

Abstract
Signaling cascades are managed in time and space by interactions between and among proteins. These interactions are often aided by adaptor proteins, which guide enzyme-substrate pairs into proximity. Miniature proteins are a class of small, well-folded protein domains possessing engineered binding properties. Here we make use of two miniature proteins with complementary binding properties to create a synthetic adaptor protein that effectively redirects a ubiquitous signaling event: tyrosine phosphorylation. We report that miniature-protein-based adaptor 3 uses templated catalysis to redirect the Src family kinase Hck to phosphorylate hDM2, a negative regulator of the p53 tumor suppressor and a poor Hck substrate. Phosphorylation occurs with multiple turnover and at a single site targeted by c-Abl kinase in the cell.
Signaling cascades are managed in time and space by interactions between and among proteins.1 In many cases, the management of these interactions is consigned to adaptor proteins that guide enzyme-substrate pairs into proximity and favor selective reaction.2-5 Here, we describe a strategy that exploits miniature proteins–small, well-folded protein domains possessing engineered binding properties–to create a synthetic adaptor protein that effectively redirects a ubiquitous signaling event: tyrosine phosphorylation. This miniature-protein based adaptor acts as a catalyst to redirect the Src family kinase Hck to phosphorylate hDM2, a negative regulator of the p53 tumor suppressor and a poor Hck substrate. Phosphorylation occurs with multiple substrate turnover and at a single site targeted by c-Abl kinase in the cell.
Our design strategy began with two previously reported miniature proteins, YY27 and 3.3.8 Miniature protein YY2 uses residues within its PPII helix to interact with the SH3 domains of certain Src family kinases, such as Hck (Figure 1A).7 This interaction disrupts the intramolecular SH3 domain interaction that downregulates kinase activity and results in kinase activation. YY2 rivals the HIV-1 protein Nef as an activator of Hck.9,10 Miniature protein 3.3 uses residues within its α-helix to bind to hDM2, thereby inhibiting its association with p53.11 This interaction frees p53 to promote the transcription of p53-dependent genes.12 We envisioned two ways that these functional domains could be combined in a synthetic adaptor; we chose the arrangement that placed YY2 on the N-terminus and 3.3 on the C-terminus to allow optimal access to the helices involved in protein binding (Figure 1A). The two miniature proteins were conjoined by a 4, 8 or 12 amino acid linker to generate adaptors 1, 2 or 3, respectively (Figure 1B). We hypothesized that these adaptor proteins would be capable of forming a ternary complex with hDM2 and Hck to promote the phosphorylation of hDM2. As hDM2 would otherwise be a poor Hck substrate, we refer to this process as ‘templated catalysis’. (Figure 1C).
Figure 1.
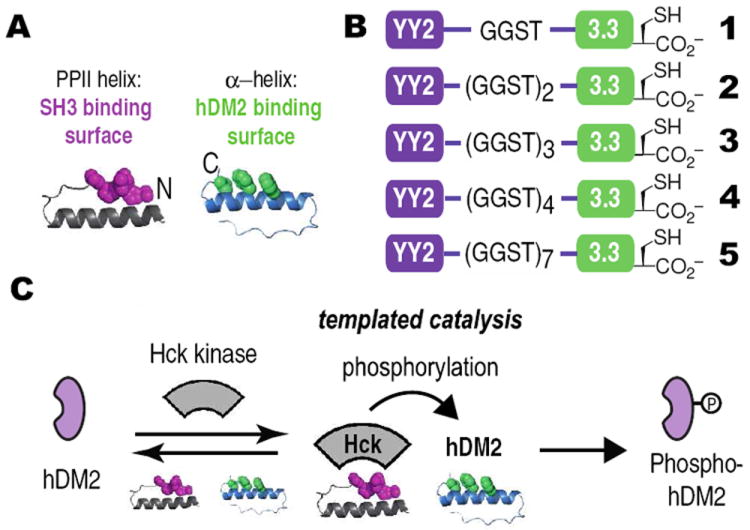
(A) Cartoon illustrating the design of a miniature protein-based adaptor protein. (B) Adaptors 1-5 illustrating differences in inter-domain linkage. (C) Proposed mechanism for templated catalysis of hDM2 phosphorylation by Hck via ternary complex formation.6
We first performed equilibrium fluorescence polarization experiments to characterize the affinity of adaptors 1-3 for hDM2 and Hck. Each adaptor was labeled on a C-terminal cysteine thiol with 5-iodoacetamidofluorescein and titrated with hDM21-188 (hDM2) or GST-Hck-SH3 (Hck-SH3) (Figure 2A,B). We confirmed that none of the miniature protein variants bound appreciably to GST (KD ≥ 18.1 μM). Adaptors 1Flu, 2Flu and 3Flu displayed affinities for hDM2 that are comparable (KD = 278 ± 83.5 nM, 76.4 ± 5.9 nM and 133 ± 8.5 nM, respectively) to that of miniature protein 3.3Flu (35 ± 3.3 nM)8 (Figure 2A). Similiarly, adaptors 1Flu, 2Flu and 3Flu displayed affinities for Hck-SH3 (KD = 3.1 ± 0.5 μM, 7.6 ± 3.0 μM and 5.0 ± 0.3 μM, respectively (Figure 2B)) that are comparable to that of YY2Flu (Kd = 1.4 ± 0.2 μM).7 In contrast, YY2Flu bound hDM2 with low affinity (Kd = 70 ± 5.8 μM) and 3.3Flu bound Hck-SH3 with low affinity (Kd = 85.3 ± 19.4 μM). These comparisons indicate that hDM2 discriminates well between the two miniature protein components (ΔΔG = 4.5 kcal•mol-1), whereas the discrimination by Hck-SH3 is more modest (ΔΔG = 1.0 kcal•mol-1); this difference in specificity influenced our choice of reaction conditions (vide infra). Nevertheless, these in vitro data indicate that incorporation of miniature proteins YY2 and 3.3 into adaptors 1-3 occurs with little or no loss in equilibrium binding affinity or selectivity.
Figure 2.
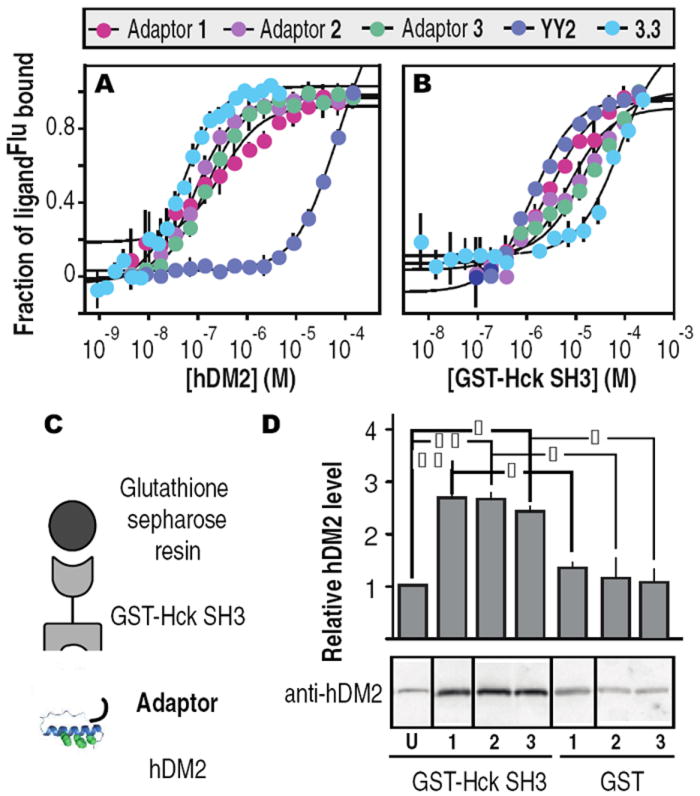
Analysis of the equilibrium affinity of adaptors 1-3 and miniature proteins YY2 and 3.3 for (A) hDM2 and (B) Hck-SH3. (C) Schematic and (D) visualization and quantification of pulldown assay used to analyze ternary complex formation.6 Adaptors 1, 2, and 3 are so indicated (15 μM each); U refers to YY2 and 3.3 (15 μM each). | p ≤ 0.05, | | p ≤ 0.01. ANOVA with Bonferroni post test
Next we performed a pulldown assay to verify formation of a ternary complex between each adaptor and both Hck-SH3 and hDM2 (Figure 2C). Ternary complex formation was performed under conditions of limiting adaptor, with 70 μM Hck-SH3, 157 μM hDM2, and 15 μM adaptor 1Flu, 2Flu, or 3Flu. Bound proteins were eluted with glutathione, analyzed by Western blot and probed with an α-hDM2 antibody. Incubation of immobilized Hck-SH3 with YY2 and 3.3 (15 μM each) retained little or no hDM2 relative to that observed when GST itself (as opposed to GST-Hck-SH3) was immobilized. However, incubation with adaptors 1-3 increased the amount of retained hDM2 by threefold (Figure 2D). These results indicate that all three adaptors can form a ternary complex with Hck-SH3 and hDM2. Although adaptors 1-3 contain different linkers and therefore differ in the relative arrangement of the two component miniature proteins, these differences have no observable effect on the fraction of hDM2 retained. We estimate that under these conditions, only about 8% of the hDM2 present will be assembled into ternary complex with Hck-SH3 and an adaptor;6 incubations at higher [hDM2] were not possible due to limited solubility.
With evidence for a ternary complex in hand, we explored the extent to which adaptors 1-3 would promote the phosphorylation of full length hDM21-491 by full length Hck (Hck). In preliminary experiments, we systematically varied the concentrations of hDM21-491, downregulated10 Hck, and adaptor 2Flu; the relative amounts of phosphorylated hDM21-491 (pY-hDM2) were assessed by Western blot using both α-pY and α-hDM2 antibodies. We observed a bell-shaped dependence on the concentration of adaptor 2 (Figure 3A) whereas no such effect was observed when [Hck] was varied. These experiments revealed that the greatest increase in adaptor 2-promoted hDM2 phosphorylation occurred in reactions containing 22 nM Hck, 5 μM hDM21-491 and 35 nM 2Flu. Under these conditions, the steady state concentration of ternary complex is approximately 140 pM.6 The presence of adaptor 2 results in a twofold increase in pY-hDM2; the increase observed in the presence of adaptor 3 was nearly threefold. By contrast, the increase in pY-hDM2 in the presence of adaptor 1 or miniature protein components 3.3 and YY2 was minimal (Figure 3B). The increase in pY-hDM2 due to adaptors 2 and 3 is not due simply to activation of Hck; incubation of hDM21-491 and Hck with adaptor 1 had no effect on the level of pY-hDM2 (Figure 3B). Moreover, adaptors conjoined by longer linkers (4 and 5) showed no improvement in pY-hDM2 yield. Similar trends in reactivity were observed when the reactions were performed in the presence of bacterial cell lysate.6 These results indicate that adaptors 2 and 3 act as a template to bring Hck and hDM2 into proximity to favor an otherwise unfavorable phosphorylation reaction.
Figure 3.
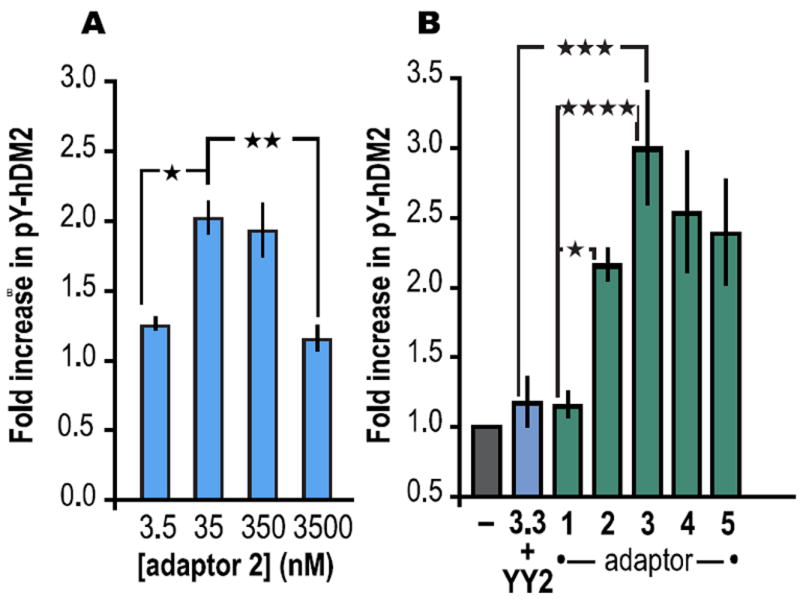
(A) Bell-shaped dependence of the fold increase in phosphorylated hDM2 (pY-hDM2) as a function of [2Flu]. (B) Fold increase in phosphorylated hDM2 (pY-hDM2) produced in vitro in the presence of adaptors 1-5 (35 nM) or miniature proteins YY2 and 3.3 (35 nM each). | p ≤ 0.05, | | p ≤ 0.01, | | | p ≤ 0.001, | | | | p ≤ 0.0001. ANOVA with Bonferroni post-test
The phosphorylation state of hDM2 regulates the activity of the tumor suppressor protein p53.14 In response to DNA damage, hDM2 is phosphorylated at Y276, Y394 and Y405 by c-Abl kinase (Figure 4A).15,16 These post-translational modifications inhibit the association and ubiquitination of p53 by hDM2 and upregulate the transcription of p53-dependent genes. hDM2 contains 14 tyrosine residues, 13 of which are accessible or located in regions of unknown structure. We used PeptideCutter17 to identify trypsin and chymotrypsin as a pair of proteolytic enzymes that would fragment hDM2 to report on the phosphorylation of 12 of these 14 positions (Y68 and Y104 could not be observed). Treatment of hDM21-491 (5 μM) with adaptor 3 (35 nM) and Hck (22 nM) was followed by proteolytic digest with chymotrypsin or trypsin.6 Peptide fragments were resolved and detected using LC/MS/MS and analyzed using BioPharmaLynx software. This analysis revealed phosphorylation at positions Y276 and Y405, but not Y394 nor any other observable tyrosine side chain.6 Thus, adaptor 3 promotes phosphorylation of hDM21-491 at two of the three sites targeted by c-Abl kinase, despite the fact that hDM2 is a poor Hck substrate.18,19 Subsequent analysis of the phosphorylation of Y276F, Y394F and Y405F hDM2 using Western blots identify Y405 as the predominant phosphorylation site (Figure 4B). Notably, the redirected specificity of Hck in the presence of adaptor 3 would not be predicted by the known in vitro preferences of either c-Abl or Src/Hck. These results suggest that other factors contribute to the unique reactivity of these two positions.
Figure 4.
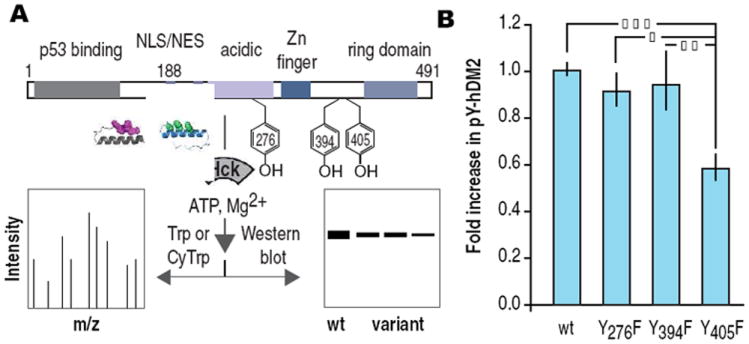
(A) Domain architecture of hDM2 showing location of tyrosine residues phosphorylated by c-Abl. Reaction schematic demonstrating experimental details. (B) Fold increase in pYhDM2 and variants thereof produced in vitro in the presence of adaptor 3 and Hck. | p ≤ 0.05, | | p ≤ 0.01, | | | p ≤ 0.001. ANOVA with Bonferroni post test
Our adaptor proteins were designed to interact simultaneously with both an enzyme (Hck) and a latent substrate (hDM2). As a result, these adaptors have the potential to affect multiple substrate turnovers. To calculate a turnover number,20 we quantified the incorporation of 32P into pY-hDM2 in the presence and absence of adaptor 3 using scintillation counting and storage phosphor autoradiography.6 Incubation of 5 μM hDM2 with 22 nM Hck, 50 μM ATP (spiked with 14-35 pmol γ-32P ATP) and 35 nM adaptor 3Flu yielded 6.7 ± 0.8 pmol pY-hDM2 (11% yield), whereas 1.9 ± 0.4 pmol (3% yield) was produced when adaptor 3 was replaced by YY2 and 3.3 (35 nM each). When calculated on the basis of [Hck]T, the turnover number in the presence of adaptor 3 is 26 (6.7 pmol ÷ 0.26 pmol), which is larger than the value of 7 calculated in the presence of 3.3 and YY2 (1.9 pmol ÷ 0.26 pmol). We note, however, that the true catalytic species of interest in this reaction is not Hck but rather its complex with adaptor 3. When calculated on the basis of [3•Hck] (estimated as 149 pM),6 the turnover number is 3700 (6.7 pmol ÷ 0.0018 pmol). This value is ten times larger than the value of 302 calculated on the basis of [YY2•Hck] (estimated at 524 pM)6 (1.9 pmol ÷ 0.0063 pmol).
In this work we apply classic principles of proximity-induced reaction21-23 to redirect a kinase to an otherwise poor substrate, effectively rewiring a cellular signaling event. In this case, phosphorylation of hDM2 should reactivate p53 and upregulate p53-dependent genes; these studies are in progress. The synthetic adaptor concept could be applied generally as part of an expanding synthetic biology toolkit.24 Miniature proteins are encodable and evolvable;25,26 moreover, the strategy does not require the replacement of endogenous cellular proteins27 and exploits orthogonal domains with less potential for unintended cross-talk.28
Supplementary Material
Acknowledgments
We thank Alexis Rovner for the expression and purification of GST, Olga Fedorova for assistance with experiments employing γ-32P-ATP and Stephen Edwards, Jon Miller, Eugene Douglass and David Spiegel for assistance with calculations. This work was supported by the NIH (GM 65453).
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Materials and experimental methods, Kd values and pulldown data. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Pawson T, Nash P. Genes Dev. 2000;14:1027. [PubMed] [Google Scholar]
- 2.Pawson T, Scott JD. Science. 1997;278:2075. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 3.Burack WR, Shaw AS. Curr Opin Cell Biol. 2000;12:211. doi: 10.1016/s0955-0674(99)00078-2. [DOI] [PubMed] [Google Scholar]
- 4.Bhattacharyya RB, Reményi A, Yeh BJ, Lim WA. Annu Rev Biochem. 2006;75:655. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 5.Good MC, Zalatan JG, Lim WA. Science. 2011;332:680. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Please see supplementary information for details.
- 7.Zellefrow CD, Griffiths JS, Saha S, Hodges AM, Goodman JL, Paulk J, Kritzer JA, Schepartz A. J Am Chem Soc. 2006;128:16506. doi: 10.1021/ja0672977. [DOI] [PubMed] [Google Scholar]
- 8.Kritzer JA, Zutshi R, Cheah M, Ran FA, Webman R, Wongjirad TM, Schepartz A. ChemBioChem. 2006;7:29. doi: 10.1002/cbic.200500324. [DOI] [PubMed] [Google Scholar]
- 9.Briggs SD, Sharkey M, Stevenson M, Smithgall TE. J Biol Chem. 1997;272:17899. doi: 10.1074/jbc.272.29.17899. [DOI] [PubMed] [Google Scholar]
- 10.Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee C-H, Kuriyan J, Miller WT. Nature. 1997;385:650. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- 11.Wade M, Wang YV, Wahl GM. Trends Cell Biol. 2010;20:299. doi: 10.1016/j.tcb.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perkins TJ, Swain PS. Mol Syst Biol. 2009;5:326. doi: 10.1038/msb.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen JT, Porter M, Amoui M, Miller WT, Zuckermann RN, Lim WA. Chem Biol. 2000;7:463. doi: 10.1016/s1074-5521(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 14.Meek DW, Knippeschild U. Mol Cancer Res. 2003;1:1017. [PubMed] [Google Scholar]
- 15.Goldberg Z, Vigt Sionov R, Berger M, Zwang Y, Peters R, Van Etten RA, Oren M, Taya Y, Haupt Y. EMBO J. 2002;21:3715. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias SS, Milne DM, Meek DW. Oncogene. 2006;25:6666. doi: 10.1038/sj.onc.1209671. [DOI] [PubMed] [Google Scholar]
- 17.Gasteiger EHC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. In: The Proteomics Protocols Handbook. Walker JM, editor. Humana Press; Totowa, NJ: 2005. [Google Scholar]
- 18.Luo W, Slebos RJ, Hill S, Li M, Brábek J, Amanchy R, Chaekady R, Pandey A, Han A-JL, Hanks SK. J Prot Res. 2008;7:3447. doi: 10.1021/pr800187n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeda H, Kawamura Y, Miura A, Mori M, Wakamatsu A, Yamamoto J-I, Isogai T, Matsumoto M, Nakayama KI, Natsume T, Nomura N, Goshima N. J Prot Res. 2010;9:5982. doi: 10.1021/pr100773t. [DOI] [PubMed] [Google Scholar]
- 20.Jacobsen EN, Finney NS. Chem Biol. 1994;1:85. doi: 10.1016/1074-5521(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 21.Corey DR, Schultz PG. Science. 1987;238:1401. doi: 10.1126/science.3685986. [DOI] [PubMed] [Google Scholar]
- 22.Gartner ZJ, Liu DR. J Am Chem Soc. 2001;123:6961. doi: 10.1021/ja015873n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis CA, Miller SJ. Angew Chem Intl Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 24.Lim WA. Nat Rev Mol Cell Biol. 2010;11:393. doi: 10.1038/nrm2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chin JW, Schepartz A. Angew Chem Intl Ed. 2001;40:3806. [PubMed] [Google Scholar]
- 26.Chin JW, Schepartz A. J Am Chem Soc. 2001;123:2929. doi: 10.1021/ja0056668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tatebayashi K, Takekawa M, Saito H. EMBO J. 2003;22:3624. doi: 10.1093/emboj/cdg353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howard PL, Chia MC, Del Rizzo S, Liu F-F, Pawson T. Proc Natl Acad Sci USA. 2003;100:11267. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.