

Understanding the intimate details of O2 activation at metal sites is of interest because of the relevance of such reactions in biological and technological processes.[1] Of particular relevance is uncovering basic chemical principles and mechanisms for taming the high oxidizing potential of the O2 molecule into highly selective oxidative transformations, especially those involving the selective hydroxylation of C–H bonds.
For the particular case of dicopper sites, three basic Cu2O2 core structures have been widely described as arising from the interaction of discrete CuI complexes and O2 (Figure 1).[2] Each specific Cu2O2 core determines particular spectroscopic and chemical properties:[2b,c] whereas end-on trans-CuII2-(μ-η1:η1-O2) species exhibit nucleophilic and basic behavior, side-on CuII2(μ-η2:η2-O2) and bis-μ-oxido dicopper(III) (CuIII2(μ-O)2) cores show electrophilic character, and can mediate tyrosinase-like phenolate ortho-hydroxylation reactions.[3] The latter reactivity has never been observed for end-on Cu2O2 species; therefore its possible biological relevance has been ignored so far.
Figure 1.
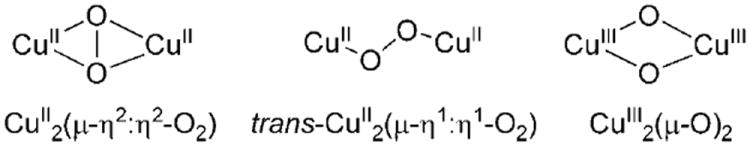
The rich and subtle chemistry exhibited by Cu2O2 cores makes unsymmetric options interesting. Actually, Cu2O2 species in systems containing distinct copper sites have been rarely observed.[2b] Herein we describe a novel dicopper complex based on a heptadentate ligand that gives rise to an unsymmetric N3CuIIN4CuII(μ-η1:η1-O2) core, which hitherto exhibits reactivity patterns not observed for symmetric analogues. This nonsymmetric peroxide species shows an exquisite selectivity in its oxygen atom transfer reactivity. It performs the selective intermolecular ortho hydroxylation of a phenolate, but fails to oxidize many common oxophilic substrates.
Reaction of m-XylN3N4 with [CuI(CH3CN)4X] (X = CF3SO3, PF6, ClO4) in acetonitrile affords the unsymmetric dinuclear copper(I) complex [CuI2(m-XylN3N4)](X)2 (1-X; Figure 2). For comparative purposes, [CuI2(m-XylN4N4)]- (ClO4)2 (2-ClO4) was also prepared. Crystallographic characterization of 1-CF3SO3 reveals that the copper ion bound to the tridentate arm adopts a highly distorted T-shape geometry, whereas the copper ion bound to the tetradentate arm has a distorted trigonal-pyramidal geometry, with structural parameters nearly superimposable with those of the two tetracoordinated copper ions in 2-ClO4.[4]
Figure 2.
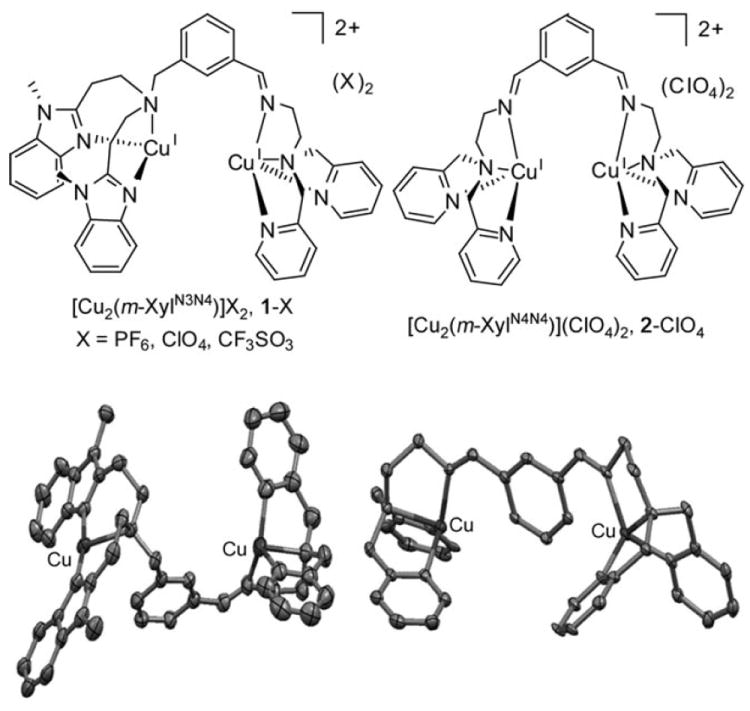
Top: Chemical diagram of 1-X (left) and 2-ClO4 (right). Bottom: Ellipsoid diagrams (30 % probability) of the cationic parts of 1-CF3SO3 (left) and 2-ClO4 (right). H atoms were omitted for clarity.
Acetone solutions of 1-X at −90°C react with O2 within seconds to form a red-brown species [Cu2(O2)(m-XylN3N4)]2+ (1-O2), characterized by a visible band at 478 nm (ε = 7800m−1 cm−1), and a broad shoulder between 575 and 700 nm (Figure 3). The visible spectrum of 1-O2 is intermediate between those of Itoh’s proposed CuII2(μ-η1:η2-O2) species[5] and reported CuII2(μ-η1:η1-O2) complexes.[2b,6] UV/Vis monitoring of this reaction shows an isosbestic point at 414 nm indicating the clean transformation of 1-CF3SO3 into 1-O2 without accumulation of any intermediate species. 1-O2 is stable for hours at −90°C, but it decomposes within seconds when warmed to room temperature.
Figure 3.

Top: UV/Vis spectra for the reaction of 1-CF3SO3 with O2 in acetone at −90°C to form 1-O2. Bottom: Resonance Raman spectra (λex = 488 nm) of frozen acetone solutions of 1-O2 from 16O2 (A), 18O2 (B), and 18O16O (C).
To gain insight into the peroxido binding mode of 1-O2, resonance Raman spectra of frozen samples of 1-O2 were obtained. Laser excitation at 488 nm (Figure 3 bottom, insets A and B) gives rise to two resonance-enhanced peaks at 832 and 520 cm−1 [Δ18O2-16O2 = 45 and 22 cm−1, respectively], characteristic of O–O and Cu–O stretching vibrations of an end-on CuII2(μ-η1:η1-O2) species.[2b] Excitation profiles indicate that the two vibrations are in resonance with the lower energy band, and no features resulting from a CuII2(μ-η2:η2-O2) species were observed. Experiments with mixed labeled O2 (Figure 3 bottom, inset C) showed a ν(O–O) region with three isotopomeric peaks at frequencies that suggest insensitivity of the bound O2 to the unsymmetrical nature of the ligand.[7]
In contrast, symmetric complex 2-ClO4 reacts at −90°C in acetone to form a different metastable purple species, 2-O2, which is characterized by two intense UV/Vis bands at λmax = 500 nm (ε = 5000m−1 cm−1) and 635 nm (ε = 3300m−1 cm−1), typical of an end-on trans-CuII2(μ-η1:η1:O2) species.[2b] The O2-binding mode was confirmed by the resonance Raman spectra of a frozen 2-O2 solution, collected with laser excitation at 488 nm (see the Supporting Information), which shows a characteristic resonance-enhanced ν(O–O) band at 826 cm−1[Δ18O2-16O2=44 cm−1]. The fact that both 1-O2 and 2-O2 give rise to ν(O–O) features of nearly the same frequency strongly suggests that the dioxygen moiety is bound in the same fashion in the two complexes. Because of the high energy of the ν(O–O) stretching frequency,[2b] and because a μ-η1:η2-O2 binding mode is unlikely for 2-O2,[8] we favor a μ-η1:η1-O2 mode for both O2 adducts.
The reactivities of 1-O2 and 2-O2 with different substrates were explored (Schemes 1 and 2). 1-O2 and 2-O2 rapidly and quantitatively react with CF3CO2H releasing H2O2 (99% yield, see the Supporting Information). Titration experiments reveal that both reactions are complete with 1 equivalent of H+ (per Cu), and no other intermediate species are detected when substoichiometric amounts of H+ are added. 1-O2 and 2-O2 react neither with thioanisole, styrene, triphenylmethane, nor with electron donors such as ferrocene. Thermal decomposition of 1-O2, by warming up acetone solutions to room temperature, in the presence of large excess of toluene (1000 equiv), did not cause toluene oxidation.[9] The addition of PPh3 (10 equiv) to 1-O2 and 2-O2 at −90°C induces fast O2 release (t1/2 ≈ 5 min) without formation of OPPh3.[10] In sum, all the above observations suggest that 1-O2 and 2-O2 are not electrophilic oxidants, and typically react like other end-on trans-CuII2(μ-η1:η1-O2) complexes.[11]
Scheme 1.
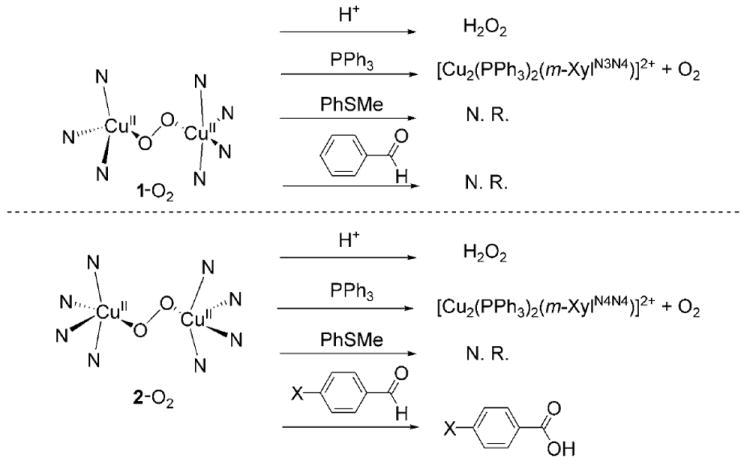
Schematic representation of selected reactivity exhibited by 1-O2 and 2-O2. (N.R. = no reaction).
Scheme 2.

Reaction of 1-O2 with the sodium salt of para-substituted phenolate.
However, substantial differences arise when the reactions of 1-O2 and 2-O2 with benzaldehydes are studied. 2-O2 reacts with benzaldehydes to generate the corresponding benzoic acids in quantitative yields. Kinetic analyses of the reactions were performed by using UV/Vis methods to monitor the decay of the spectral features of 2-O2. The 2-O2 decay rate can be fitted to single exponential processes, and the measured kobs values are linearly dependent on substrate concentration (see the Supporting Information). Reaction of 2-O2 against a series of para-substituted benzaldehydes was studied and the corresponding decay rate constants were extracted by using UV/Vis methods to monitor the reactions. Plotting the decay rate of 2-O2 against the corresponding Hammett substituent constants (σ+) affords a linear correlation which gives ρ value of 1.4 (R2 = 0.98, see the Supporting Information), consistent with a nucleophilic oxidizing species that attacks the carbonyl moiety. In contrast, 1-O2 fails to react with benzaldehyde. Thus we conclude that 1-O2 is unreactive in oxygen atom transfer reactions to common substrates which are either electrophilic or nucleophilic in nature.
Strikingly, addition of p-Cl-C6H4ONa (3 equiv, Scheme 2) to a solution of 1-O2 at −90°C causes rapid conversion into a short-lived (t1/2 ≈ 1 min) yellow-brown species 3Cl (λmax = 470 nm, ε > 6000m−1 cm−1, see the Supporting Information). The resemblance in the UV/Vis spectral features of 1-O2 and 3Cl strongly suggests that the CuII2(μ-η1:η1-O2) core is retained, but the instability of 3Cl has thus far precluded its Raman characterization. Surprisingly, after complete decomposition of the 3Cl species, acidic work-up and subsequent HPLC/MS analyses show the formation of p-chlorocatechol in 39 % yield with respect to 1-O2. Similar addition of p-chlorophenolate to 2-O2 causes fast bleaching of its spectral features, without accumulation of any intermediate species, and without any sign of phenolate ortho hydroxylation.
Kinetic analysis indicates that the decay of 3Cl is a first-order process. The analogous species 3X (X = F, Me, H, and OMe) were generated by the addition of 3 equivalents of p-X-C6H4ONa to 1-O2 at −90°C in acetone, and their corresponding UV/Vis decay rates were fitted to a single exponential function by nonlinear regression methods. Product analysis after 3X (X = F, Me, H, and OMe) decomposition reveals that the corresponding catechol is formed in 34 %, 34 %, 36 %, and 14% yields, respectively. A Hammett plot (log (kobs) for 3X versus σ+) affords a linear correlation which gives a ρ value of −0.6 (R2 = 0.98, see the Supporting Information), consistent with an electrophilic oxidizing species that attacks the aromatic ring in the rate-determining step of the reactions. In line with this reactivity, no catechol product was formed when electron-poor phenolates (X = CN, NO2, and CO2Me) were used as substrates. In contrast, substrate hydroxylation does not appear to be only determined by the electron-releasing nature of the substrate because the electron-rich, sterically more demanding 2,4-di-tert-butyl-catecholate was neither hydroxylated nor oxidized to the corresponding diphenol coupled product. We conclude that hydroxylation occurs exclusively for non-electron-poor, sterically unhindered phenolate substrates. Furthermore 1-O2 differs from any other CuII2(μ-η1:η1-O2) intermediate in its capacity to carry out electrophilic arene hydroxylation.[2c] We propose that the difference in the reactivity of 1-O2 and any previously reported end-on CuII2(μ-η1:η1-O2) species (including 2-O2) stems from the possibility that phenolate can initially bind at the N3Cu site, as proposed in a symmetric m-xylyl-bridged bis-tridentate CuII2(μ-η2:η2-O2) system.[3b] Indeed, in the present example, the substrate binding event can be understood as playing a selective peroxide-activation role, since 1-O2 by itself lacks oxygen atom transfer reactivity.
The selective oxygen atom transfer reactivity exhibited by 1-O2 was additionally substantiated by DFT computational methods.[12] The computed structure of 1-O2 (see the Supporting Information) reveals a CuII2(μ-η1:η1-O2) complex with a Cu⋯Cu distance of 4.31 Å and structural parameters in good agreement with a crystallographically characterized example.[13] We have also found that the CuIII2(μ-O)2 isomer is 36.8 kcal mol−1 higher in energy.[12] In addition, attempts to perform geometry optimizations on side-on CuII2(μ-η2:η2-O2) isomeric cores proved unsuccessful.[14] Therefore, consistent with the experimental observations, the end-on CuII2(μ-η1:η1-O2) is the most stable species.[14-16] Phenolate binding to 1-O2 retains the CuII2(μ-η1:η1-O2) core as the most stable isomer (in agreement with our formulation of 3X based on its UV/Vis spectrum), and causes an elongation of the Cu⋯Cu distance up to 4.50 Å. Interestingly, the phenolate π system is adjacent to the peroxide oxygen atom bound to the other Cu in 3Me (Figure 4), offering a plausible pathway for a σ* electrophilic attack of the peroxide moiety on the aromatic ring.[17] Most remarkably, the computed activation barrier for this reaction is only 14.7 kcalmol−1, and no intermediates regarding the isomerization to side-on CuII2(μ-η2:η2-O2) or CuIII2(μ-O)2 cores are found along this attack, thereby strongly suggesting that the trans end-on peroxido core is a competent species for executing the aromatic C–H hydroxylation event.[12]
Figure 4.
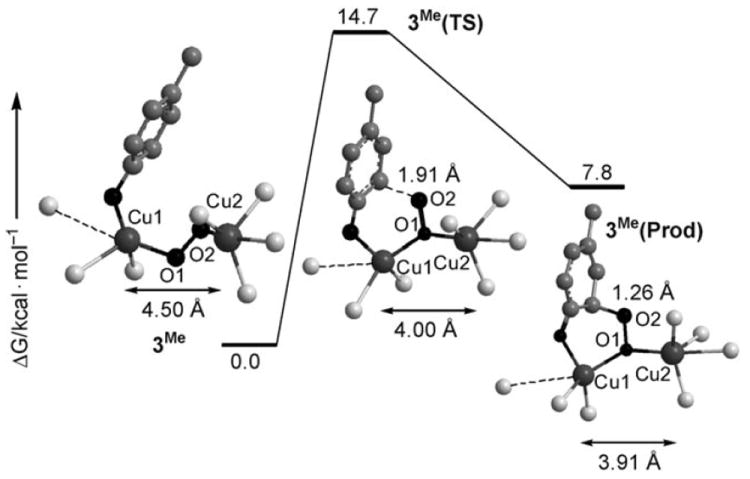
Stationary points along the reaction pathway of the ortho hydroxylation of para-methyl phenolate into 4-methyl catechol from 3Me.
In conclusion, our study of O2 activation at a novel asymmetric dicopper complex 1-O2 has hitherto uncovered reactivity patterns thus far not observed for symmetric analogues. 1-O2 is basically unreactive in oxygen atom transfer reactions. However, it has an available coordination site that selectively binds phenolate and mediates its ortho hydroxylation, therefore functionally mimicking tyrosinase through a unique pathway. Furthermore, coordination of the phenolate substrate turns on the unprecedented electrophilic reactivity of the asymmetric end-on trans-peroxido core. The combined experimental and computational evidence indicate that the ortho hydroxylation of a phenolate by a Cu2O2 species can occur by adjacent binding of phenolate and O2 at a common N3Cu site without requiring the peroxido to be side-on bound, thus offering a conceptually new understanding of O2 activation at dicopper sites.
Experimental Section
See the Supporting Information for the full experimental details for the synthesis, spectroscopic, and crystallographic characterization of 1-X (X = CF3SO3, PF6, ClO4) and 2-ClO4, the experimental procedures for the generation, characterization, and reactivity studies of 1-O2 and 2-O2, and the computational details on the DFT calculations.
Supplementary Material
Footnotes
This work was supported financially by MCYT of Spain (projects CTQ2006-05367/BQU and CTQ2009-08464/BQU to M.C.), by the U.S. NIH (grant GM38767 to L.Q.), and by the Italian MIUR (Prin project to L.L.). I.G.B and A.C. thank MICINN for PhD grants. M.T.-S. thanks the CSIC for the JAE-DOC contract. We thank STR-UdG for technical support. M.C. also thanks the Generalitat Catalunya for an ICREA-Academia award and for project 2009 SGR-637.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.200906749.
Contributor Information
Isaac Garcia-Bosch, Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Catalonia (Spain).
Dr. Anna Company, Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Catalonia (Spain).
Jonathan R. Frisch, Department of Chemistry and Center for Metals in Biocatalysis, University of Minnesota, 207 Pleasant Street SE, Minneapolis, MN 55545 (USA)
Dr. Miquel Torrent-Sucarrat, Institut de Química Avançada de Catalunya, IQAC–CSIC, Catalonia (Spain)
Mar Cardellach, Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Catalonia (Spain).
Dr. Ilaria Gamba, Department of General Chemistry, University of Pavia, 27100 Pavia (Italy)
Dr. Mireia Güell, Institut de Química Computacional, Universitat de Girona, Catalonia (Spain)
Prof. Luigi Casella, Email: bioinorg@unipv.it, Department of General Chemistry, University of Pavia, 27100 Pavia (Italy).
Prof. Lawrence Que, Jr., Email: larryque@umn.edu, Department of Chemistry and Center for Metals in Biocatalysis, University of Minnesota, 207 Pleasant Street SE, Minneapolis, MN 55545 (USA).
Dr. Xavi Ribas, Email: xavi.ribas@udg.edu, Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Catalonia (Spain).
Dr. Josep M. Luis, Email: josepm.luis@udg.edu, Institut de Química Computacional, Universitat de Girona, Catalonia (Spain).
Dr. Miquel Costas, Email: miquel.costas@udg.edu, Departament de Química, Universitat de Girona, Campus de Montilivi, 17071 Girona, Catalonia (Spain).
References
- 1.For an special issue on dioxygen activation by metalloenzymes and models, see Nam W. Acc Chem Res. 2007;40(7) doi: 10.1021/ar700027f.; Punniyamurthy T, Velusamy S, Iqbal J. Chem Rev. 2005;105:2329–2364. doi: 10.1021/cr050523v.; Sawyer DT. Oxygen Chemistry. Oxford University Press; Oxford: 1991. .
- 2.a) Itoh S. In: Comprehensive Coordination Chemistry II. Que L Jr, Tolman WB, editors. Vol. 8. Elsevier; Amsterdam: 2004. pp. 369–393. [Google Scholar]; b) Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; c) Lewis EA, Tolman WB. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; d) Hatcher LQ, Karlin KD. J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]; e) Battaini G, Granata A, Monzani E, Gullotti M, Casella L. Adv Inorg Chem. 2006;58:185–233. [Google Scholar]
- 3.a) Itoh S, Kumei H, Taki M, Nagatomo S, Kitagawa T, Fukuzumi S. J Am Chem Soc. 2001;123:6708–6709. doi: 10.1021/ja015702i. [DOI] [PubMed] [Google Scholar]; b) Palavicini S, Granata A, Monzani E, Casella L. J Am Chem Soc. 2005;127:18031–18036. doi: 10.1021/ja0544298. [DOI] [PubMed] [Google Scholar]; c) Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]; d) Company A, Palavicini S, Garcia-Bosch I, Mas-Ballesté R, Que L, Jr, Rybak-Akimova EV, Casella L, Ribas X, Costas M. Chem Eur J. 2008;14:3535–3538. doi: 10.1002/chem.200800229. [DOI] [PubMed] [Google Scholar]; e) Herres-Pawlis S, Verma P, Haase R, Kang P, Lyons CT, Wasinger EC, Flörke U, Henkel G, Stack TDP. J Am Chem Soc. 2009;131:1154–1169. doi: 10.1021/ja807809x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CCDC 755536 (1CF3SO3) and 755537 (2ClO4) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 5.Tachi Y, Aita K, Teramae S, Tani F, Naruta Y, Fukuzumi S, Itoh S. Inorg Chem. 2004;43:4558–4560. doi: 10.1021/ic049524g. [DOI] [PubMed] [Google Scholar]
- 6.Börzel H, Comba P, Hagen KS, Kerscher M, Pritzkow H, Schatz M, Schindler S, Walter O. Inorg Chem. 2002;41:5440–5452. doi: 10.1021/ic011114u. [DOI] [PubMed] [Google Scholar]
- 7.The CuO2 local symmetry cannot necessarily be established by isotopomeric O2 fragments. Kinsinger CR, Gherman BF, Gagliardi L, Cramer CJ. J Biol Inorg Chem. 2005;10:778–789. doi: 10.1007/s00775-005-0026-0..
- 8.Bis-tetradentate ligands most often give rise to μ-η1-η1 isomers: see [2b] and references within.
- 9.Aliphatic C–H oxidation of toluene mediated by CuII2(μ-η1:η1-O2) species has been recently reported: Lucas HR, Li L, Narducci Sarjeant AA, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2009;131:3230–3245. doi: 10.1021/ja807081d.; Würtele C, Sander O, Lutz V, Waitz T, Tuczek F, Schindler S. J Am Chem Soc. 2009;131:7544–7545. doi: 10.1021/ja902327s..
- 10.1H and 31P NMR spectra of the final mixture are identical to those observed after mixing 1ClO4 and 10 equivalents PPh3 in CD3CN under argon. See the Supporting Information for details.
- 11.Paul PP, Tyeklar Z, Jacobson RR, Karlin KD. J Am Chem Soc. 1991;113:5322–5332. [Google Scholar]
- 12.DFT geometries were optimized at the B3LYP level in junction of the SDD basis set and associated ECP for Cu, 6-311G(d) basis set for the atoms bond to Cu, and 6-31G basis set for the other atoms, as implemented in the Gaussian 03 program (see the Supporting Information). The energies were additionally refined by single-point calculations using cc-pVTZ basis set for Cu and the atoms bond to Cu, and cc-pVDZ basis set for the other atoms. Final free energies given in this work include energies computed at the B3LYP/cc-pVTZ&cc-pVDZ//B3LYP/SDD&6-311G(d)&6-31G level of theory together with zero-point energies, thermal corrections, and entropy calculated at the B3LYP/SDD&6-311G(d)&6-31G level. The same qualitative results were also obtained with the BLYP and OPBE methods.
- 13.Tyeklár Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD. J Am Chem Soc. 1993;115:2677–2689. [Google Scholar]
- 14.CuII2(μ-η1:η1-O2) to CuII2(μ-η2:η2-O2)isomerization is precedented; Halfen JA, Young VG, Jr, Tolman WB. J Am Chem Soc. 1996;118:10920–10921.; Jung B, Karlin KD, Zuberbühler AD. J Am Chem Soc. 1996;118:3763–3764..
- 15.For recent computational studies evaluating CuII2(μ-η1:η2-O2)species as arene hydroxylating species, see: Siegbahn PEM. J Biol Inorg Chem. 2003;8:567–576. doi: 10.1007/s00775-003-0449-4.; Sander O, Henß A, Näther C, Würtele CC, Holthausen MC, Schindler S, Tuczek F. Chem Eur J. 2008;14:9714–9729. doi: 10.1002/chem.200800799.; Inoue T, Shiota Y, Yoshizawa K. J Am Chem Soc. 2008;130:16890–16897. doi: 10.1021/ja802618s..
- 16.Because of the energy proximity of the asymmetric CuII2(μ-η1:η2-O2) species (+ 1 kcal mol−1), we have also considered its ability to bind phenolate, and found that upon phenolate coordination the species also converts into a CuII2(μ-η1:η1-O2) core.
- 17.Decker H, Dillinger R, Tuczek F. Angew Chem. 2000;112:1656–1660. doi: 10.1002/(sici)1521-3773(20000502)39:9<1591::aid-anie1591>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2000;39:1591–1595. doi: 10.1002/(sici)1521-3773(20000502)39:9<1591::aid-anie1591>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.