

Abstract
Fanconi anemia (FA) is a rare, genetically heterogeneous autosomal recessive disorder associated with progressive aplastic anemia, congenital abnormalities, and cancer. FA has a very high incidence in the Afrikaner population of South Africa, possibly due to a founder effect. Previously we observed allelic association between polymorphic markers flanking the FA group A gene (FANCA) and disease chromosomes in Afrikaners. We genotyped 26 FA families with microsatellite and single nucleotide polymorphic markers and detected five FANCA haplotypes. Mutation scanning of the FANCA gene revealed association of these haplotypes with four different mutations. The most common was an intragenic deletion of exons 12–31, accounting for 60% of FA chromosomes in 46 unrelated Afrikaner FA patients, while two other mutations accounted for an additional 20%. Screening for these mutations in the European populations ancestral to the Afrikaners detected one patient from the Western Ruhr region of Germany who was heterozygous for the major deletion. The mutation was associated with the same unique FANCA haplotype as in Afrikaner patients. Genealogical investigation of 12 Afrikaner families with FA revealed that all were descended from a French Huguenot couple who arrived at the Cape on June 5, 1688, whereas mutation analysis showed that the carriers of the major mutation were descendants of this same couple. The molecular and genealogical evidence is consistent with transmission of the major mutation to Western Germany and the Cape near the end of the 17th century, confirming the existence of a founder effect for FA in South Africa.
Fanconi anemia (FA) is an autosomal recessive disorder that is associated with progressive bone marrow failure, multiple congenital abnormalities, and a high incidence of cancer. Seven complementation groups (A–G) have been described, and five of the corresponding genes have been cloned (1–6). The condition is rare in most populations, with the heterozygote frequency estimated to be about 1 in 300 (7). The two notable exceptions to this are Ashkenazi Jews and the white Afrikaans-speaking population of South Africa. In the Ashkenazim, almost all cases of FA have been shown to be caused by a single splice site mutation in the FANCC gene, IVS4+4A>T (8), and the heterozygote frequency is about 1 in 90 (9, 10). In South Africa, the incidence of FA in children aged under 16 years was reported to be 1 in 22,000 in the Orange Free State and Northern Cape region (11). The prevalence in the Afrikaners of the Southern Transvaal province was at least 1 in 22,000 live births, which would produce an expected heterozygote frequency of 1 in 77 (12). It was proposed that the high prevalence of FA in the Afrikaners was consistent with random genetic drift and a founder effect (12). There is strong precedent for founder effects in this population [reviewed by Jenkins (13)]. The best documented of these is variegate porphyria, with most patients being descended from a 17th century Dutch immigrant (14), and more than 95% of families carrying the R59W mutation in the protoporphyrin oxidase gene (15). The existence of founder effects in the Afrikaner can be explained by the history of the formation of this population (16). The establishment of a settlement at the Cape by the Dutch East India company in 1652 was followed by the arrival of Dutch, German, and French Huguenot settlers in the latter part of the 17th century. However, there was very little immigration in the next century, and the population expanded about 2,500-fold over a period of 300 years in relative social isolation.
Our earlier linkage studies in 21 Afrikaner families with FA detected no recombination between the disease phenotype and the polymorphic marker D16S303, which is closely linked to the FANCA gene (17). We also observed highly significant allelic association between this marker and disease chromosomes, which supported the suggestion of a founder effect for FA in this population. We then identified the FANCA gene by both functional complementation and positional cloning (2, 3). We have now screened this gene for mutations in Afrikaner FA patients and report the existence of multiple founder mutations in the FANCA gene in this population. Our molecular and genealogical studies of South African and European FA patients also provide pointers as to the likely origin of the most common of these mutations.
Materials and Methods
Patients and Families.
Patients were ascertained from referrals to the Departments of Human Genetics in Bloemfontein and Johannesburg for analysis of sensitivity to the DNA crosslinking agents diepoxybutane or mitomycin C, which is the definitive confirmatory test for FA (18). Only patients with increased sensitivity of peripheral blood lymphocytes to these agents were included in the study. For the purposes of this study, Afrikaner patients were defined as individuals with at least one maternal and one paternal Afrikaans-speaking grandparent. Blood samples were obtained from FA patients and their families with informed consent.
Genealogical Studies.
Families with at least one child affected with FA, as defined by hypersensitivity to diepoxybutane, and where genealogical evidence from at least 4–5 generations was available, were selected for the study. Twelve families met these criteria. Genealogical evidence on the 4–5 generations preceding the index case, and dating back to 1880–1910, was obtained primarily from the families themselves, whereas information before this was obtained from death notices and books on Afrikaner genealogy. Death notices are retained by the Master of the Supreme Court of South Africa. Records of individuals in the former Orange Free State province who have died since 1960 are kept in Bloemfontein, and the records of individuals from the former Transvaal province who have died since 1975 are kept in Pretoria. Records of individuals who died before these dates are held in the state archives of the same cities. This information was used to complement, and find a connection with, data recorded in the literature on Afrikaner genealogy. The cyrillic 2r computer program was used to store all relevant data and to draw family trees.
Haplotype Analysis.
DNA was extracted and typed with fluorescently labeled microsatellite polymorphisms flanking the FANCA gene, as described (17). Markers used were D16S305, D16S413, D16S3023, D16S3026, D16S3121, and D16S303. Single nucleotide polymorphisms (SNPs) associated with the major microsatellite haplotypes were typed by automated fluorescent sequencing on an Applied Biosystems 377 automated DNA sequencer.
Mutation Analysis.
The full coding sequence of the FANCA gene was scanned for mutations by using a two-step protocol (19). In step 1, the FANCA coding region was amplified by reverse transcription and PCR in six overlapping fragments and scanned for point mutations by fluorescent chemical cleavage of mismatch analysis. In step 2, multiplex fluorescent dosage analysis was used to detect large intragenic deletions in the homozygous or heterozygous state. Small mutations were characterized by fluorescent sequencing of amplified products in both forward and reverse directions. Single exons were amplified as described (19, 20).
Deletion Breakpoint Cloning.
The genomic sequence of the introns flanking the major deletion mutation in FANCA was not available. It therefore was obtained by construction of vectorette libraries from P1 clones containing the entire FANCA gene (the kind gift of J. Pronk, Free University of Amsterdam, The Netherlands), using published protocols (21, 22). Intron sequences adjacent to exons 11 and 32 then were amplified from the libraries with the vectorette primer 224 and FANCA-specific primers from exon 11 or exon 32. Vectorette PCR products then were sequenced, and the sequences were used to design new primers to obtain further vectorette products, and thus additional intron 11 and 31 sequences. DNA from patients who were homozygous for the major deletion was amplified with forward and reverse primers from exons 11 and 32, respectively, which generated a breakpoint fragment of 2.2 kb. This fragment then was sequenced from each end, and the data from the vectorette and breakpoint products were assembled into contigs until divergence between control and patient sequences was obtained. The sequence from this region was analyzed by using blast (23).
PCR Assays for Mutation Screening.
The major deletion of exons 12–31 was detected by amplification of patient and parental genomic DNA with PCR primers 5′ and 3′ to the breakpoints. The 5′ primer was placed at IVS11+200 (AAGAAAATTCAGAATTATGAGTGG) and the 3′ primer at IVS31–970 (CACATCATTTTTGCCTCACAAG). A genomic fragment containing exons 12–14 was included as an internal control for a nondeletion allele (CCC ACA ACT TTT TGA TCT CTG and GCTGACAGCAAGGTTGCTCAC). A hot start biplex PCR was performed with these primers in 20-μl reactions at an annealing temperature of 60°C for 35 cycles, using 50–100 ng of genomic DNA. The PCR products from the normal and deleted alleles were 1.3 kb and 715 bp, respectively. The deletion of exons 11–17 was detected in the heterozygous state by fluorescent PCR dosage analysis (19), except that the exon 31 PCR product for multiplex 4 was incorrectly listed as 268 bp; it is in fact 308 bp. Heterozygosity for the 3398delA mutation was detected by heteroduplex analysis, as follows: 50–100 ng genomic DNA was amplified in a 25-μl reaction with the exon 34 primers 34F (CAAAACTGAATCCACAGCAGCC) and 34R (CAGGAAGCTGACAGGAGGATC) for 30 cycles at an annealing temperature of 60°C. The 220-bp product was electrophoresed on a 10% polyacrylamide gel.
Results
Identification of the Major Founder Mutation.
We genotyped 26 FA families from the Afrikaner population with four microsatellite markers in a 2-cM region flanking the FANCA gene and detected five different haplotypes (see Fig. 1), with type I being the most common. These haplotypes were further characterized by the analysis of six SNPs within intron 31 (see below). Patients homozygous for haplotype I then were screened for mutations in FANCA. A single large intragenic deletion was detected, which removed exons 12–31 from the FANCA gene (Fig. 2A, lanes 1, 2, 5, and 6). The breakpoints of the deletion then were characterized in detail by vectorette PCR and sequencing (see Materials and Methods). Sequencing of a 2.2-kb breakpoint PCR fragment and comparison with wild-type sequence (Fig. 2B) revealed divergence ≈570 bp into intron 11. Sequence 3′ to this point in the patient was derived from intron 31. The breakpoint occurs in a region of intron 11 that is almost identical to part of intron 31. A blast search of the breakpoint sequence revealed a high degree of homology with several subfamilies of Alu repeat sequences (underlined in Fig. 2B). Sequencing of this region in several type I homozygotes showed that they had the identical breakpoint, which removes ≈40 kb from the gene. The localization of the type I deletion breakpoints allowed us to design a biplex PCR assay to detect the presence of the type I deletion in the presence of a wild-type allele (Fig. 2C). Sequencing of wild-type and patient DNA also revealed the presence of six SNPs in intron 31: IVS31–259G/T, IVS31–114C/A, IVS31–97T/C, IVS31–57A/C, IVS31–23G/A, and IVS31–4T/C. All patients homozygous for the type I deletion were also homozygous for the T/A/C/C/A/C IVS31 SNP haplotype (see Fig. 1).
Figure 1.

Haplotypes at the FANCA locus in Afrikaner FA patients. TACCAC and GCTAGT represent two SNP haplotypes in intron 31 of the FANCA gene.
Figure 2.

(A) PCR analysis of the type I deletion. Lanes 1–4, biplex PCR of exons 11 and 12; lanes 5–8, biplex PCR of exons 31 and 32. The type I homozygotes in lanes 1, 2, 5, and 6 lack exons 12 and 31; lanes 3, 4, 7, and 8 are normal controls. (B) Sequence analysis of the type I deletion breakpoint. IVS11 and 31 show experimentally determined sequences from introns 11 and 31 in a 5′ to 3′ direction. The sequence determined from the type 1 breakpoint PCR product is shown between the intronic sequences. The dotted box shows homology between IVS11 sequence and the 5′ end of the type 1 sequence. The dashed box shows homology between the type 1 sequence and IVS31. The overlap between these two regions of homology is the likely recombination site. The double-underlined italic bold motif in the type 1 sequence was found to be highly homologous to Alu repeat sequences of various subfamilies. An unidentified nucleotide is designated N in the IVS31 sequence. (C) PCR analysis of the type I deletion. The larger product is a 1.3-kb fragment amplified from exons 12–14 of the wild-type FANCA gene, and the smaller product is the 715-bp fragment from the type I deletion. Lanes 1, 2, 5, and 8, type I heterozygotes; lanes 6 and 7, controls; lanes 3 and 4, type I homozygotes.
Afrikaner FANCA Mutation Profile.
FA patients who were compound heterozygotes for haplotypes I and II were analyzed for the presence or absence of specific exons. These patients lacked exons 12–17, and sequencing of reverse transcription–PCR products in the region showed that exons 11–17 were deleted. The extent of this deletion was confirmed by multiplex fluorescent dosage analysis of genomic DNA (19). The mutation associated with haplotype III was detected by amplification of each of the 43 exons of FANCA from genomic DNA and sequencing. This revealed a deletion of a single nucleotide in exon 34, 3398delA. Chemical cleavage analysis of the single patient who was a compound heterozygote for haplotypes I and IV detected a 14-bp deletion in one allele of exon 9, 795–808del, which was associated with haplotype IV. One patient was a compound heterozygote for haplotypes I and V. This patient lacked exons 12–31 and had two copies of exons 11 and 32, indicating homozygosity for the exon 12–31 deletion. The telomeric end of haplotype V is allele 11 of the marker D16S303, and this patient was homozygous for the six SNPs in intron 31 that are associated with haplotype 1 but are not present in haplotypes II–IV (Fig. 1). This finding suggested that haplotype V originated from haplotype I by recombination between the FANCA gene and the proximal microsatellite marker D16S3121. This was confirmed by PCR analysis from intron 11 to intron 31, which produced the expected 715-bp breakpoint product.
A total of 46 Afrikaner FA patients were screened for the four FANCA mutations described above and for other intragenic deletions using the fluorescent dosage assays. The prevalence of the mutations detected is given in Table 1, which shows that the type I deletion is the major FA mutation in this population, with types I–III accounting for about 80% of all FA alleles. Two additional mutations were detected by the dosage assay. One patient was a compound heterozygote for the type I deletion and a deletion that included exons 10–12. Another patient had the type I deletion in combination with a deletion that included exons 10–17. The endpoints of these deletions were not defined.
Table 1.
Frequency of FANCA gene mutations in Afrikaner FA patients
| Mutation | No. alleles | Frequency, % |
|---|---|---|
| Del E12–31 | 55 | 59.8 |
| Del E11–17 | 12 | 13.0 |
| 3398delA | 6 | 6.5 |
| 795–808del | 1 | 1.1 |
| Del E10–12* | 1 | 1.1 |
| Del E10–17* | 1 | 1.1 |
| Unknown† | 16 | 17.4 |
Deletion endpoints undefined.
No FANCA mutation detected; complementation group unknown.
Molecular Analysis of the Origin of the Founder Mutation.
Because the Afrikaner population was formed by European emigration mainly from The Netherlands, Germany, and France (16), it is likely that the major FANCA mutations originate from one or more of these countries. We therefore screened samples from 52 FA-A patients (26 French, 18 German, and 8 Dutch) from these countries for the four mutations. Only one of these mutations was detected: a single patient from the Western Ruhr region of Germany was a compound heterozygote for the type I deletion of exons 12–31, with a deletion of exons 21–28 on the other FANCA allele. This family then was typed for the microsatellite polymorphisms and the FANCA intron 31 SNPs and was found to have a haplotype identical to that in the Afrikaner patients with the exon 12–31 deletion (Fig. 1). This identity actually extended over the larger interval of D16S305–D16S303, which is a distance of 6.3 cM. We have not detected this haplotype or mutation in extensive studies of other FA patients from Europe, Asia, Africa, and the Middle East (19, 20).
Genealogical Search for a Common Ancestor.
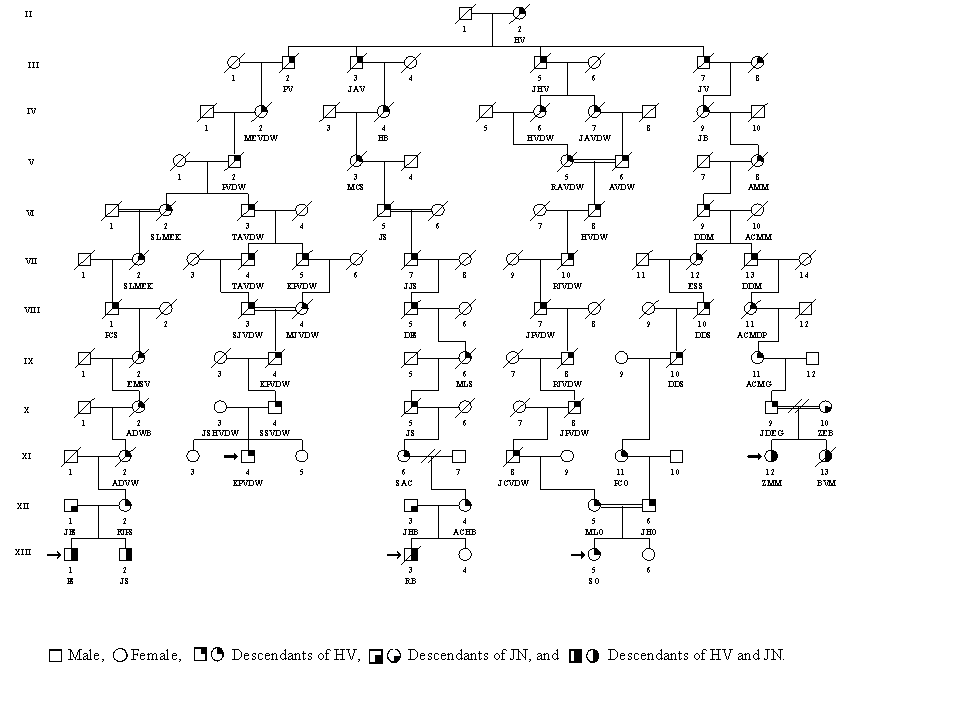
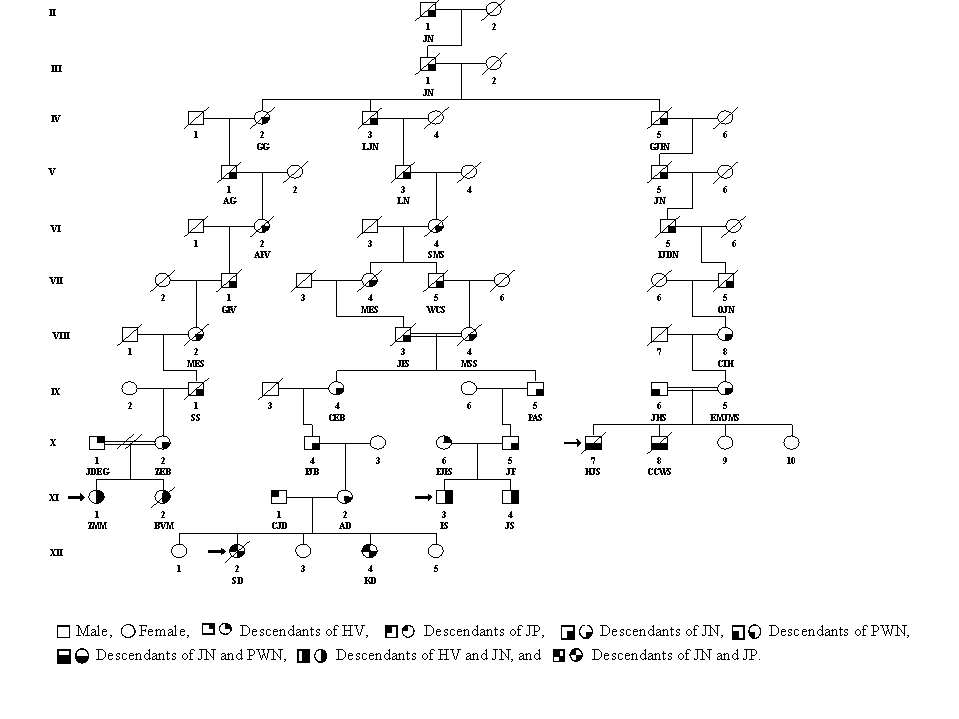
The genealogy of 12 Afrikaner FA families was investigated to determine whether they shared a common ancestor. Initially, several interfamilial relationships were established between pairs of patients. For example (Fig. 3), CCWS (XI-2) and JG (XII-1) shared paternal ancestors MECS (VIII-1) and HAS (VIII-2), three and four generations back, respectively. The genealogical information showed that one particular surname was common in all 12 families. This finding ultimately led to the establishment of linkage between all 12 families. The linkage of seven of the families to one common ancestor is shown in Fig. 3. All 12 families shared common ancestors in the persons of GN and his wife JDLB, a French Huguenot couple who came to the Cape via Amsterdam, arriving on June 5, 1688. They had 10 children, four of whom (JN, JP, PWN, and HV) were the ancestors of 19 of the 24 parents in the 12 FA families (see Table 2). Genealogical information was incomplete for four of the remaining five parents and was unobtainable from one individual who had been adopted. Pedigrees linking the other five families to GN and JDLB can be found in Figs. 4–6, which are published as supplemental material on the PNAS web site, www.pnas.org. DNA was available from 11 of the 12 patients and from 20 of their parents. These samples were tested for the four mutations associated with haplotypes I-IV, and the results are shown in Table 2. The type I mutation was present in at least one chromosome of all 11 patients analyzed, and all 10 of the type I carrier parents whose ancestry could be traced were descended from GN and JDLB.
Figure 3.

Possible segregation of the defective FANCA gene in the descendants of PWN.
Table 2.
Genealogy and mutations in 12 Afrikaner FA families
| Patient | Mutations | Paternal ancestor | Paternal mutation | Maternal ancestor | Maternal mutation |
|---|---|---|---|---|---|
| BS | I/I | nf | I | PWN | I |
| CCWS | I/III | PWN | I | JN | III |
| DMK | I/II | JP | I | PWN | II |
| JG | I/III | PWN | I | nf | III |
| KPVDW | I/II | HV | II | nf | I |
| OS | I/I | nf | I | PWN | I |
| PS | I/II | JN | — | HV | — |
| RB | I/I | PWN | I | HV | I |
| RP | — | nf | — | PWN | — |
| SD | I/IV | JP | IV | JN | I |
| SO | I/III | HV | I | HV | III |
| ZMM | I/III | HV | III | JN | I |
Dash indicates mutation unknown. nf, ancestor not found. I–IV, Afrikaner FANCA mutations associated with haplotypes I–IV.
Discussion
Genotyping of FA families from the Afrikaner population of South Africa with four microsatellite markers flanking the FANCA gene led to the detection of five different haplotypes, which might be associated with five different mutations. Mutation screening showed that these five haplotypes were in fact associated with four mutations. The type I mutation, a deletion of exons 12–31, accounted for about 60% of the mutant FA alleles, and the type II and III mutations for an additional 20%. These data confirm the hypothesis of Rosendorff et al. (12) that the high incidence of FA in this population is a result of a founder effect. The presence of multiple founder mutations in FA is reminiscent of the situation in familial hypercholesterolemia (FH), which is five times more frequent in the Afrikaner population than in the populations of Europe or the United States (24). One mutation in the low density lipoprotein receptor gene accounts for about 67% of FH in the Afrikaner, with another two mutations contributing an additional 10–30% (25, 26). Thus for both FA and FH the disease is caused by multiple founders, but a single mutation provides the major contribution to the high incidence of the disorder in this population. The relatively homogeneous mutation spectrum for FA in the Afrikaner population will greatly simplify the molecular diagnosis of this condition. Also, in view of the high incidence of FA in this population, carrier detection could be offered to couples if one prospective parent had a family history of this devastating disease. The development of a simple PCR assay for the major mutation will allow the carrier frequency to be determined accurately.
The fact that both of the two most common mutations are large intragenic deletions of the FANCA gene is not surprising, given our recent finding of a high frequency of deletions in this gene in European patients (19). In that study we showed a highly significant correlation between the number of deletion breakpoints and the number of Alu repeat sequences in a given intron and suggested that the deletions might be caused by Alu-mediated recombination. Detailed molecular characterization of the type I deletion in this study (Fig. 2B) showed that the breakpoint occurred at two homologous sequences in introns 11 and 31 of the FANCA gene, and that these sequences were themselves highly homologous with several subfamilies of Alu repeats. A similar mechanism has been detected in deletions of exons 16–17 and exons 18–21 of FANCA (27, 28). Indeed, Alu and other repeat-mediated recombinations appear to be a relatively common cause of pathological mutation in humans (29, 30).
The identification of three founder mutations in the Afrikaner population allowed us to address the question of the possible historical and geographic origin of these mutations. The type I mutation was detected in one FA-A patient from the Western Ruhr region of Germany and was associated with exactly the same microsatellite and SNP haplotype as that seen in the Afrikaner patients. Extensive linkage and mutation analysis of FA patients from Europe and elsewhere during the past 5 years (3, 17, 19, 20) has failed to detect any additional examples of this haplotype or mutation. This is not surprising, because a consequence of the extensive heterogeneity of the FANCA mutation spectrum is that most mutations are rare (19, 20, 31). It is therefore possible that the major founder mutation was introduced into South Africa from the Western Ruhr region of Germany. This hypothesis is consistent with the history of the formation of the Afrikaner population, because Rhinelanders were a substantial component of the crew of the ships of the Dutch East India Company, and about one-third of the original European population of the Cape were of German origin (16). However, our genealogical analysis of 12 Afrikaner FA families revealed that at least one parent in all of these families was descended from the French Huguenot couple GN and JDLB, who fled France as a result of the religious persecution that followed the revocation of the Edict of Nantes in 1685. In seven of the 12 families, both parents were descended from this couple, as were all of the traceable carriers of the type I mutation. Because GN was born in Rouen and JDLB in Saumur, the genealogical evidence supports a French origin for the major founder mutation. The fact that this mutation was not detected in 26 French FA-A patients does not exclude a French origin, given the rarity of individual FANCA mutations. The molecular and genealogical findings could be reconciled by the assumption that one of two related Huguenot families who carried this mutation migrated to the Rhineland and that the other went to the Cape. Most merchants and industrialists in the Ruhr region were Calvinists, many of whom were of French Huguenot origin (32). Both scenarios are consistent with our observation that the haplotype in the German patient was conserved for microsatellite markers in a 6-cM region flanking the FANCA gene, because this implies a relatively recent separation of the German and South African versions of this mutation.
In conclusion, this study confirms the original proposal of Rosendorff et al. (12) that the high incidence of FA in the Afrikaner population is the result of a founder effect, but demonstrates that, as in familial hypercholesterolaemia, it is composed of multiple founder mutations. Molecular evidence points to a German origin for the major mutation, but the genealogical data support a French Huguenot origin and are consistent with transmission of the mutation to both Western Germany and the Cape near the end of the 17th century. Accurate molecular diagnosis of FA will now be possible for the majority of cases in this South African population.
Supplementary Material
Acknowledgments
We thank the FA families for their help in providing blood samples and information, the Huguenot Memorial Museum (Franschhoek, South Africa) and The Huguenot Society of Great Britain and Ireland for their assistance, Trefor Jenkins for critical comments on the manuscript, and Elizabeth Manners for its preparation. We thank the Fanconi Anemia Research Fund (U.S.), the European Union Biomed program, and Fanconi Anaemia Breakthrough (U.K.) for their support.
Abbreviations
- FA
Fanconi anemia
- SNP
single nucleotide polymorphism
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Strathdee C A, Duncan A M, Buchwald M. Nat Genet. 1992;1:196–198. doi: 10.1038/ng0692-196. [DOI] [PubMed] [Google Scholar]
- 2.Lo Ten Foe J R, Rooimans M A, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen D F, Savoia A, et al. Nat Genet. 1996;14:320–323. doi: 10.1038/ng1196-320. [DOI] [PubMed] [Google Scholar]
- 3.The Fanconi Anaemia/Breast Cancer Consortium. Nat Genet. 1996;14:324–328. doi: 10.1038/ng1196-324. [DOI] [PubMed] [Google Scholar]
- 4.de Winter J P, Waisfisz Q, Rooimans M A, van Berkel C G M, Bosnoyan-Collins L, Alon N, Carreau M, Bender O, Demuth I, Schindler D, et al. Nat Genet. 1998;20:281–283. doi: 10.1038/3093. [DOI] [PubMed] [Google Scholar]
- 5.de Winter J P, Rooimans M A, van der Weel L, van Berkel C G M, Alon N, Bosnoyan-Collins L, de Groot J, Zhi Y, Waisfisz Q, Pronk J C, et al. Nat Genet. 2000;24:15–16. doi: 10.1038/71626. [DOI] [PubMed] [Google Scholar]
- 6.Swift M. Nature (London) 1971;230:370–373. doi: 10.1038/230370a0. [DOI] [PubMed] [Google Scholar]
- 7.De Winter J P, Léveillé F, van Berkel C G M, Rooimans M A, van der Weel L, Steltenpool J, Demuth I, Morgan N V, Alon N, Bosnoyan-Collins L, et al. Am J Hum Genet. 2000;67:1306–1308. doi: 10.1016/s0002-9297(07)62959-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitney M A, Saito H, Jakobs P M, Gibson R A, Moses R E, Grompe M. Nat Genet. 1993;4:202–205. doi: 10.1038/ng0693-202. [DOI] [PubMed] [Google Scholar]
- 9.Whitney M A, Jakobs P, Kaback M, Moses R E, Grompe M. Hum Mutat. 1994;3:339–341. doi: 10.1002/humu.1380030402. [DOI] [PubMed] [Google Scholar]
- 10.Verlander P C, Kaporis A, Liu Q, Zhang Q, Seligsohn U, Auerbach A D. Blood. 1995;86:4034–4038. [PubMed] [Google Scholar]
- 11.Smith S, Marx M P, Jordaan C J, van Niekerk C H. In: Fanconi Anemia: Clinical, Cytogenetic, and Experimental Aspects. Schroeder-Kurth T M, Auerbach A D, Obe G, editors. Heidelberg: Springer; 1989. pp. 34–46. [Google Scholar]
- 12.Rosendorff J, Bernstein R, Macdougall L, Jenkins T. Am J Med Genet. 1987;27:793–797. doi: 10.1002/ajmg.1320270408. [DOI] [PubMed] [Google Scholar]
- 13.Jenkins T. J Med Genet. 1990;27:760–769. doi: 10.1136/jmg.27.12.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dean G. The Porphyrias: A Story of Inheritance and Environment. 2nd Ed. London: Pitman Medical; 1971. [Google Scholar]
- 15.Meissner P N, Dailey T A, Hift R J, Ziman M, Corrigall A V, Roberts A G, Meissner D M, Kirsch R E, Dailey H A. Nat Genet. 1996;13:95–97. doi: 10.1038/ng0596-95. [DOI] [PubMed] [Google Scholar]
- 16.Nurse G T, Weiner J S, Jenkins T. The Peoples of Southern Africa and their Affinities. Oxford: Clarendon; 1985. pp. 186–217. [Google Scholar]
- 17.Pronk J C, Gibson R A, Savoia A, Wijker M, Morgan N V, Melchionda S, Ford D, Temtamy S, Ortega J J, Jansen S, et al. Nat Genet. 1995;11:338–340. doi: 10.1038/ng1195-338. [DOI] [PubMed] [Google Scholar]
- 18.Auerbach A D, Ghosh R, Pollio P C, Zhang M. In: Fanconi Anemia: Clinical, Cytogenetic, and Experimental Aspects. Schroeder-Kurth T M, Auerbach A D, Obe G, editors. Heidelberg: Springer; 1989. pp. 71–82. [Google Scholar]
- 19.Morgan N V, Tipping A J, Joenje H, Mathew C G. Am J Hum Genet. 1999;65:1330–1341. doi: 10.1086/302627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wijker M, Morgan N V, Herterich S, van Berkel C G M, Tipping A J, Gross H J, Gille J J P, Pals G, Savino M, Altay C, et al. Eur J Hum Genet. 1999;7:52–59. doi: 10.1038/sj.ejhg.5200248. [DOI] [PubMed] [Google Scholar]
- 21.Riley J F, Butler R, Ogilvie D, Finniear R, Jenner D, Powell S, Anand R, Smith J C, Markham A F. Nucleic Acids Res. 1990;18:2887–2890. doi: 10.1093/nar/18.10.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts R G, Coffey A J, Bobrow M, Bentley D R. Genomics. 1992;13:942–950. doi: 10.1016/0888-7543(92)90005-d. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 24.Leitersdorf E, van der Westhuyzen D R, Coetzee G A, Hobbs H H. J Clin Invest. 1989;84:954–961. doi: 10.1172/JCI114258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotze M J, Langenhoven E, Warnich L, du Plessis L, Retief A E. Ann Hum Genet. 1991;55:115–121. doi: 10.1111/j.1469-1809.1991.tb00404.x. [DOI] [PubMed] [Google Scholar]
- 26.Graadt van Roggen F, van der Westhuyzen D R, Marais A D, Gever W, Coetzee G A. Hum Genet. 1999;88:204–208. doi: 10.1007/BF00206073. [DOI] [PubMed] [Google Scholar]
- 27.Levran O, Doggett N A, Auerbach A D. Hum Mutat. 1998;12:145–152. doi: 10.1002/(SICI)1098-1004(1998)12:3<145::AID-HUMU2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 28.Centra M, Memeo E, d'Apolito M, Savino M, Ianzano L, Notarangelo A, Liu J M, Doggett N A, Zelante L, Savoia A. Genomics. 1998;51:467–477. doi: 10.1006/geno.1998.5353. [DOI] [PubMed] [Google Scholar]
- 29.Purandare S M, Patel P I. Genome Res. 1997;7:773–786. doi: 10.1101/gr.7.8.773. [DOI] [PubMed] [Google Scholar]
- 30.Deininger P L, Batzer M A. Mol Genet Metabol. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 31.Levran O, Erlich T, Magdalena N, Gregory J J, Batish S D, Verlander P C, Auerbach A D. Proc Natl Acad Sci USA. 1997;94:13051–13056. doi: 10.1073/pnas.94.24.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagarra E. A Social History of Germany 1648–1914. London: Methuen; 1977. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}