

Abstract
ATP-binding cassette transporter A1 (ABCA1) is a cholesterol transporter that transfers excess cellular cholesterol onto lipid-poor apolipoproteins. Given its critical role in cholesterol homeostasis, ABCA1 has been studied as a therapeutic target for Alzheimer’s disease. Transcriptional regulation of ABCA1 by liver X receptor has been well characterized. However, whether ABCA1 expression is regulated at the posttranscriptional level is largely unknown. Identification of a novel pathway that regulates ABCA1 expression may provide new strategy for regulating cholesterol metabolism and amyloid β (Aβ) levels. Since ABCA1 has an unusually long 3′ untranslated region, we investigated whether microRNAs could regulate ABCA1 expression. We identified miR-106b as a novel regulator of ABCA1 expression and Aβ metabolism. miR-106b significantly decreased ABCA1 levels and impaired cellular cholesterol efflux in neuronal cells. Furthermore, miR-106b dramatically increased levels of secreted Aβ by increasing Aβ production and preventing Aβ clearance. Alterations in Aβ production and clearance were rescued by expression of miR-106b-resistant ABCA1. Taken together, our data suggest that miR-106b affects Aβ metabolism by suppressing ABCA1 expression.
Keywords: microRNA, miR-106b, ABCA1, Cholesterol, Amyloid β, Aβ, Alzheimer’s disease, Liver X receptor, LXR, Lipid
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. Abnormal accumulation of amyloid β (Aβ) peptide in the brain is associated with pathologic cascades that eventually lead to AD (Hardy and Selkoe, 2002). Aβ peptides are generated by sequential cleavages of amyloid precursor protein (APP) by β-site APP-cleaving enzyme1 (BACE1) and γ-secretases. Mutations in presenilin (PSEN), the major component of γ-secretase, are responsible for the majority of early-onset autosomal dominant familial AD. Given the critical role of Aβ in AD pathogenesis, strategies to modulate Aβ production and clearance are actively being pursued as AD therapies.
ATP-binding cassette transporter A1 (ABCA1), a member of the ABC transmembrane transporter family, plays an important role in cholesterol transport by transferring cholesterol and phospholipid onto lipid-poor apolipoprotein A1 (apoA-I) and apolipoprotein E (apoE) to form high-density lipoprotein (HDL) particles (Hirsch-Reinshagen et al., 2009). Several key proteins implicated in AD pathogenesis, such as APP, BACE1, and PSEN1, are membrane proteins and their trafficking and proteolytic activity are altered by cholesterol dysregulation. Due to its critical role in maintaining cholesterol homeostasis, ABCA1 has been considered as a promising therapeutic target for AD (Kim et al., 2009).
Several lines of data strongly support that ABCA1 has a critical role in Aβ metabolism and accumulation. ABCA1 induction by liver X receptor (LXR) agonist decreased Aβ production in various cell lines (Kim et al., 2007; Koldamova et al., 2003; Sun et al., 2003). Genetic studies with AD mouse models have demonstrated that the deletion of ABCA1 increases Aβ deposition, while overexpression of ABCA1 dramatically reduces Aβ deposition (Hirsch-Reinshagen et al., 2004; Koldamova et al., 2005; Wahrle et al., 2005; Wahrle et al., 2008; Wahrle et al., 2004). Recently, Donkin et al. also indicated that ABCA1 is required for the beneficial effect of LXR agonist in AD mouse model (Donkin et al., 2010). However, the exact mechanisms underlying the regulation of Aβ metabolism by ABCA1 have not been clearly described. Nonetheless, identification of novel pathway that regulates ABCA1 expression may represent a new AD treatment strategy.
MicroRNAs (miRNAs) are small non-coding RNA molecules comprised of 21–23 nucleotides, regulate expression of protein-coding genes. By binding to their target messenger RNAs (mRNAs), miRNAs repress translation or degrade target mRNAs (Huntzinger and Izaurralde, 2011). Strong alterations in miRNA levels are often observed under disease conditions and the functional effects of miRNAs on target genes become much more evident. miRNAs have been shown to regulate various biological processes, such as oncogenesis, development, and brain functions, including neuronal differentiation, synaptic plasticity, and memory formation (Cohen et al., 2011; Kosik and Krichevsky, 2005; Smalheiser and Lugli, 2009). In particular, previous studies demonstrated that miRNAs contribute to neurodegenerative diseases (Hebert and De Strooper, 2009; Nelson et al., 2008). For example, several miRNAs have been identified to regulate APP (Hebert et al., 2009; Liu et al., 2010; Patel et al., 2008; Vilardo et al., 2010) and BACE1 (Boissonneault et al., 2009; Hebert et al., 2008; Wang et al., 2008). In this study, we identified that miR-106b functions as a novel regulator of Aβ production and clearance through the suppression of ABCA1 expression.
Materials and Methods
Cell culture
Mouse neuroblastoma Neuro2a cells and human hepatocyte HepG2 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10 % fetal bovine serum (FBS) and 1 % penicillin/streptomycin at 37 °C in a humidified 5% CO2 incubator. All transient transfections were performed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s guide. Synthetic miR-106b and scrambled negative control were from Insight Genomics and miRCURY LNA™ miR-106b inhibitor and negative control were from Exiqon. pcDNA3.1-mouse ABCA1 open reading frame (ORF) without 3′ untranslated region (UTR) and pAG3-Swedish mutant (K670N/M671L) APPsw ORF without 3′ UTR were kind gifts from Dr. David M. Holtzman (Washington University, St. Louis, USA) and Dr. Todd Golde (University of Florida, Gainesville, USA), respectively.
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNAs were extracted from Neuro2a cells using TRIzol® Reagent (Invitrogen) and reverse transcribed with Mir-X™ miRNA First-Strand Synthesis Kit (Clontech, Mountain View, CA) following the manufacturer’s guide. Quantitative PCR was performed with ABI 7500 (Applied Biosystems, Carlsbad, CA) using TaqMan Universal PCR Master Mix with default thermal cycling program. TaqMan assays for mouse ABCA1 and GAPDH endogenous control (Applied Biosystems) were used. Endogenous GAPDH was used as a normalization control. Relative mRNA levels were calculated by comparative Ct method using ABI 7500 software (version 2.0.5).
Luciferase reporter assay
cDNA fragments corresponding to the entire 3′ UTR of human Abca1 were amplified by RT-PCR from total RNA extracted from HepG2 cells. The PCR products were directionally cloned downstream of the Renilla luciferase open reading frame in the psiCHECK™-2 vector (Promega, Madison, WI). This vector also contains a constitutively expressed firefly luciferase gene that was used to normalize results against efficiency of transfection (Rayner et al., 2010). Neuro2a cells were plated at a density of 4 × 104 cells per well in a 96-well plate a day before transfection. 0.12 μg psiCHECK™-2-human ABCA1 3′ UTR luciferase reporter vector were transfected. After 8hr, media were changed to fresh DMEM/10% FBS and the cells were allowed to recover overnight. 24hr after the first transfection, cells were transfected again with miR-106b or negative control at a final concentration of 75 nM for 24hr. Luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega). Renilla luciferase activity was normalized with the corresponding firefly luciferase activity. Each transfection condition was performed in sextuplicate and experiments were repeated twice.
Immunocytochemistry and quantitative image analysis
Primary hippocampal neurons from E18–E19 Sprague Dawley rats were cultured at 150 cells/mm2 density. Neurons were cotransfected with GFP vector and scrambled negative control, or GFP vector and miR-106b (75 nM) at 14 day in vitro (DIV) by Lipofectamin 2000 (2 μg of DNA per well). Forty eight hours post- transfection (DIV 16), cells were fixed and immunostained with anti-ABCA1 antibody [AB.H10] (ab18180, Abcam). Images were collected using a Zeiss LSM510 confocal microscope. Confocal z-series image stacks encompassing entire neurons were analyzed by using MetaMorph® software (Molecular Devices, Sunnyvale, CA). To measure ABCA1 levels, dendrites from hippocampal neurons were carefully traced and surface fluorescence intensities were determined for the traced region using MetaMorph®. Each transfection condition was performed in triplicate. Experiments and quantitative analyses were independently repeated twice with two different batches of primary neurons (n=33–39).
Cholesterol Efflux Assays
Neuro2a cells were plated in 12-well plates (1 × 106 cells/well) and transfected with either miR-106 or scrambled negative control (Insight Genomics, Falls Church, VA) for 24hr. Cells were loaded with 0.5μCi/ml 3H-cholesterol (Perkin Elmer, Waltham, MA) for an additional 24hr. LXR agonist TO901317 (Cayman Chemical, Ann Arbor, MI) was treated at 1μM, where it is indicated. Cells were washed twice with PBS and incubated with 2% fatty-acid free BSA (FAFA, Sigma) in media in the presence of ACAT inhibitor (2 μM) for 2h prior to the addition of 50μg/ml human apoA-I (Meridian Life Science, Industrial Park Road, ME) in FAFA-media or 10% FBS with or without the indicated treatments. Supernatants were collected after 6h, radioactivity was counted and expressed as a percentage of total cell 3H-cholesterol content (total effluxed 3H-cholesterol+cell-associated 3H-cholesterol) (Rayner et al., 2010). Each assay was performed in triplicate and experiments were repeated three times.
Measurement of secreted Aβ levels
Neuro2a cells were plated at a density of 1.25 × 105 cell per well in a 24-well plate one day before transfection. Neuro2A cells were first transfected with 0.3 μg of pAG3-Swedish mutant (K670N/M671L) APPsw ORF without 3′ UTR for 8h and allowed to recover overnight. Then, miR-106b or a scrambled negative control was transfected into cells. Forty eight hours post-transfection, media were changed. 6h after media change, cells were collected by centrifugation at 1500 × g for 5 min after being washed in PBS. Media were collected on ice with EDTA containing protease inhibitor cocktail (Roche, Madison, WI). Cell debris in media was removed by centrifugation at 6000 × g for 10 min. In experiments with mouse ABCA1 (mABCA1) ORF plasmid without 3′ UTR, Neuro2A cells were first co-transfected with 0.3 μg of each hAPPsw and mABCA1 ORF construct and all following procedures were same as above. Cell lysates and media were applied for western blot analysis of APP, BACE1, CTFβ in cells, and Aβ in medium. The levels of APP and BACE1 were normalized with the actin level and the levels of CTFβ and Aβ were normalized with the cellular APP level. All experiments were performed in duplicate or triplicate and repeated independently three times.
Aβ clearance assays
Neuro2a cells were plated at a density of 1.25 × 105 cells per well in a 24-well plate a day before transfection. Neuro2A cells were transfected with miR-106b or scrambled negative control. Forty eight hours post-transfection, cells were incubated in fresh DMEM/10%FBS medium or DMEM/N2 medium containing 20 nM Aβ40 (American Peptide Company, Sunnyvale, CA) for 24hr. Aβ peptides were dissolved in neat trifluoroacetic acid, and then dried under an inert atmosphere of nitrogen. The peptide was washed in hexafluoroisopropanol twice, then evaporated using a nitrogen gas. The peptide is stored at −80 °C until use. The peptides were dissolved in dimethyl sulfoxide. In experiments with mABCA1 ORF plasmid without 3′ UTR, Neuro2A cells were first transfected with 0.3 μg of mABCA1 ORF construct for 8h and recovered overnight. Then, miR-106b or scrambled negative control was introduced into cells and following procedures were same as above. Media were used for Western blot analysis of Aβ. The levels of Aβ were normalized with total protein levels. All experiments were performed in duplicate or triplicate and repeated independently three times.
Western blot analysis
Cells were lysed in RIPA buffer (Millipore, Billerica, MA) with EDTA-free protease inhibitor cocktail (Roche) on ice for 30 min. The lysates were spun down at 18,000 × g for 15 min and the supernatants were collected. Protein concentration was determined by BCA protein assay (Thermo Fisher, Rockford, IL). Equal amounts of total protein for each lysate were separated on 4–15 % TGX™ (Tris-Glycine eXtended) gels (Bio-Rad, Hercules, CA) and transferred to 0.45 μm pore size PVDF membranes (Millipore). For Aβ detection, same volumes of media were run on 16.5% Tris-Tricine gel (Bio-Rad) and transferred to 0.2 μm pore size nitrocellulose membranes (Bio-Rad). Nitrocellulose membranes were boiled for 10 minutes in PBS, as previously reported (McGowan et al., 2005). All membranes were blocked with 4% non-fat dry milk in TBS/T (Tris buffered saline with 0.125% Tween-20). All blots were probed with mouse anti-ABCA1 antibody (HJ1, a kind gift from Dr. David M. Holtzman), rabbit anti-APP antibody (Invitrogen), rabbit anti-BACE1 antibody (D10E5, Cell Signaling Technology, Boston, MA), mouse anti-Aβ antibody (82E1, IBL International, Japan), or mouse anti-actin antibody (Sigma, St Louis, MO) at room temperature for 1h. After secondary antibody incubation, membranes were developed using Lumigen TMA-6 ECL detection kit (Lumigen INC, Southfield, MI).
Statistical analysis
To determine the statistical significance (*p<0.05, **p<0.01, ***p<0.001), we first tested whether our data sets passed the equal variance test (Levene Median test) and normality test (Kolmogorov-Smirnov test) (SigmaStat 3.5., San Jose, CA). After confirmation that the data did not violate the assumptions of parametric testing, a two-tailed Student’s t-test was used (GraphPad Prism 5, La Jolla, CA). All data are shown as mean ± standard error of the mean (SEM).
Results
Identification of miR-106b as a regulator of ABCA1 expression
ABCA1 (ABCA1) gene has an unusually long 3′ UTR (> 3.3 kilobases), raising a possibility that it might be regulated by post-transcriptional regulations, such as miRNA-mediated gene repression. To identify potential miRNAs that directly regulate ABCA1 gene expression, we utilized several miRNA target prediction algorithms, such as Targetscan (http://www.targetscan.org/), PicTar (http://pictar.mdc-berlin.de/), and miRanda (http://www.microrna.org/microrna/home.do) (Betel et al., 2008; Krek et al., 2005; Lewis et al., 2005). Given the high false-positive rates of individual prediction algorithm (Peter, 2010), we focused on miRNAs that are commonly predicted by all algorithms. Among several predicted candidates, we further narrowed down the candidates based on their relative binding locations within 3′ UTR and evolutionary sequence conservation among mammals. While a seed region (nucleotides 2 to 8 of miRNA) matching is often sufficient to confer mRNA recognition, we also analyzed whether there are 3′ supplementary pairing sites in miRNA. miR-106b was predicted to have a binding site at the near end of 3′ UTR of ABCA1 with an 8mer perfect match (an exact match to positions 2–8 of the mature miRNA followed by an adenosine nucleotide) (Fig. 1A) (Bartel, 2009). In addition, there were several supplementary pairing sites at the 3′ side of miR-106b. Furthermore, miR-106b seed region was well conserved among mammals, such as human, rat, and mouse (Fig. 1A).
Fig. 1. miR-106b suppresses endogenous ABCA1 expression by directly targeting 3′ UTR of ABCA1.

(A) Schematic diagram of ABCA1 mRNA containing conserved putative target site of miR-106b. The seed match in italic is indicated in the grey box. miR-106b binding region within 3′ UTR of ABCA1 is well conserved in human, rat, and mouse. (B) Decrease of endogenous ABCA1 levels by miR-106b in mouse neuroblastoma Neuro2a cells and human hepatocyte HepG2 cells. Cells were transfected with miR-106b or scrambled negative control. Forty eight hours post- transfection, cells were harvested for analyses. (C) ABCA1 protein levels were normalized by β-actin levels and quantified as a percentage of control. (D) Quantitative RT-PCR analysis of ABCA1 mRNA levels in Neuro2a cells. The levels of ABCA1 mRNA were not changed by miR-106b. GAPDH mRNA levels were used as a normalization control. (E) miR-106b suppressed expression of luciferase with full-length hABCA1 3′ UTR. Luciferase reporter construct used here contains the full-length 3′ UTR of hABCA1 at the downstream of luciferase. Luciferase reporter assay was done as described in the “Materials and Methods”. Data are shown as a percentage of negative control. Values are mean ± SEM. (*, p<0.05; **, p<0.01; ***, p<0.001 compared to the control).
To experimentally validate its effect on endogenous ABCA1 expression, we transfected synthetic miR-106b or scrambled negative control RNAs to Neuro2a mouse neuronal cells and HepG2 human hepatocytes. miR-106b significantly decreased ABCA1 protein levels in both mouse and human cell types (Fig. 1B). Approximately 45% and 40 % reductions in ABCA1 protein levels were observed in Neuro2a and HepG2 cells, respectively (Fig. 1C). Repression of gene expression by miRNAs could be due to translational repression or mRNA decay. Initially, it has been assumed that target protein reduction by miRNA is primarily through translational inhibition in animals. However, recent studies suggest that miRNA can also cause mRNA decay (Huntzinger and Izaurralde, 2011). To determine the relative contribution of two mechanisms, we analyzed ABCA1 mRNA levels after transfection of miR-106b and scrambled negative control RNAs. miR-106b had no effect on ABCA1 mRNA level (Fig. 1D). These data suggest that miR-106b may act by interfering translation of ABCA1 mRNA, rather than inducing mRNA decay.
However, we cannot exclude the possibility that miR-106b affects ABCA1 gene expression through an indirect consequence of targeting other gene. To determine whether miR-106b directly affects ABCA1 expression by targeting its 3′ UTR region, we performed a luciferase reporter assay with the full-length hABCA1 3′ UTR. First, we generated a reporter construct containing the entire 3′ UTR of hABCA1 at the downstream of luciferase. We co-transfected this construct with miR-106b or scrambled negative control sequence to Neuro2a cells. Compared to control, miR-106b significantly reduced luciferase activity (Fig. 1E). Collectively, our data suggest that miR-106b directly regulates ABCA1 expression by binding to the 3′ UTR of ABCA1 and repressing its translation.
Since the organization of membrane microdomains and kinetics of ABCA1 protein synthesis and degradation might be different between dividing, undifferentiated neuronal cell lines and matured primary neurons, we examined whether miR-106b can regulate ABCA1 expression in fully differentiated primary neurons. Rat primary hippocampal neurons at DIV14 were transfected with scrambled negative control or miR-106b along with GFP expressing plasmid. Compared to scrambled negative control (Fig. 2A), miR-106b dramatically decreased ABCA1 levels in primary hippocampal neurons (Fig. 2B). Quantitative analysis using MetaMorph® indicated approximately 60% reduction in ABCA1 protein levels by miR-106b (Fig. 2C). Taken together, our data demonstrate that miR-106b suppresses ABCA1 expression in cells derived from human, rat, and mouse.
Fig. 2. miR-106b suppresses endogenous ABCA1 expression in rat primary neurons.

Primary hippocampal neurons (DIV 14) were cotransfected with GFP vector and scrambled negative control (A), or GFP vector and 75 nM miR-106b (B). Forty eight hours post-transfection, cells were fixed and immunostained with anti-ABCA1 antibody. (C) The levels of ABCA1 on the dendrites were measured using MetaMorph® software. Data are shown as a percentage of negative control. Values are mean ± SEM. (***, p<0.001 compared to the control).
miR-106b decreases cellular cholesterol efflux
ABCA1 transports cellular cholesterol from cell surface to lipid-poor apolipoproteins, including apoA-I, generating high-density lipoproteins (Linsel-Nitschke and Tall, 2005). Given the significant reduction of ABCA1 protein levels by miR-106b (Fig. 1B), we hypothesized that miR-106b may impair cellular cholesterol efflux. To test this hypothesis, we transfected Neuro2a cells with miR-106b or scrambled negative control and then incubated the cells with radioactively labeled 3H-cholesterol. Since apoA-I is the most abundant apolipoproteins in the cerebrospinal fluid (Koch et al., 2001), we assessed the effect of miR-106b on cholesterol efflux using apoA-I as a cholesterol acceptor. miR-106b significantly reduced apoA-I-mediated cholesterol efflux under basal conditions (without an induction of ABCA1 gene expression by LXR agonist) (Fig. 3A).
Fig. 3. miR-106b decreases cellular cholesterol efflux.
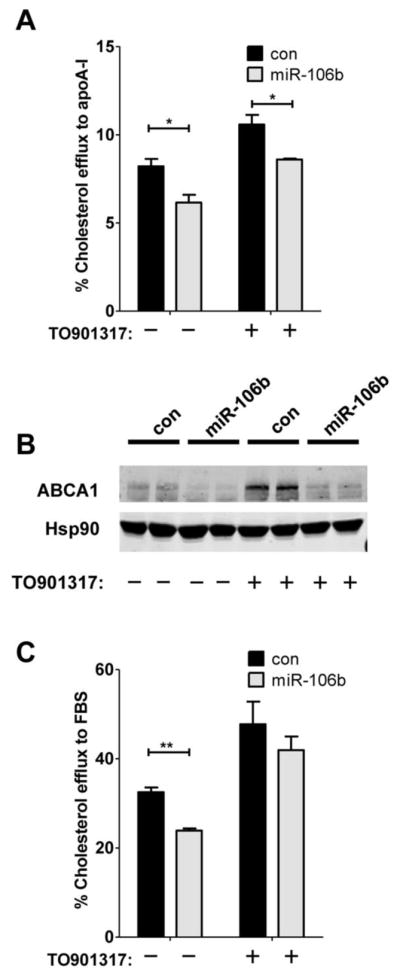
Neuro2a cells were transfected with miR-106b or scrambled negative control for 24hr and then incubated the cells with radioactively labeled 3H-cholesterol. After 24hr, cells were incubated with human apoA-I (A) or FBS (C) as a cholesterol acceptor with or without LXR agonist (TO901317), as described in the “Materials and Methods”. Data is shown as a percentage of total cellular 3H-cholesterol content (total effluxed 3H-cholesterol+cell-associated 3H-cholesterol). (B) ABCA1 protein levels were monitored by western blot analysis. While TO091317 treatment significantly increased ABCA1 protein levels compared with no treatment, miR-106b dramatically repressed TO901317-mediated ABCA1 induction.
Activation of LXR with its agonist is known to increase ABCA1 transcription and facilitate cholesterol efflux (Hirsch-Reinshagen and Wellington, 2007). Therefore, selective LXR agonists are currently being investigated as promising therapeutics for metabolic diseases and AD (Fan et al., 2009; Faulds et al., 2010). We tested whether miR-106b could still impair cholesterol efflux in the presence of LXR agonist (T0901317). Transfection of Neuro2a cells with miR-106b significantly attenuated cholesterol efflux to apoA-I (Fig. 3A). The functional effects of miR-106b on cholesterol efflux were consistent with the levels of ABCA1 protein (Fig. 3B). While activation of LXR considerably increased ABCA1 protein levels compared with no T0901317 treatment, miR-106b dramatically repressed LXR-mediated ABCA1 induction (Fig. 3B).
To investigate the influence of miR-106 on cholesterol efflux in the presence of several apolipoproteins, we measured the extent of cholesterol efflux using FBS as a cholesterol acceptor. FBS contains high levels of apoAs, apoBs, apoCs, apoE and other minor species of apolipoproteins. As reported previously (Marquart et al., 2010), FBS functioned as a very efficient cholesterol acceptor. Approximately four-fold increase in cholesterol efflux was observed with FBS (Fig. 3C) in the absence and presence of T0901317, compared with using apoA-I as an acceptor (Fig. 3A). miR-106b significantly reduced cholesterol efflux to FBS in the absence of LXR activation (Fig. 3C). However, FBS-mediated cholesterol efflux was not significantly suppressed by miR-106b in the presence of T0901317. Taken together, these data demonstrate that the reduction of ABCA1 by miR-106b is sufficient to impair cellular cholesterol efflux.
miR-106b increases levels of secreted Aβ
Previous in vitro and in vivo studies demonstrated that ABCA1 influences Aβ levels (Bu, 2009; Fan et al., 2009; Hirsch-Reinshagen and Wellington, 2007; Kim et al., 2009; Koldamova and Lefterov, 2007). Therefore, we hypothesized that miR-106b might increase Aβ level through repression of ABCA1 expression. To test this hypothesis, we transfected Neuro2a cells with APPsw along with miR-106b or scrambled negative control and measured Aβ levels in the media (Fig. 4A). miR-106b significantly increased the levels of secreted Aβ, compared to negative control (Fig. 4B). To gain insight into the potential mechanism by which miR-106b increases Aβ levels in the media, we analyzed APP, BACE1, and APP C-terminal fragment β (CTFβ) by western blot analysis (Fig. 4A). While there were no noticeable changes in the levels of cellular APP and BACE1 proteins, miR-106b significantly increased CTFβ, the cleavage product of APP by β-secretase. These data suggest that an increase of Aβ levels by miR-106b may be due to a facilitation of APP processing toward Aβ production.
Fig. 4. miR-106b increases levels of secreted Aβ.
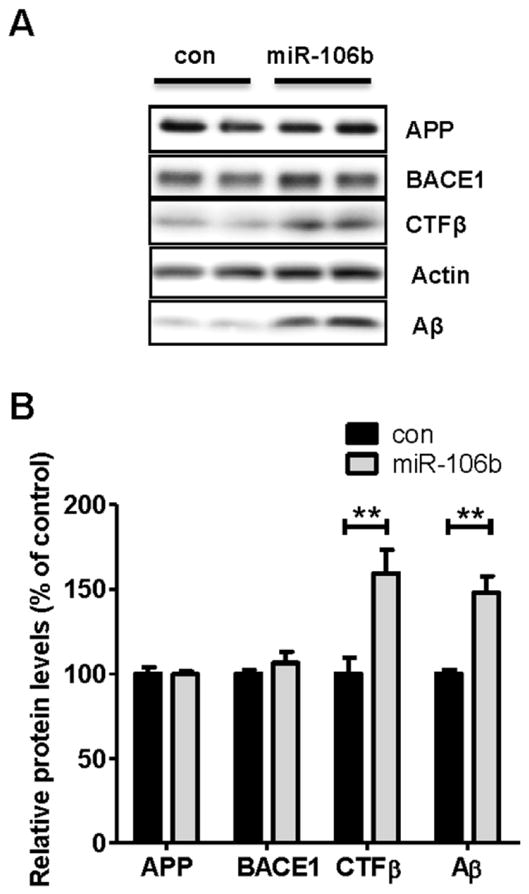
Neuro2a cells were first transfected with hAPPsw construct for 8h and allowed to recover overnight. miR-106b or scrambled negative control was transfected into cells. Forty eight hours post-transfection, cells were incubated in fresh medium. 6h after media change, cells and media were collected and analyzed. (A) Western blot analysis of APP, BACE1, CTFβ, and Aβ in Neuro2a cells transfected with miR-106b or negative control. miR-106b did not alter levels of APP and BACE1 but significantly increased the levels of cellular CTFβ and secreted Aβ. Aβ levels were measured in media and all other proteins were detected in cell lysates. (B) Quantification of APP, BACE1, CTFβ, and Aβ levels. The levels of APP and BACE1 were normalized with the actin levels. The levels of CTFβ and Aβ were normalized with the APP levels. Data is shown as a percentage of control and values are mean ± SEM. (**p<0.01 compared to the control).
Alteration in APP processing by miR-106b is rescued by ABCA1 ORF expression
Individual miRNAs can modulate the expression of many genes. While miR-106b significantly decreased ABCA1 protein levels (Fig. 1) and cholesterol efflux (Fig. 3), an increase of secreted Aβ (Fig. 4) could be due to a downregulation of other target genes, rather than ABCA1. We reasoned that, if miR-106b increases Aβ levels by repressing ABCA1 expression, this effect will be rescued by expression of miR-106b-resistant ABCA1. We first tested whether miR-106b-resistant ABCA1 could affect APP processing and subsequent Aβ production. Since miR-106b targets 3′ UTR of ABCA1, we generated a plasmid construct that expresses ABCA1 ORF without the 3′ UTR, thereby creating a miR-106b-resistant ABCA1. Expression of the miR-106b-resistant ABCA1 significantly decreased the levels of secreted Aβ in the media, compared with an empty vector transfection (Fig. 5A, 5B). CTFβ levels were also decreased by the expression of ABCA1 ORF. The levels of cellular APP and BACE1 proteins were not altered by ABCA1 ORF (Fig. 5A, 5B). To determine whether miR-106b-resistant ABCA1 can rescue miR-106’s effect on Aβ and CTFβ, we expressed ABCA1 ORF in the presence of scrambled negative control and miR-106b. Increases of Aβ and CTFβ levels by miR-106b were rescued by miR-106b-resistant ABCA1 ORF (Fig. 5C, 5D). These results suggest that the increase of Aβ by miR-106b is likely due to a repression of ABCA1, rather than other targets.
Fig. 5. Alterations in CTFβ and Aβ by miR-106b were rescued by overexpression of ABCA1 ORF.
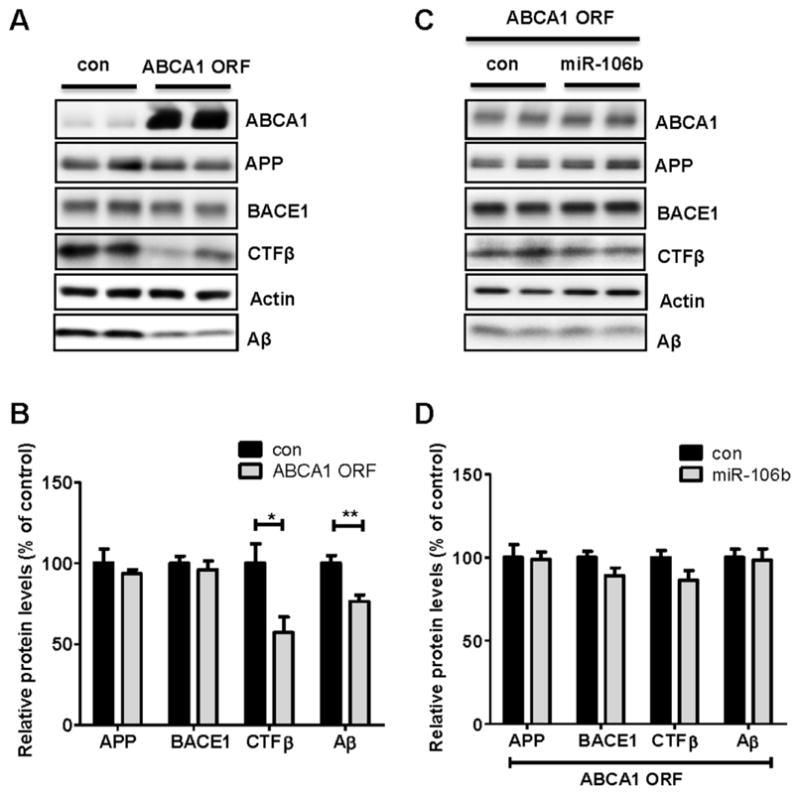
(A) ABCA1 decreased levels of cellular CTFβ and secreted Aβ. Neuro2a cells were first transfected with hAPPsw for 8h. Twenty four hours after the initial transfection, mABCA1 ORF construct or negative control vector was transfected into cells. After 48hr, cells were incubated in fresh medium. 6h later, cells and media were collected for analyses. Aβ levels in media and all other protein levels in cell lysates were analyzed by western blot. (B) Each protein level was quantified after normalization as a percentage of control. (C) Overexpression of ABCA1 ORF rescued the altered levels of CTFβ and secreted Aβ by miR-106b. Cells were first co-transfected with hAPPsw and mABCA1 ORF and then miR-106b or scrambled negative control was transfected into cells. Following procedures were same as above. (D) The levels of APP and BACE1 were normalized with the actin levels and the levels of CTFβ and Aβ were normalized with the APP levels. Values are mean ± SEM. (*, p<0.05; **, p<0.01 compared to the control).
Impairment of Aβ clearance by miR-106b is rescued by ABCA1 ORF expression
Increased levels of Aβ in the media could be due to increased Aβ production or an impairment of extracellular Aβ clearance. Given the miR-106b-mediated alteration in APP processing (Fig. 4), increased Aβ production appears to be responsible for the increase of extracellular Aβ levels by miR-106b. In order to assess the effect of miR-106b on Aβ clearance, we transfected Neuro2a cells with scrambled negative control or miR-106 and then incubated cells with exogenously added Aβ40. After 24hr of incubation, levels of Aβ40 remained in the media were measured by Western blot analysis. miR-106b significantly reduced Aβ clearance, compared to the negative control (Fig. 6A, 6B).
Fig. 6. Reduction of Aβ clearance by miR-106b is rescued by ABCA1 ORF expression.
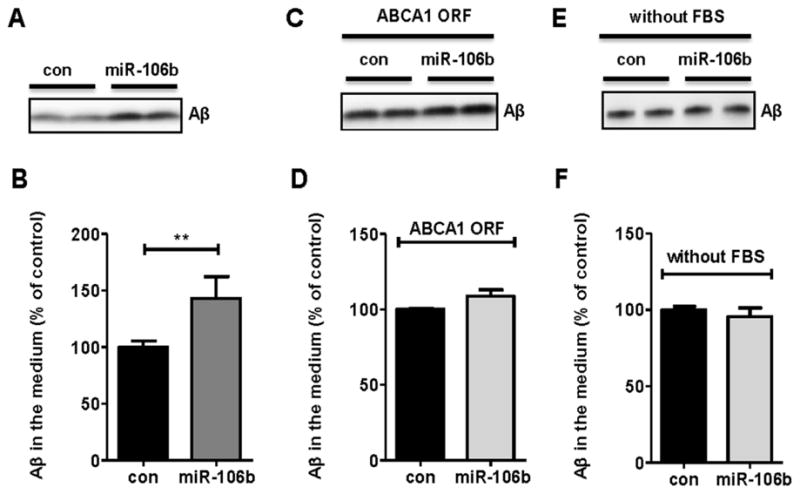
(A) Reduction of Aβ clearance by miR-106b. Neuro2a cells were transfected with miR-106b or scrambled negative control. After 48hr, cells were incubated in fresh DMEM/10% FBS media containing 20 nM Aβ40. Twenty four hours later, both cells and media were collected for analyses. Aβ levels in media were analyzed by western blot. (B) Aβ levels were quantified after normalization as a percentage of control. (C) Overexpression of ABCA1 ORF rescued impairment of Aβ clearance by miR-106b. Cells were first transfected with mABCA1 ORF for 8hr. Twenty four hours after transfection, scrambled negative control or miR-106b was transfected into cells. Following procedures were same as above. (D) The levels of Aβ were normalized with total protein levels. (E) No change of Aβ clearance by miR-106b under FBS free condition. Neuro2a cells were transfected with scrambled negative control or miR-106b. After 48hr, cells were incubated in fresh DMEM/N2 media containing 20 nM Aβ40. Aβ clearance was not altered by miR-106b in the media without FBS (F). Values represent mean ± SEM. (**, p<0.01 compared to the control).
Since the reduced Aβ clearance could be due to downregulation of other target genes, rather than ABCA1, we directly tested the role of ABCA1 in mediating Aβ clearance in the neuronal cells that overexpress miR-106b. If miR-106b impairs Aβ clearance by repressing other target gene, this effect will not be rescued by expression of miR-106b-resistant ABCA1 construct that lacks the 3′ UTR sequence (ABCA1 ORF). To test this possibility, we expressed miR-106b-resistant ABCA1 ORF in the presence of scrambled negative control or miR-106b. miR-106b-resistant ABCA1 ORF was able to rescue the impaired Aβ clearance (Fig. 6C, 6D). Interestingly, when we performed Aβ clearance assay without FBS in the cell culture media, miR-106b did not affect Aβ clearance (Fig. 6E, 6F). Collectively, our results suggest that the decrease of Aβ clearance by miR-106b is likely mediated through a repression of ABCA1 and ABCA1-mediated Aβ clearance depends on the presence of FBS.
Discussion
Accumulation of Aβ in the brain is hypothesized to trigger pathogenic cascades that eventually lead to AD (Hardy and Selkoe, 2002; Holtzman et al., 2011). Identification of a novel pathway that regulates Aβ metabolism may lead to better understanding of pathogenic mechanisms. Dysregulation of cholesterol metabolism has been implicated in the pathogenesis of several neurodegenerative diseases, including AD (Di Paolo and Kim, 2011; Hirsch-Reinshagen and Wellington, 2007). Many studies have suggested that cholesterol efflux and membrane lipids affect Aβ production by modulating APP trafficking as well as β- and γ-secretase activity (Osenkowski et al., 2008; Simons et al., 1998; Wray and Noble, 2009; Xiong et al., 2008).
Due to its role in the regulation of cholesterol efflux, ABCA1 has been extensively studied as a therapeutic target in AD. Most of studies demonstrated that ABCA1 induction decreases the levels of Aβ and CTFβ (Kim et al., 2007; Koldamova et al., 2003; Sun et al., 2003). These results appear to be consistent with our finding. In our current study, we demonstrated that ABCA1 overexpression decreased Aβ production whereas miR-106b-mediated ABCA1 repression increased Aβ levels in the media. In vivo studies using genetically modified mouse models support in vitro cell culture data. We and others demonstrated that modulation of ABCA1 affects Aβ levels and amyloid accumulation in vivo. ABCA1 deficiency leads to an increase in Aβ and amyloid deposition (Hirsch-Reinshagen et al., 2005; Koldamova et al., 2005; Wahrle et al., 2005) whereas ABCA1 overexpression ameliorated Aβ accumulation (Donkin et al., 2010; Wahrle et al., 2008).
Most previous studies suggested that the main mechanism by which ABCA1 affects extracellular Aβ level is through regulation of Aβ production, not through Aβ clearance (Di Paolo and Kim, 2011; Hirsch-Reinshagen and Wellington, 2007). Interestingly, we found that miR-106b impairs extracellular Aβ clearance by decreasing ABCA1 levels. In line with previous studies, we also failed to detect an increase of Aβ clearance by ABCA1 overexpression (data not shown). It appears that reduction of ABCA1 level below a certain endogenous level leads to an impairment of Aβ clearance, whereas an increase of ABCA1 level above endogenous level does not affect Aβ clearance in neuronal cells. The mechanism underlying alteration of Aβ clearance by miR-106b remains unclear. A recent in vitro study demonstrated that the presence of fully lipidated apoE is required for effective degradation of Aβ in microglia (Jiang et al., 2008; Terwel et al., 2011). This finding suggests that ABCA1 may affect Aβ clearance by facilitating apoE lipidation. In addition, uptake of Aβ through apolipoprotein receptors may be another Aβ clearance pathway (Bu, 2009; Fuentealba et al., 2010; Kanekiyo et al., 2011). Further studies are warranted to fully elucidate how miR-106b affects Aβ clearance.
Recent studies reported that ABCA1 expression was increased in the hippocampus of AD cases as well as an AD mouse model (Akram et al., 2010; Kim et al., 2010). Induction of ABCA1 in AD brains was mainly due to increased neuronal expression (Kim et al., 2010) and levels of ABCA1 highly correlated with severity of cognitive impairment (Akram et al., 2010). Given the reduction of amyloid deposition observed in our ABCA1 transgenic mice (Wahrle et al., 2008), it is tempting to speculate that the upregulation of ABCA1 in AD brains might be a compensatory attempt to decrease Aβ secretion and facilitate Aβ clearance through the increase of cellular cholesterol efflux. Interestingly, miR-106b levels were significantly decreased in brains of AD cases and AD mouse model (Hebert et al., 2009; Wang et al., 2011; Wang et al., 2009). Given our observation of miR-106-mediated ABCA1 repression, it may be interesting to investigate whether increase of ABCA1 is in part due to a reduction of miR-106b.
Previous studies have shown that miR-106b targets several genes implicated in cell cycles, such as cyclin-dependent kinase inhibitor 1A (p21), E2F transcription factor, TAF5-like RNA polymerase II, and phosphatase and tensin homolog (Ivanovska et al., 2008; Pichiorri et al., 2008; Poliseno et al., 2010; Trompeter et al., 2011). In addition, the miR-106b cluster is known to regulate stem cell proliferation and neuronal differentiation (Brett et al., 2011; Li et al., 2011; Peck and Schulze, 2011). Most interestingly, APP is another miR-106b target gene. In previous study, miR-106b significantly decreased APP levels in HeLa cervical cancer cell line (Hebert et al., 2009). When we analyzed the effect of miR-106b on mouse endogenous APP in Neuro2a cells, we also obtained similar data. miR-106b significantly decreased APP levels by ~40% (Scrambled negative control: 100±4.285 % versus miR-106b: 59.92±7.252 %, p=0.0031, n=4). In our study, we measured Aβ generated from a plasmid expressing APP ORF without 3′ UTR. Since we were primarily interested in the effect of miR-106b on ABCA1-mediated Aβ alteration, we used an APP construct without potential miRNA target sites (3′ UTR). Therefore, we were able to exclude a potentially confounding contribution by miRNA-mediated APP downregulation. While this experimental strategy allowed us to clearly understand the effect of miR-106b on Aβ through ABCA1 suppression, our study cannot address whether the reduction of miR-106b observed in AD cases is detrimental or beneficial for AD pathogenesis. Since miR-106b suppresses the expression of APP as well as ABCA1 and both proteins have opposite effects on Aβ levels, the in vivo significance of miR-106b modulation on Aβ accumulation and AD pathologies will need further study using animal models.
In the current study, we identified ABCA1 as a novel target of miR-106b. Suppression of ABCA1 expression by miR-106b impaired cellular cholesterol efflux and increased the levels of secreted Aβ. Modulation of Aβ levels by miR-106 was through a facilitation of APP processing toward Aβ production and an impaired Aβ clearance. Since expression of ABCA1 ORF rescued the phenotypes triggered by miR-106b, ABCA1 is likely a critical mediator of miR-106b function. In conclusion, our study identified a novel miRNA that regulates Aβ metabolism and suggests that miR-106b might have important implications to the pathogenesis of AD.
Acknowledgments
We are grateful to Peter T. Nelson (University of Kentucky) for technical advice, Jason Eriksen (University of Houston) for discussion, David Holtzman (Washington University) and Todd Golde (University of Florida) for valuable reagents. This work was supported by NIH grants P30NS069329 (J.K.), R01HL107953 (C. F.-H.), and AG034253(H.H).
Abbreviations
- ABCA1
ATP-binding cassette transporter A1
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- APP
amyloid precursor protein
- ApoA-I
apolipoprotein A1
- ApoE
apolipoprotein E
- BACE1
β-site APP-cleaving enzyme1
- CTFβ
C-terminal fragment β
- HDL
high-density lipoprotein
- LXR
liver X receptor
- miR
microRNA
- ORF
open reading frame
- PSEN
presenilin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akram A, et al. Increased expression of cholesterol transporter ABCA1 is highly correlated with severity of dementia in AD hippocampus. Brain Res. 2010;1318:167–77. doi: 10.1016/j.brainres.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D, et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008;36:D149–53. doi: 10.1093/nar/gkm995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissonneault V, et al. MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem. 2009;284:1971–81. doi: 10.1074/jbc.M807530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett JO, et al. The microRNA cluster miR-106b~25 regulates adult neural stem/progenitor cell proliferation and neuronal differentiation. Aging. 2011;3:108–24. doi: 10.18632/aging.100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE, et al. MicroRNA regulation of homeostatic synaptic plasticity. Proc Natl Acad Sci U S A. 2011;108:11650–5. doi: 10.1073/pnas.1017576108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–96. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donkin JJ, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–54. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, et al. Greasing the wheels of Abeta clearance in Alzheimer’s disease: the role of lipids and apolipoprotein E. Biofactors. 2009;35:239–48. doi: 10.1002/biof.37. [DOI] [PubMed] [Google Scholar]
- Faulds MH, et al. Molecular biology and functional genomics of liver X receptors (LXR) in relationship to metabolic diseases. Curr Opin Pharmacol. 2010;10:692–7. doi: 10.1016/j.coph.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Fuentealba RA, et al. Low-Density Lipoprotein Receptor-Related Protein 1 (LRP1) Mediates Neuronal Abeta42 Uptake and Lysosomal Trafficking. PLoS ONE. 2010;5:e11884. doi: 10.1371/journal.pone.0011884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease:progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009;32:199–206. doi: 10.1016/j.tins.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Hebert SS, et al. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol Dis. 2009;33:422–8. doi: 10.1016/j.nbd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Hebert SS, et al. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/{beta}-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–20. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, et al. Why lipids are important for Alzheimer disease? Mol. Cell Biochem. 2009;326:121–9. doi: 10.1007/s11010-008-0012-2. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, et al. The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem. 2005;280:43243–56. doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, Wellington CL. Cholesterol metabolism, apolipoprotein E, adenosine triphosphate-binding cassette transporters, and Alzheimer’s disease. Curr Opin Lipidol. 2007;18:325–32. doi: 10.1097/MOL.0b013e32813aeabf. [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V, et al. Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem. 2004;279:41197–207. doi: 10.1074/jbc.M407962200. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, et al. Alzheimer’s Disease: The Challenge of the Second Century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- Ivanovska I, et al. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol. 2008;28:2167–74. doi: 10.1128/MCB.01977-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, et al. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–93. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekiyo T, et al. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011;31:1644–51. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WS, et al. Increased ATP-binding cassette transporter A1 expression in Alzheimer’s disease hippocampal neurons. J Alzheimers Dis. 2010;21:193–205. doi: 10.3233/JAD-2010-100324. [DOI] [PubMed] [Google Scholar]
- Kim WS, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein-E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–61. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- Koch S, et al. Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res. 2001;42:1143–51. [PubMed] [Google Scholar]
- Koldamova R, Lefterov I. Role of LXR and ABCA1 in the Pathogenesis of Alzheimer’s Disease - Implications for a New Therapeutic Approach. Curr Alzheimer Res. 2007;4:171–178. doi: 10.2174/156720507780362227. [DOI] [PubMed] [Google Scholar]
- Koldamova R, et al. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280:43224–35. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- Koldamova RP, et al. 22R-hydroxycholesterol and 9-cis-retinoic acid induce ATP-binding cassette transporter A1 expression and cholesterol efflux in brain cells and decrease amyloid beta secretion. J Biol Chem. 2003;278:13244–13256. doi: 10.1074/jbc.M300044200. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Krichevsky AM. The Elegance of the MicroRNAs: A Neuronal Perspective. Neuron. 2005;47:779–82. doi: 10.1016/j.neuron.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Krek A, et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- Lewis BP, et al. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Li Z, et al. Small RNA-mediated regulation of iPS cell generation. The EMBO journal. 2011;30:823–34. doi: 10.1038/emboj.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193–205. doi: 10.1038/nrd1658. [DOI] [PubMed] [Google Scholar]
- Liu W, et al. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2010.04.034. in press. [DOI] [PubMed] [Google Scholar]
- Marquart TJ, et al. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–32. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–9. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, et al. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008;18:130–8. doi: 10.1111/j.1750-3639.2007.00120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osenkowski P, et al. Direct and Potent Regulation of {gamma}-Secretase by Its Lipid Microenvironment. J Biol Chem. 2008;283:22529–22540. doi: 10.1074/jbc.M801925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel N, et al. MicroRNAs can regulate human APP levels. Mol Neurodegener. 2008;3:10. doi: 10.1186/1750-1326-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck B, Schulze A. A role for the cancer-associated miR-106b~25 cluster in neuronal stem cells. Aging. 2011;3:329–31. doi: 10.18632/aging.100302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME. Targeting of mRNAs by multiple miRNAs: the next step. Oncogene. 2010;29:2161–4. doi: 10.1038/onc.2010.59. [DOI] [PubMed] [Google Scholar]
- Pichiorri F, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:12885–90. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliseno L, et al. Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29. doi: 10.1126/scisignal.2000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner KJ, et al. MiR-33 Contributes to the Regulation of Cholesterol Homeostasis. Science. 2010;328:1570–3. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, et al. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A. 1998;95:6460–4. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalheiser NR, Lugli G. microRNA regulation of synaptic plasticity. Neuromolecular Med. 2009;11:133–40. doi: 10.1007/s12017-009-8065-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, et al. Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J Biol Chem. 2003;278:27688–94. doi: 10.1074/jbc.M300760200. [DOI] [PubMed] [Google Scholar]
- Terwel D, et al. Critical Role of Astroglial Apolipoprotein E and Liver X Receptor-{alpha} Expression for Microglial A{beta} Phagocytosis. J Neurosci. 2011;31:7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompeter HI, et al. MicroRNAs MiR-17, MiR-20a, and MiR-106b act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS ONE. 2011;6:e16138. doi: 10.1371/journal.pone.0016138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardo E, et al. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J Biol Chem. 2010;285:18344–51. doi: 10.1074/jbc.M110.112664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, et al. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–42. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- Wahrle SE, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, et al. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–93. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wang W-X, et al. The Expression of MicroRNA miR-107 Decreases Early in Alzheimer’s Disease and May Accelerate Disease Progression through Regulation of {beta}-Site Amyloid Precursor Protein-Cleaving Enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, et al. Patterns of microRNA expression in normal and early Alzheimer’s disease human temporal cortex: white matter versus gray matter. Acta Neuropathol. 2011;121:193–205. doi: 10.1007/s00401-010-0756-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. miR-34a, a microRNA up-regulated in a double transgenic mouse model of Alzheimer’s disease, inhibits bcl2 translation. Brain Res Bull. 2009;80:268–73. doi: 10.1016/j.brainresbull.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Wray S, Noble W. Linking amyloid and tau pathology in Alzheimer’s disease: the role of membrane cholesterol in Abeta-mediated tau toxicity. J Neurosci. 2009;29:9665–7. doi: 10.1523/JNEUROSCI.2234-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, et al. Cholesterol retention in Alzheimer’s brain is responsible for high [beta]- and [gamma]-secretase activities and A[beta] production. Neurobiol Dis. 2008;29:422–437. doi: 10.1016/j.nbd.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]