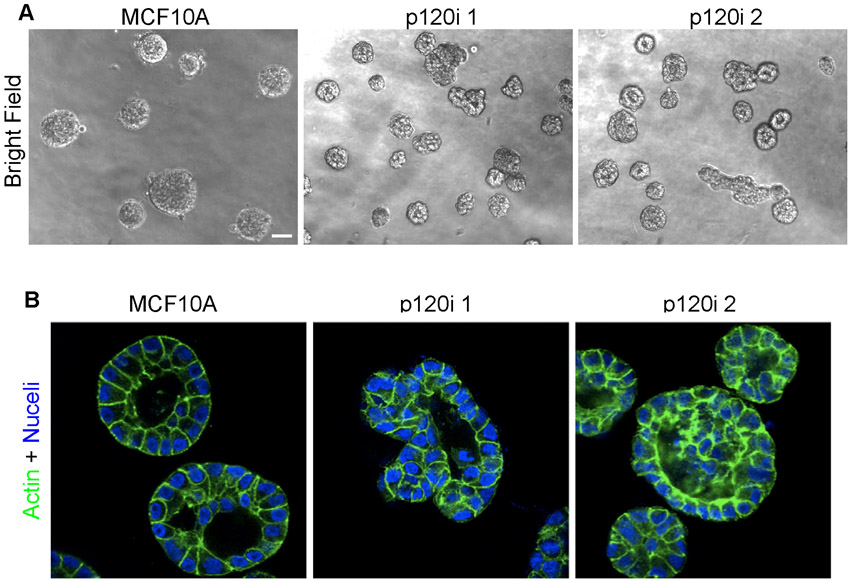
To clarify the mechanism of the underlying requirement for p120, we utilized RNA interference (RNAi) to stably knock down p120 in the nontumorigenic human mammary cell line MCF10A (Fig. 8). Although experiments in vitro do not necessarily recapitulate the complexity of in vivo morphogenesis, MCF10A cells have nonetheless been frequently used for mechanistic modeling of collective migration and other phenomena associated with mammary development (Debnath et al., 2002; Simpson et al., 2008). Fig. 8A illustrates the extent of p120 knockdown in two independently derived MCF10A clones. Similar to what was seen in the mammary epithelium, E-cadherin levels are significantly reduced by p120 depletion and are efficiently rescued by forced expression of either p120 isoform 1A or 3A (Fig. 8A).
Fig. 8.

p120 is required for collective migration. (A) Lysates from parental MCF10A cells, p120 knockdown monoclonal lines (p120i 1 and 2) and polyclonal cells expressing control, human p120 isoform 1A or 3A vectors were analyzed by immunoblotting as indicated (tubulin provided a loading control). (B) Two-dimensional morphology. Subconfluent cells were imaged by bright-field microscopy. (C) Wound-healing assays. Images are from time-lapse videos (supplementary material Movies 1-4) using the indicated cell lines. Arrows indicate single-cell migration events. Scale bar: 50 μm. (D) Quantification of wound healing. Cells from A were assayed at 6 or 12 hours. Mean with s.e.m. for five independent experiments. *P<0.05 compared with parental MCF10A cells and #P<0.05 compared with p120i + empty vector, Mann-Whitney tests.
In 2D cell cultures, parental MCF10A cells formed tightly adherent colonies, whereas cell-cell adhesion was completely disrupted by p120 knockdown (Fig. 8B). The phenotype was efficiently rescued by either p120 isoform 1A or 3A, as expected if the cell-cell adhesion defects are the result of p120 depletion (Fig. 8B).
We then examined the effects of p120 depletion on 3D acinar morphogenesis using a previously described Matrigel system (supplementary material Fig. S1) (Debnath et al., 2003). Whereas parental MCF10A cells formed simple lumen-containing acini, p120 loss resulted in disorganization of acinar structure and poorly defined lumens (supplementary material Fig. S1). In general, these structures closely resembled those of p120 null ducts as illustrated in Fig. 3.
Collective cell migration is required for TEB invasion through the mammary fat pad. To assess the role of p120 in collective cell migration, we conducted p120 knockdown/add-back experiments using wound-healing assays as a readout. Fig. 8C shows selected images from time-lapse movies (supplementary material Movies 1-4). As also observed in vivo, p120 depletion did not alter cell proliferation (data not shown). However, although TEB outgrowth was delayed in the p120 KO gland, wound closure in vitro by p120 knockdown MCF10A cells was not impaired, and individual p120 knockdown cells in fact migrated faster than their parental counterparts (Fig. 8D). Thus, the delay in TEB outgrowth is unlikely to stem from a migration defect per se. Instead, the data suggest that p120-deficient cells simply fail to participate in the collective migration process due to loss of cell-cell adhesion (Fig. 8C, supplementary material Movie 2).
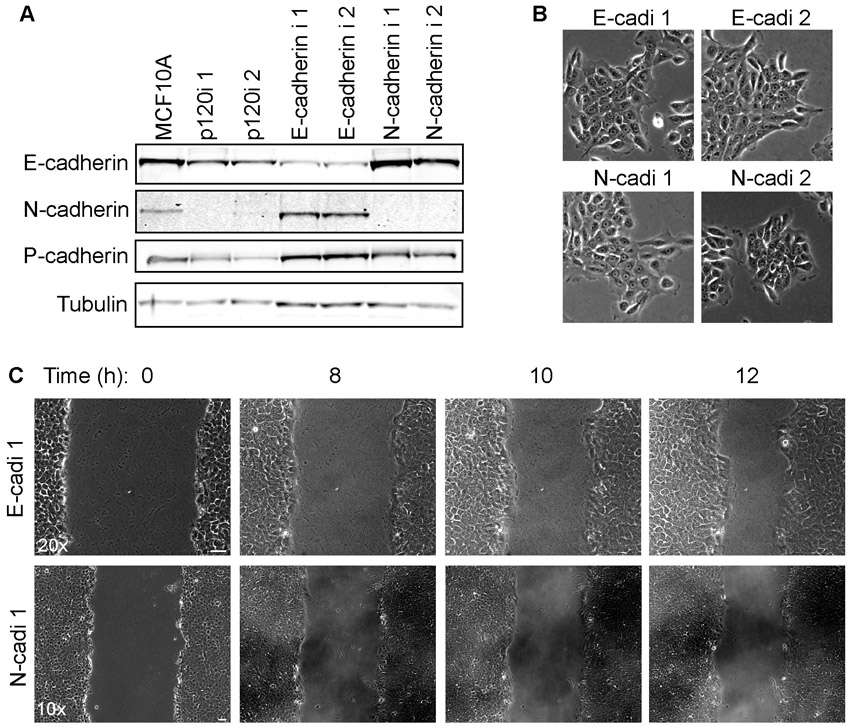
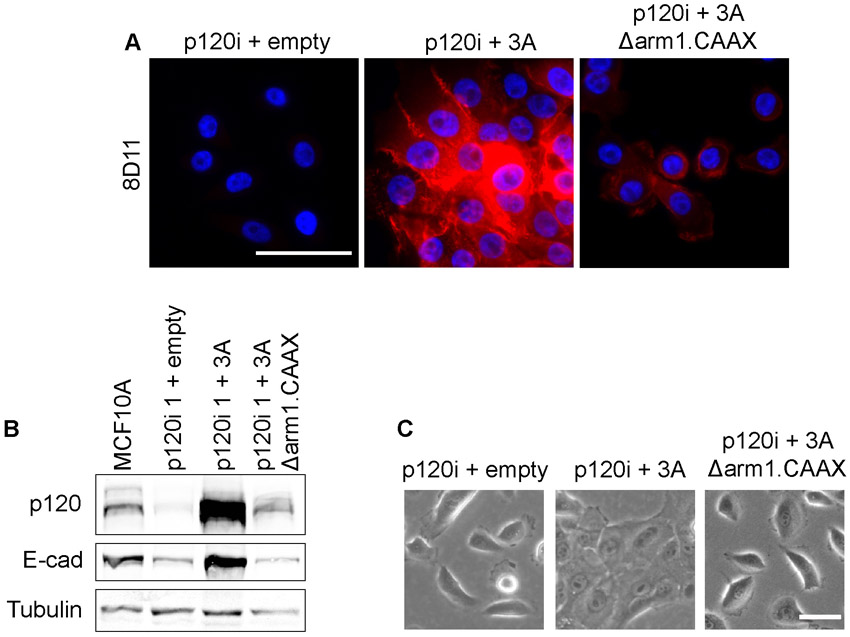
To determine whether the role of p120 in collective migration is dependent on cadherin association, we tested a mutant form of p120 isoform 3 that is driven to the membrane by a CAAX box but which cannot bind cadherins (3A Δarm1.CAAX) (Wildenberg et al., 2006) (supplementary material Fig. S3). In contrast to p120 3A, the mutant did not rescue collective migration, suggesting that the role of p120 in this process is cadherin dependent (Fig. 8C, supplementary material Movies 3, 4). Depletion of E-cadherin or N-cadherin (cadherin 2) individually from MCF10A cells did not disrupt collective migration, suggesting that one can stand in for the other. p120 depletion, however, is effective because it reduces all classical cadherins (supplementary material Fig. S2).
Collectively, these observations predict that branching morphogenesis will be impaired or blocked altogether if p120 is unavailable. Thus, we examined the effects of p120 depletion in vitro and in vivo on branching morphogenesis (Fig. 9). PMECs were formed into mammospheres and induced to branch using FGF2 (McCaffrey and Macara, 2009; Ewald et al., 2008). In the absence of p120, branching was reduced and often failed entirely (Fig. 9A-C). p120 depletion is illustrated in supplementary material Fig. S4. Note that when branching was observed, p120 was invariably retained (i.e. GFP negative) (Fig. 8D). When transplanted into cleared fat pads, p120i PMECs were unable to reconstitute the gland (Fig. 9E). Control cells formed clearly identifiable ducts and TEBs, whereas p120-depleted cells manifested as thin strands and small cell groups without discernible structure.
Fig. 9.

p120 is necessary for in vitro mammary branching and in vivo gland reconstitution. (A-D) PMECs infected with either control GFP or p120i GFP virus were subjected to branching assays. (A) Representative images of control and p120-depleted branched mammospheres. Yellow dashed lines indicate that TEB-like structures always retain p120. (B) Quantification of percentage branched mammospheres from five independent experiments. (C) Quantification of branches per mammosphere. Median values are listed. Three independent experiments were performed. (D) Percentage of GFP-positive branches per mammosphere. Three independent experiments were performed. (B-D) Student’s t-test. (E) In vivo mammary reconstitution assays. Representative fluorescent images of whole-mounts are shown. The mammary fat pad is outlined. Data are mean percentage outgrowth ± s.e.m. Paired t-test, n=6. Scale bars: 50 μm in A; 1 mm in E.
DISCUSSION
The effects of p120 KO in different organ systems are highly variable. Here, we show that p120 plays an essential role in the morphogenesis of the mammary gland. Ductal architecture is rapidly compromised and p120 null cells disappear altogether within a few weeks. In the TEB, p120 null cells are virtually unable to participate in the dynamic rearrangements required for invasion and morphogenesis. Functional analyses in vitro reveal severe defects in cell-cell adhesion and a striking failure of collective migration. Thus, it appears that mammary gland development depends on p120 because the TEB, which is the main functional unit of mammary development, is effectively disabled by p120 ablation.
In our current mouse model, the severity of the phenotype is largely masked by the mosaic nature of the p120 KO. The phenotype ultimately manifests as little more than a delay in ductal penetration, but in fact reflects massive sorting and elimination of p120 null cells, such that very few remain 3 weeks after p120 ablation. From then on, the ‘knockout’ gland is essentially p120 positive and morphogenesis proceeds normally. The strong selective pressure for cells that have retained p120 suggests that if the knockout had been complete, the gland would not have formed at all. Anecdotal evidence from previous studies of p120 ablation in the salivary gland using a different MMTV-Cre mouse suggests that this is, in fact, the case. Indeed, although the vast majority of these animals died shortly after birth, females that survived into adulthood were completely devoid of mammary ductal trees (supplementary material Fig. S5) (Davis and Reynolds, 2006). Our in vivo PMEC assays confirm these findings, as p120i cells are unable to form a mammary gland (Fig. 9E).
Interestingly, in vitro p120 depletion in different epithelial cell types results in a wide spectrum of adhesion phenotypes. For example, mammary MCF10A cells separate completely from one another in 2D cultures, whereas similarly cultured MDCK cells lacking p120 form colonies that are essentially indistinguishable from those of parental controls (Fig. 8B) (Dohn et al., 2009; Simpson et al., 2008). More common is a spectrum of adhesive defects that fall between these extremes (Davis et al., 2003). Similarly, in vivo p120 KO phenotypes are surprisingly diverse. For example, although intercellular adhesive defects are not observed after p120 KO in the epidermis, a massive inflammatory response is induced by cell-autonomous signaling defects associated with NFκB activity (Perez-Moreno et al., 2006). In the prostatic epithelium, cadherin expression is nearly eliminated by p120 ablation and glandular morphology appears to be virtually unaffected (A.B.R., unpublished). In salivary gland and intestinal epithelium, cadherin depletion is more moderate following p120 KO (i.e. ∼50% depletion relative to control epithelium), but nonetheless causes obvious adhesion defects with extensive cell shedding (Davis and Reynolds, 2006; Smalley-Freed et al., 2010). Cell- and tissue-specific contexts are clearly crucial and contribute along with other factors to the ultimate effect of p120 ablation.
Our in vivo data reveal that the TEB is extraordinarily sensitive to p120 ablation. Interestingly, an unbiased in vitro RNAi screen for proteins affecting MCF10A cell motility identified both p120 and P-cadherin as central mediators of collective migration (Simpson et al., 2008). This result highlights the often overlooked fact that p120 stabilizes all classical cadherins, and implies an activity for P-cadherin that might not be shared by E- and/or N-cadherin (at least in MCF10A cells). Similarly, p120 is required for cadherin-dependent collective invasion in an A431 squamous carcinoma cell model (Macpherson et al., 2007). Eric Sahai’s group has recently proposed that collective migration is controlled in part by an E-cadherin/DDR1/Par3-Par6 complex that functions to limit actomyosin contractility as needed at adherens junctions through mechanisms involving p190ARhoGAP (Grlf1) and RhoE (Rnd3) (Hidalgo-Carcedo et al., 2011). Although not directly included as part of the Sahai model, p120 is likely to play a role. We have previously demonstrated that interactions between p120, RhoA and p190RhoGAP function to limit contractility at N-cadherin-based adherens junctions in NIH3T3 cells (Wildenberg et al., 2006). Thus, one possibility is that p120 functions in the Sahai model as part of the machinery that enables collective migration by suppressing RhoA. Indeed, p120-depleted MCF10A cells are highly contractile and demonstrate readouts indicative of high Rho activity (data not shown). Alternatively, p120 ablation might simply override the normal mechanisms for modulating collective migration by depleting E-cadherin to levels that cannot sustain cell-cell adhesion. These models are not necessarily mutually exclusive. Exactly how E-cadherin levels are controlled by p120 is not well understood and could conceivably be related to novel concepts proposed by Sahai and colleagues.
Although TEB defects associated with p120 ablation could in principle stem from events unrelated to loss of E-cadherin [e.g. dysregulation of Kaiso (Zbtb33) activity] (Daniel and Reynolds, 1999), the evidence overall points strongly to E-cadherin depletion as the dominant, if not the sole, driver of the phenotype. p120 is required for the stability of all classical cadherins, including the E- and P-cadherins found in luminal and basal cells, respectively. Accordingly, E-cadherin neutralizing antibodies selectively disrupt the body cell layer, whereas those for P-cadherin disrupt only the cap cell layer (Daniel et al., 1995). TEB activity stalls in either case, indicating that both layers must be intact for the TEB to function normally. Notably, the cell-cell adhesion defects associated with cadherin blocking are morphologically almost indistinguishable from those induced by p120 ablation, and both mechanisms clearly act through disabling cadherins. Thus, the effects of p120 ablation on cadherin loss are sufficiently severe in the TEB that secondary and/or less obvious consequences of p120 ablation, if present, go undetected. For example, cell polarity proteins interact functionally with cadherin complexes (Qin et al., 2005; Navarro et al., 2005; Zhan et al., 2008), but might be largely disabled in the context of severely compromised cell-cell adhesion.
Surprisingly, p120 family members were unable to compensate for loss of p120, despite evidence that they can effectively rescue cadherin stability and cell adhesion in vitro (Davis et al., 2003). Fig. 6 illustrates clearly the significant presence of all three family members in p120 KO tissue. It is unclear whether this failure to rescue p120 ablation extends to other organs. In most epithelial tissues, including the epidermis, gastrointestinal tract and salivary glands, cadherin levels are reduced but not decimated to the extent observed in the mammary gland. In fact, on average, p120-ablated tissues tend to retain 25-50% of cadherin levels found in control tissue (Davis and Reynolds, 2006; Perez-Moreno et al., 2006; Smalley-Freed et al., 2010). In vivo correlations, where available, appear to support the in vitro data in that p120 family members have been found in tissues in which p120 ablation does not result in complete cadherin loss (Marciano et al., 2011; Perez-Moreno et al., 2006). However, whether endogenous p120 family members compensate for p120 loss in vivo has yet to be directly demonstrated in any tissue (e.g. by in vivo double KO). In the TEB, the near complete absence of both cadherins and junctional β-catenin following p120 ablation indicates that these potential compensatory mechanisms are either insufficient or inactive.
Although p120 knockdown in vitro induced severe distortions in MCF10A mammosphere morphology, the cells themselves were healthy and persisted indefinitely. By contrast, p120-ablated cells in the developing mammary gland were rapidly lost and rarely observed past week 6. Interestingly, detached cells were frequently TUNEL and cleaved caspase 3 positive. Thus, although the exact mechanism of cell death is unclear, our detachment and apoptosis data imply a form of anoikis (Wang et al., 2003; Gilmore, 2005).
In contrast to several other tissues (Perez-Moreno et al., 2006; Smalley-Freed et al., 2010), we did not observe significant inflammation in the p120 KO mammary gland. It might be that recognition and removal of p120 null cells does not require de novo influx of immune cells. Rather, in the greater scheme of active TEB invasion, efficient removal of p120 null cells by already present tissue-resident macrophages might be sufficiently routine to go largely unnoticed. Tissue-resident macrophages are known to actively participate in TEB-proximal stromal remodeling and were in fact detected at normal levels (Gouon-Evans et al., 2002; Ingman et al., 2006). Additionally, these cells might also be cleared by neighboring mammary epithelial cells via efferocytosis, a phagocytic process recently shown to be important during involution of the mammary gland (Monks et al., 2008; Sandahl et al., 2010).
In conclusion, we demonstrate for the first time that p120 is essential for mammary gland development. The explanation is likely to lie in the extraordinary sensitivity of the TEB to p120 loss and the dependence of TEB function on collective migration, a phenomenon based on dynamically regulated cell-cell adhesion. Our work extends previous observations on the role of p120 in collective migration (Hidalgo-Carcedo et al., 2011; Macpherson et al., 2007; Simpson et al., 2008) to a highly relevant in vivo setting and is in line with prior anecdotal evidence that mammary development essentially fails altogether in the absence of p120 (Davis and Reynolds, 2006). Given the unique morphogenetic status of the TEB, it will be interesting to extend these studies to p120 KO in breast cancer models as well as fully developed mammary epithelium.
Supplementary Material
Acknowledgments
We acknowledge the assistance of the Human Tissue Acquisition and Shared Resource Core at Vanderbilt (National Institutes of Health P30 CA68485) and the Vanderbilt Flow Cytometry Core Lab (National Institutes of Health P30 CA68485 and DK058404). We thank Elizabeth Koehler and Dr Gregory Ayers for guidance on statistical analysis; Dr Ian Macara and Dr Joanne Montalbano for assistance with mammosphere technology; and members of the A.B.R. laboratory and Dr Rebecca Muraoka-Cook for helpful discussions of this work.
Footnotes
Funding
This work was supported by the National Institutes of Health [NIH R01 CA111947 and NIH R01 CA55724 to A.B.R.; NIH R01 CA085492 to H.L.M.; NIH 2PO1CA099031-06A1 to W.J.M.]; by the Department of Defense [Predoctoral Trainee Award BC083306 to S.J.K.]; by the Terry Fox Foundation [#020002 to W.J.M.]; by the Canadian Institutes of Health Research [MOP 93525 and MOP 89751 to W.J.M.]; and by German Cancer Aid (to I.H.). Funding was also received through the Vanderbilt Cancer Center Support Grant [NIH P30 CA068485] and a Pilot Grant to A.B.R. through the Vanderbilt Breast SPORE [NIH P50 CA98131]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.072769/-/DC1
References
- Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. (2000). Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2, 637–644 [DOI] [PubMed] [Google Scholar]
- Andrechek E. R., Hardy W. R., Siegel P. M., Rudnicki M. A., Cardiff R. D., Muller W. J. (2000). Amplification of the neu/erbB-2 oncogene in a mouse model of mammary tumorigenesis. Proc. Natl. Acad. Sci. USA 97, 3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrechek E. R., White D., Muller W. J. (2005). Targeted disruption of ErbB2/Neu in the mammary epithelium results in impaired ductal outgrowth. Oncogene 24, 932–937 [DOI] [PubMed] [Google Scholar]
- Bartlett J. D., Dobeck J. M., Tye C. E., Perez-Moreno M., Stokes N., Reynolds A. B., Fuchs E., Skobe Z. (2010). Targeted p120-catenin ablation disrupts dental enamel development. PLoS ONE 5, e12703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier W. (1995). E-cadherin as a tumor (invasion) suppressor gene. BioEssays 17, 97–99 [DOI] [PubMed] [Google Scholar]
- Cardiff R. D., Wellings S. R. (1999). The comparative pathology of human and mouse mammary glands. J. Mammary Gland Biol. Neoplasia 4, 105–122 [DOI] [PubMed] [Google Scholar]
- Daniel C. W., Strickland P., Friedmann Y. (1995). Expression and functional role of E- and P-cadherins in mouse mammary ductal morphogenesis and growth. Dev. Biol. 169, 511–519 [DOI] [PubMed] [Google Scholar]
- Daniel J. M., Reynolds A. B. (1999). The catenin p120(ctn) interacts with Kaiso, a novel BTB/POZ domain zinc finger transcription factor. Mol. Cell. Biol. 19, 3614–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M., Reynolds A. (2006). Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev. Cell 10, 21–31 [DOI] [PubMed] [Google Scholar]
- Davis M. A., Ireton R. C., Reynolds A. B. (2003). A core function for p120-catenin in cadherin turnover. J. Cell Biol. 163, 525–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J., Mills K. R., Collins N. L., Reginato M. J., Muthuswamy S. K., Brugge J. S. (2002). The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40 [DOI] [PubMed] [Google Scholar]
- Debnath J., Muthuswamy S. K., Brugge J. S. (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268 [DOI] [PubMed] [Google Scholar]
- Dohn M. R., Brown M. V., Reynolds A. B. (2009). An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 184, 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G., Abdallah W. M., Foley J. M., Jackson K. W., Clarke M. F., Kawamura M. J., Wicha M. S. (2003). In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia L. P., Yamamoto M., Zang K., Reichardt L. F. (2006). p120 catenin regulates dendritic spine and synapse development through Rho-family GTPases and cadherins. Neuron 51, 43–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald A. J., Brenot A., Duong M., Chan B. S., Werb Z. (2008). Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev. Cell 14, 570–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallin W. J. (1998). Evolution of the “classical” cadherin family of cell adhesion molecules in vertebrates. Mol. Biol. Evol. 15, 1099–1107 [DOI] [PubMed] [Google Scholar]
- Gilmore A. P. (2005). Anoikis. Cell Death Differ. 12, 1473–1477 [DOI] [PubMed] [Google Scholar]
- Gouon-Evans V., Lin E. Y., Pollard J. W. (2002). Requirement of macrophages and eosinophils and their cytokines/chemokines for mammary gland development. Breast Cancer Res. 4, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasl-Kraupp B., Ruttkay-Nedecky B., Koudelka H., Bukowska K., Bursch W., Schulte-Hermann R. (1995). In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21, 1465–1468 [DOI] [PubMed] [Google Scholar]
- Gumbiner B. M. (2005). Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6, 622–634 [DOI] [PubMed] [Google Scholar]
- Hennighausen L., Robinson G. W. (2005). Information networks in the mammary gland. Nat. Rev. Mol. Cell Biol. 6, 715–725 [DOI] [PubMed] [Google Scholar]
- Herrenknecht K., Ozawa M., Eckerskorn C., Lottspeich F., Lenter M., Kemler R. (1991). The uvomorulin-anchorage protein alpha catenin is a vinculin homologue. Proc. Natl. Acad. Sci. USA 88, 9156–9160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo-Carcedo C., Hooper S., Chaudhry S. I., Williamson P., Harrington K., Leitinger B., Sahai E. (2011). Collective cell migration requires suppression of actomyosin at cell-cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat. Cell Biol. 13, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinck L., Silberstein G. B. (2005). Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res. 7, 245–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann I., Schlechter T., Kuhn C., Hergt M., Franke W. W. (2009). Protein p0071-an armadillo plaque protein that characterizes a specific subtype of adherens junctions. J. Cell Sci. 122, 21–24 [DOI] [PubMed] [Google Scholar]
- Hulpiau P., van Roy F. (2009). Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 41, 349–369 [DOI] [PubMed] [Google Scholar]
- Hulsken J., Birchmeier W., Behrens J. (1994). E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J. Cell Biol. 127, 2061–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingman W. V., Wyckoff J., Gouon-Evans V., Condeelis J., Pollard J. W. (2006). Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev. Dyn. 235, 3222–3229 [DOI] [PubMed] [Google Scholar]
- Ireton R. C., Davis M. A., van Hengel J., Mariner D. J., Barnes K., Thoreson M. A., Anastasiadis P. Z., Matrisian L., Bundy L. M., Sealy L., et al. (2002). A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 159, 465–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Fisher A. J., Bellinger G., Ramabhadran R., Morris J. K., Lee K.-F., Stern D. F. (2004). ErbB2 is required for ductal morphogenesis of the mammary gland. Proc. Natl. Acad. Sci. USA 101, 17138–17143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouros-Mehr H., Slorach E. M., Sternlicht M. D., Werb Z. (2006). GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 127, 1041–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P., Ewald A. J., Martin G. R., Werb Z. (2008). Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev. Biol. 321, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson I. R., Hooper S., Serrels A., Mcgarry L., Ozanne B. W., Harrington K., Frame M. C., Sahai E., Brunton V. G. (2007). p120-catenin is required for the collective invasion of squamous cell carcinoma cells via a phosphorylation-independent mechanism. Oncogene 26, 5214–5228 [DOI] [PubMed] [Google Scholar]
- Marciano D. K., Brakeman P. R., Lee C. Z., Spivak N., Eastburn D. J., Bryant D. M., Beaudoin G. M., 3rd, Hofmann I., Mostov K. E., Reichardt L. F. (2011). p120 catenin is required for normal renal tubulogenesis and glomerulogenesis. Development 138, 2099–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariner D. J., Davis M. A., Reynolds A. B. (2004). EGFR signaling to p120-catenin through phosphorylation at Y228. J. Cell Sci. 117, 1339–1350 [DOI] [PubMed] [Google Scholar]
- McCaffrey L. M., Macara I. G. (2009). The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev. 12, 1450–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea P. D., Gumbiner B. M. (1991). Purification of a 92-kDa cytoplasmic protein tightly associated with the cell-cell adhesion molecule E-cadherin (uvomorulin). Characterization and extractability of the protein complex from the cell cytostructure. J. Biol. Chem. 266, 4514–4520 [PubMed] [Google Scholar]
- McIlroy D., Tanaka M., Sakahira H., Fukuyama H., Suzuki M., Yamamura K., Ohsawa Y., Uchiyama Y., Nagata S. (2000). An auxiliary mode of apoptotic DNA fragmentation provided by phagocytes. Genes Dev. 14, 549–558 [PMC free article] [PubMed] [Google Scholar]
- Monks J., Smith-Steinhart C., Kruk E. R., Fadok V. A., Henson P. M. (2008). Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biol. Reprod. 78, 586–594 [DOI] [PubMed] [Google Scholar]
- Nagafuchi A., Takeichi M., Tsukita S. (1991). The 102 kd cadherin-associated protein: similarity to vinculin and posttranscriptional regulation of expression. Cell 65, 849–857 [DOI] [PubMed] [Google Scholar]
- Navarro C., Nola S., Audebert S., Santoni M. J., Arsanto J. P., Ginestier C., Marchetto S., Jacquemier J., Isnardon D., LeBivic A., et al. (2005). Junctional recruitment of mammalian Scribble relies on E-cadherin engagement. Oncogene 27, 4330–4339 [DOI] [PubMed] [Google Scholar]
- Noren N. K., Liu B. P., Burridge K., Kreft B. (2000). p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 150, 567–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oas R. G., Xiao K., Summers S., Wittich K. B., Chiasson C. M., Martin W. D., Grossniklaus H. E., Vincent P. A., Reynolds A. B., Kowalczyk A. P. (2010). p120-Catenin is required for mouse vascular development. Circ. Res. 106, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M., Mailleux A. A., Mouneimne G., Normand G., Schnitt S. J., King R. W., Cibas E. S., Brugge J. S. (2007). A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979 [DOI] [PubMed] [Google Scholar]
- Parsa S., Ramasamy S., Delanghe S., Gupte V., Haigh J., Medina D., Bellusci S. (2008). Terminal end bud maintenance in mammary gland is dependent upon FGFR2b signaling. Dev. Biol. 317, 121–131 [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M., Davis M. A., Wong E., Pasolli H. A., Reynolds A. B., Fuchs E. (2006). p120-catenin mediates inflammatory responses in the skin. Cell 124, 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y., Capaldo C., Gumbiner B. M., Macara I. G. (2005). The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J. Cell Biol. 171, 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A. B., Daniel J. M., Mo Y. Y., Wu J., Zhang Z. (1996). The novel catenin p120cas binds classical cadherins and induces an unusual morphological phenotype in NIH3T3 fibroblasts. Exp. Cell Res. 225, 328–337 [DOI] [PubMed] [Google Scholar]
- Richert M. M., Schwertfeger K. L., Ryder J. W., Anderson S. M. (2000). An atlas of mouse mammary gland development. J. Mammary Gland Biol. Neoplasia 5, 227–241 [DOI] [PubMed] [Google Scholar]
- Rimm D. L., Koslov E. R., Kebriaei P., Cianci C. D., Morrow J. S. (1995). Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc. Natl. Acad. Sci. USA 92, 8813–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandahl M., Hunter D. M., Strunk K. E., Earp H. S., Cook R. S. (2010). Epithelial cell-directed efferocytosis in the post-partum mammary gland is necessary for tissue homeostasis and future lactation. BMC Dev. Biol. 10, 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson K., Selfors L., Bui J., Reynolds A., Leake D., Khvorova A., Brugge J. (2008). Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 10, 1027–1038 [DOI] [PubMed] [Google Scholar]
- Smalley-Freed W. G., Efimov A., Burnett P. E., Short S. P., Davis M. A., Gumucio D. L., Washington M. K., Coffey R. J., Reynolds A. B. (2010). p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J. Clin. Invest. 120, 1824–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K., Strickland P., Valdes A., Shin G. C., Hinck L. (2003). Netrin-1/neogenin interaction stabilizes multipotent progenitor cap cells during mammary gland morphogenesis. Dev. Cell 4, 371–382 [DOI] [PubMed] [Google Scholar]
- Stairs D. B., Bayne L. J., Rhoades B., Vega M. E., Waldron T. J., Kalabis J., Klein-Szanto A., Lee J.-S., Katz J. P., Diehl J. A., et al. (2011). Deletion of p120-Catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell 19, 470–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht M. D. (2006). Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 8, 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht M. D., Kouros-Mehr H., Lu P., Werb Z. (2006). Hormonal and local control of mammary branching morphogenesis. Differentiation 74, 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. (1991). Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455 [DOI] [PubMed] [Google Scholar]
- Takeichi M. (1995). Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7, 619–627 [DOI] [PubMed] [Google Scholar]
- Takeichi M., Hatta K., Nose A., Nagafuchi A., Matsunaga M. (1989). Cadherin-mediated specific cell adhesion and animal morphogenesis. CIBA Found. Symp. 144, 243–249 [DOI] [PubMed] [Google Scholar]
- Thoreson M. A., Anastasiadis P. Z., Daniel J. M., Ireton R. C., Wheelock M. J., Johnson K. R., Hummingbird D. K., Reynolds A. B. (2000). Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 148, 189–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught D., Chen J., Brantley-Sieders D. M. (2009). Regulation of mammary gland branching morphogenesis by EphA2 receptor tyrosine kinase. Mol. Biol. Cell 20, 2572–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter B., Schlechter T., Hergt M., Berger I., Hofmann I. (2008). Differential expression pattern of protein ARVCF in nephron segments of human and mouse kidney. Histochem. Cell Biol. 130, 943–956 [DOI] [PubMed] [Google Scholar]
- Walter B., Krebs U., Berger I., Hofmann I. (2010). Protein p0071, an armadillo plaque protein of adherens junctions, is predominantly expressed in distal renal tubules. Histochem. Cell Biol. 133, 69–83 [DOI] [PubMed] [Google Scholar]
- Wang P., Valentijn A. J., Gilmore A. P., Streuli C. H. (2003). Early events in the anoikis program occur in the absence of caspase activation. J. Biol. Chem. 278, 19917–19925 [DOI] [PubMed] [Google Scholar]
- Wildenberg G. A., Dohn M. R., Carnahan R. H., Davis M. A., Lobdell N. A., Settleman J., Reynolds A. B. (2006). p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127, 1027–1039 [DOI] [PubMed] [Google Scholar]
- Xiao K., Allison D. F., Buckley K. M., Kottke M. D., Vincent P. A., Faundez V., Kowalczyk A. P. (2003). Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J. Cell Biol. 163, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S., Pokutta S., Drees F., Weis W. I., Nelson W. J. (2005). Deconstructing the cadherin-catenin-actin complex. Cell 123, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap A. S. (1998). The morphogenetic role of cadherin cell adhesion molecules in human cancer: a thematic review. Cancer Invest. 16, 252–261 [DOI] [PubMed] [Google Scholar]
- Yap A. S., Niessen C. M., Gumbiner B. M. (1998). The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J. Cell Biol. 141, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L., Rosenberg A., Bergami K. C., Yu M., Xuan Z., Jaffe A. B., Allred C., Muthuswamy S. K. (2008). Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 135, 865–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}